

Abstract
There is growing interest in the application of molecular profiling, including sequencing, genotyping, and/or mRNA expression profiling, to the analysis of patient tumors with the objective of applying these data to inform therapeutic choices for patients with advanced cancers. Multiple clinical trials that are attempting to validate this personalized or precision medicine approach are in various stages of development and execution. Although preliminary data from some of these efforts have fueled excitement about the value and utility of these studies, their execution has also provoked many questions about the best way to approach complicating factors such as tumor heterogeneity and the choice of which genetic mutations to target. This commentary highlights some of the challenges confronting the clinical application of molecular tumor profiling and the various trial designs being utilized to address these challenges. Randomized trials that rigorously test patient response to molecularly targeted agents assigned based on the presence of a defined set of mutations in putative cancer-driving pathways are required to address some of the current challenges and to identify patients likely to benefit from this approach.
Tumors carry genetic mutations that confer growth and/or survival advantages, making such mutations attractive targets for the development of anticancer therapies. However, tumor cells can carry hundreds of mutations in numerous molecular pathways, making it difficult to assign clinical significance to a particular mutation that can have no impact on or confer sensitivity or resistance to a particular therapeutic modality. Genetic variations in the host may also influence drug efficacy or toxicity by affecting exposure to active drug metabolites (1–3).
The development of agents targeted to molecular aberrations in cancer cells, so-called molecularly targeted agents, has evolved in concert with efforts to perform genetic sequencing for the identification of mutations that could guide selection of therapies more likely to be active. Recently, the application of molecular profiling (MP) (including sequencing, genotyping, and/or mRNA expression profiling) in clinical trials has been validated, in part, by the enhanced efficacy of some targeted agents for specific subsets of molecularly profiled patients with refractory solid tumors (4,5). Certain genetic mutations are considered “druggable” based on their growth-promoting potential and the availability of agents that target them; the underlying hypothesis is that assigning treatment based on the presence of specific targets in tumors will provide improved clinical benefit over current chemotherapy/radiotherapy-based medical practice. However, apart from certain notable successes, such as BRAF/MEK inhibitors for patients with melanoma-carrying BRAFV600E mutations (6), treatment with agents that inhibit the growth-enhancing properties of genetic abnormalities in tumors has not been routinely demonstrated to result in clinical benefit (7,8). Reasons for this could include suboptimal modulation of the targeted pathway, upregulation of compensatory pathways, or lack of sufficient understanding of the complex tumor biology needed to assign clinical significance to such abnormalities, such as the roles of passenger vs driver mutations. Tumor heterogeneity, including intratumoral, between primary and sites of metastatic disease and within different metastatic lesions also contributes to the difficulties in successfully treating advanced tumors (9).
On the other hand, there is emerging evidence that mutation-specific T-cell responses can occur in patients with solid tumors, which has implications for designing immune-based therapeutic approaches as well as incorporating advanced MP technologies into patient selection for specific immune therapies such as adoptive transfer of tumor-infiltrating lymphocytes (10). Furthermore, clinical utilization of MP data requires analytical validation of a laboratory assay and the ability to perform the analysis within a clinically relevant timeframe (such as two to three weeks) (4,5,7). Eventual application of the assay in clinical trials will establish its clinical validity (the ability of a test to accurately measure or predict a particular clinical attribute or outcome) and utility (the ability of a test to reliably define an attribute or diagnosis that affects therapeutic options) such that it will provide clinically meaningful results.
Several trial designs have been employed to address the efficacy of MP-assigned targeted agents (Table 1 and Figure 1) (11). Basket trials evaluate the effect of a single drug on a single mutation in a variety of cancers, such as vemurafenib in BRAFV600E-positive cancers (NCT01524978) (12). Umbrella trials examine the effect of different drugs on different mutations either in a single cancer, such as the Biomarker-Integrated Approaches of Targeted Therapy for Lung Cancer Elimination trial for non–small cell lung cancer (13), or in a variety of tumor types (14). Studies that interrogate “exceptional” responders aim to determine the underlying aberration that explains the unusual degree or duration of clinical benefit derived from an otherwise relatively ineffective treatment (15).
Table 1.
Characteristics of the three types of molecular profiling trials*
| Basket or bucket trials | Umbrella trials | Exceptional responder trials | |
|---|---|---|---|
| Histology/drug | Variety of cancer types Single drug targeting a single mutation |
Single cancer or variety of cancer types Multiple drugs targeting multiple mutations |
Any cancer type and drug where a patient had an unusually robust clinical benefit |
| Advantages | Access to study drug for rare cancers with a given mutation Evaluate targeting a given mutation in a variety of cancers Takes advantage of specific screening strategies for certain cancers May demonstrate clinical activity in patients carrying specific mutations which can inform future trials May facilitate FDA approval of biomarker-drug combinations |
Evaluates variety of mutations and drugs in the context of one trial Provides access to study drug for rare cancers with a variety of mutations May provide hints of clinical activity in patients carrying specific mutations which can inform future trials Screens large numbers of patients for multiple targets, reducing the screen failure rate May facilitate FDA approval of biomarker-drug combinations Adaptive designs |
Likelihood of finding a molecular characteristic that could account for the response Informs patient selection for future trials |
| Disadvantages | Evaluates only one drug/ mutation pair at a time | Unable to draw definitive conclusions for a given drug/mutation pair in trials enrolling patients with a variety of cancers Large patient numbers, resource intense |
Molecular characteristics linked to clinical activity must be tested subsequently in a larger number of patients before drawing definitive conclusions Need access to broad array of testing platforms to evaluate potential characteristics that may account for the response |
*FDA = US Food and Drug Administration.
Figure 1.
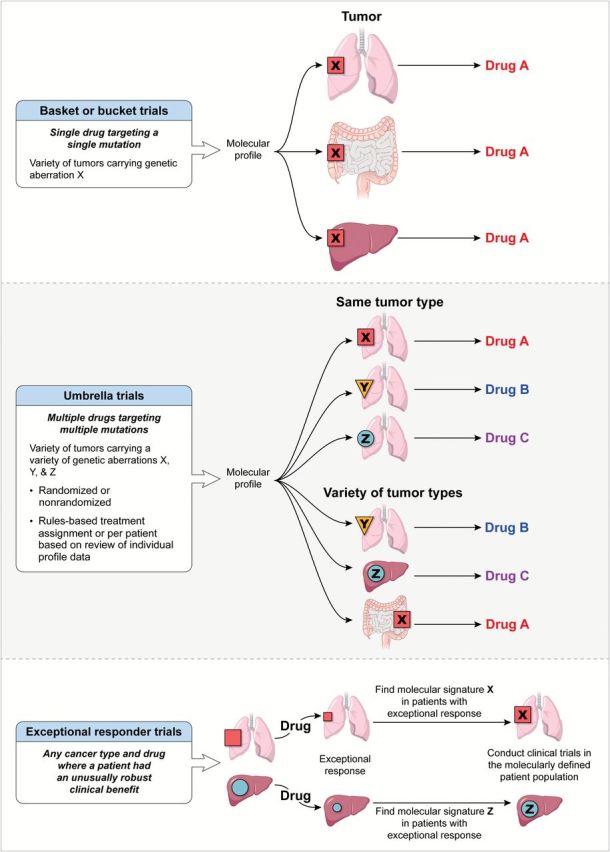
Clinical trial designs utilizing molecular profiling.
Umbrella trials can be divided based on trial design into randomized (eg, the National Cancer Institute Molecular Profiling Based Assignment of Cancer Therapy trial [NCI-MPACT; NCT01827384] or nonrandomized trials (such as the NCI Molecular Analysis for Therapy Choice [MATCH] trial and the WINTHER trial [NCT01856296]). Randomized trials compare the effectiveness of treatment assignments based on the results of MP to a control arm in which treatment may be either assigned by physician choice (eg, the SHIVA trial [NCT01771458]) or picked randomly from a list of treatment choices (eg, NCI-MPACT). Umbrella trials can also be divided into two types based on the algorithm utilized for treatment assignment. In one type, treatment decisions are based on assessment of MP data from individual patients by the treating physician or a multidisciplinary group of experts convened for such a purpose, with ad hoc assignment of treatments from either approved agents or ongoing trials at that institution, or from a list of treatments that are included in the protocol. Multidisciplinary tumor boards that are convened to analyze individual patient MP data and determine treatment assignments are usually composed of individuals with expertise in such disciplines as clinical oncology (including investigators from early drug development and disease-specific groups), genomics, experts in signal transduction pathways, genetic counselors, bioethicists, pathologists, bioinformatics experts, and biostatisticians. In the case of individual patient review for assignment of therapy, tumor boards offer the advantage of individualizing treatment decisions based on their unique analysis of a given patient’s tumor. Information from the trial and the literature can inform such decisions. However, questions such as how to “validate” the performance of such a tumor board, whether decisions are made using the same general rules on different days, and assignment bias from the availability of appropriate agents and other parameters that affect the eventual determination of clinical benefit, such as performance status, age, and end-organ function, may remain.
The second type of umbrella trial utilizes a rules-based approach for treatment assignment. This offers the advantage of predefining treatment based on the presence of a molecular aberration, ensures availability of agents within the context of a trial, and negates assignment bias because all patients with a predefined actionable mutation of interest (aMOI) are assigned a given treatment with an intent-to-treat analysis to determine benefit. For these trials, mutations of interest are deemed actionable, and concordant therapy is determined while the study is being designed, as in the NCI-MPACT trial. Once accruing patients, data from the study and from the literature can be reviewed and the rules revised; the updated treatment assignment rules then apply to all patients subsequently accrued.
Fundamental issues to address in all clinical trials that use MP data to assign therapy should include assigning clinical significance to a molecular aberration, including determining whether a given aberration is a driver or a passenger mutation. Trials, such as the WINTHER trial use genetic sequencing as well as gene expression to guide patient selection, further adding to the complexity of data interpretation and assignment of clinical significance. Another relevant issue is the percentage of cells in a given tumor sample that would be required to demonstrate the biological significance of the genetic mutation (eg, the majority or a small fraction, such as for cancer “stem cells”). Additionally, how many biopsies need to be obtained and, in cases of low tumor purity, should techniques such as micro- or macrodissection be used to separate tumor from the surrounding healthy tissue and stroma? Finally, if more than one aberration is present, in what sequence should they be targeted, and should combinations of therapy (with appropriate agents at safe doses) be considered?
Determining whether assigning treatment based on MP provides superior clinical benefit requires accounting for both the patient population and type of mutation, eg, patients with mutations known to be sensitive to a targeted therapy, such as treating patients with BRAF V600E–mutated melanoma with vemurafenib vs those with the same mutation in a previously unexplored disease context. Clinical study designs must therefore clearly define the question being asked, the patient population, mutations of interest, and the agents presumed to work on or downstream of the given mutations, as in the recently launched Lung-MAP (S1400) trial, a biomarker-driven multiarm master phase II/III trial in squamous cell lung cancer (NCT02154490) (16).
Choosing valid clinical endpoints for a study is also crucial. In single-arm, nonrandomized studies, time to tumor progression (TTP) or progression-free survival (PFS) for a given patient can be compared with TTP/PFS from prior therapy such that each patient is his/her own control, as in the Molecular Screening for Cancer Treatment Optimization trial (MOSCATO 01) (NCT01566019) or the WINTHER trial. For TTP/PFS to be a valid endpoint, however, it must be defined using the same modality and at the same predefined intervals as the previous therapy, something often not specified (14,17,18). Response rates for single-arm trials or comparing TTP or PFS between arms in randomized trials may provide better endpoints. Because MP characteristics may be prognostic of better or worse outcome (independent of treatment), the use of historical controls, not restricted by such MP characteristics, may be problematic. The NCI-MPACT study is an example of a randomized trial comparing the response rate and four-month PFS following treatment with agents chosen based on the presence of specific mutations in patient tumors (Arm A) to the treatment with agents chosen from the complementary set of agents not identified to work on the mutations of interest (Arm B) (Figure 2). If the results demonstrate a benefit for patients randomly assigned to Arm A, this trial would provide definitive data in favor of the application of genetic sequencing in early phase trials. Absence of difference between Arms A and B could indicate a lack of benefit of the application of genetic sequencing for the chosen mutations, or that ineffective agents were selected, or that the selected mutations are not driving cancer progression. To address concern about the risk of selecting ineffective agents, all the drugs chosen for this trial modulate their purported targets and have at least a phase II dose established.
Figure 2.
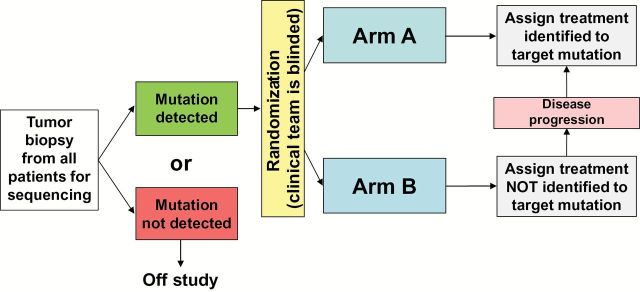
National Cancer Institute–Molecular Profiling based Assignment of Cancer Therapy (NCI-MPACT) study design. This trial requires a fresh tumor biopsy to determine the presence or absence of one of the predefined actionable mutations of interest (aMOIs) for study eligibility. Patients with one of the specified aMOIs are randomized 2:1 into Arm A (treatment regimen prospectively identified to target the identified mutation or pathway) or Arm B (treatment regimen assigned from the complementary set not prospectively identified to target one of their mutations). Pathways and drugs: mutations in the RAS/RAF pathway: trametinib (MEK inhibitor); mutations in the PI3K pathway: everolimus (mTOR inhibitor); mutations in DNA repair genes: either veliparib (PARP inhibitor) with temozolomide or carboplatin with AZD-1775 (wee1 inhibitor). To prevent assignment bias, treating physicians and staff are blinded to the sequencing data during the course of the initial therapy. A total of 180 evaluable patients (defined as patients who receive treatment on study) will be needed to discriminate between tumor response rates of 20% vs 5% and the comparison of progression-free survival (PFS) will have 90% power to detect an overall increase of 80% in median PFS by means of the log-rank test conducted at the one-sided .01 significance level.
One of the concerns in determining clinical benefit for a given treatment is that physician bias will affect assessment. Double-blind, randomized clinical trials could address this concern; however, such trials should be carefully planned taking patient acceptability and expectations into consideration. Justification for blinding the study needs to be carefully explained to potential patients during the informed consent process. A further consideration is that patients as well as their treating physicians may need to be blinded to the MP results prior to disease progression, to avoid preferential drop-out on the control arm and protect the randomization (as is done in the NCI-MPACT study). A plan for eventually sharing the MP data with the patient, discussing the results, and providing genetic counseling if necessary should be formulated and described in the informed consent documents. It is essential that processes for maintaining patient confidentiality and handling incidental findings be clearly defined prior to study initiation (19). Bioethicists, geneticists, patient advocates, and clinicians need to engage in ongoing discussions regarding their obligations to provide continued information and counseling based on evolving knowledge. Assigning treatment based on MP data is still investigational in most settings, raising concerns about performing tumor biopsies and the risks associated with the procedure. In addition, sampling a small piece of tissue at one time point limits the ability to address tumor heterogeneity and does not allow access to longitudinal samples to study alterations over time, which likely contribute to the evolution of drug resistance. Next-generation sequencing of circulating tumor cells and circulating tumor DNA has shown promise in clinical trials and could provide a relatively noninvasive way to access tumor cells/DNA over multiple time points in the course of the disease (20).
Table 2.
Selected ongoing trials using genetic sequencing to assign therapy to patients with refractory cancers*
| Trial name | Mutation/target |
|---|---|
| Basket trials | |
| Phase 1b trial of BGJ398/BYL719 in solid tumors (NCT01928459) |
Advanced or metastatic solid tumors expressing PIK3CA mutations (with or without FGFR1-3 alterations) Randomized: No |
| A study of vemurafenib in patients with BRAF V600 mutation-positive cancers (NCT01524978) | Solid tumors and multiple myelomas (except melanoma and papillary thyroid cancer) carrying BRAF V600 mutations Randomized: No |
| Umbrella trials – individual review | |
| Molecular profiling protocol (NCT00530192) |
Metastatic, refractory cancer Randomized: No |
| Molecular Screening for Cancer Treatment Optimization (MOSCATO) 01 (NCT01566019) |
Advanced, refractory solid tumors Randomized: No |
| Umbrella trials – rules-based | |
| Biomarker-Integrated Approaches of Targeted Therapy for Lung Cancer Elimination (BATTLE) (NCT00409968, NCT00411671, NCT00411632, NCT00410059, and NCT00410189) | Non–small cell lung cancer carrying one or more of the following: EGFR mutation or copy number change, KRAS/BRAF mutation, change in VEGF/VEGFR-2 or RXRs/Cyclin D1 expression, or change in CCND1 copy number Randomized: Yes |
| A randomized phase II trial comparing therapy based on tumor molecular profiling versus conventional therapy in patients with refractory cancer – SHIVA (NCT01771458) | Recurrent or metastatic solid tumors with abnormalities in KIT, ABL, or RET; AKT, mTORC1/2, PTEN, or PI3K; BRAF V600E; PDGFRA/B or FLT-3; EGFR; HER-2; SRC, EPHA2, LCK, or YES; or AR
Randomized: Yes |
| Lung-MAP: S1400 biomarker-targeted second-line therapy in treating patients with recurrent stage IIIB-IV squamous cell lung cancer (NCT02154490) |
Advanced squamous cell lung cancer not responding to routine therapies with mutations in PI3KCA, CDK4, CDK6, CCND1, CCND2, CCND3, FGFR1, FGFR2, FGFR3, or HGF/c-MET
Randomized: Yes |
| NCI-MPACT: Molecular Profiling-Based Assignment of Cancer Therapy for Patients With Advanced Solid Tumors (NCT01827384) |
Solid tumors that have progressed following at least one line of standard therapy and contain one or more of the predefined actionable mutations in the DNA repair, PI3K, or RAS/RAF genetic pathways Randomized: Yes |
| National Cancer Institute Molecular Analysis for Therapy Choice (NCI-MATCH) (not yet open) |
Solid tumors and lymphomas that have progressed following at least one line of standard therapy with actionable mutations in one or more of the over 300 predetermined genes Randomized: Yes |
| Exceptional responders trials | |
| Molecular profiling in tissue samples from patients with cancer who are exceptional responders to treatment (NCT02243592) | Retrospective analysis of tumor tissue from patients with complete responses (or 6+ month partial responses) to an agent found to be effective for less than 10% of the study population with the patient’s tumor type Randomized: No |
*NCI = National Cancer Institute.
Conclusions
Providing therapy targeted to cure an individual patient remains the goal of oncologists worldwide. Matching molecularly targeted agents to specific mutations has resulted in dramatic responses in certain tumor types; however, these successes have been limited. Advances in technology, greater understanding of cancer biology, and the ability to design agents that target specific pathways is driving the increasing application of MP in clinical trials. In addition to genetic sequencing, epigenetic changes can substantially affect various cell growth and survival pathways important for carcinogenesis and propagation of established cancers. The ability to obtain a more comprehensive analysis of gene expression and its downstream effects in proteins could also help identify drug targets and may lead to more effective therapeutic strategies. However, this will require development of validated multianalyte assays that can be incorporated into trials to assess the utility of such an approach.
The availability of laboratory resources to perform MP and the associated costs represent a barrier to the widespread application of MP technologies for treatment assignment, especially in situations where the mutation of interest is infrequently detected. Because the incidence of various mutations can be low, umbrella trials that analyze a panel of molecular abnormalities and assign various types of therapies depending on the MP data represent the most efficient and cost-effective approach to identify and treat patients. There will always be challenges inherent in this approach, but the application of new technologies with improved drugs in conjunction with rigorous, randomized clinical trial designs is likely to be the most effective way to improve cancer treatment.
Funding
This work was supported by federal funds from the National Cancer Institute, National Institutes of Health , under Contracts No. HHSN261200800001E.
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government. We thank Dr. Andrea Regier Voth, Leidos Biomedical Research, Inc., for editorial assistance in the preparation of this manuscript.
References
- 1. Province MA, Goetz MP, Brauch H, et al. CYP2D6 Genotype and Adjuvant Tamoxifen: Meta-Analysis of Heterogeneous Study Populations. Clin Pharmacol Ther. 2014;95(2):216–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ramchandani RP, Wang Y, Booth BP, et al. The role of SN-38 exposure, UGT1A1*28 polymorphism, and baseline bilirubin level in predicting severe irinotecan toxicity. J Clin Pharmacol. 2007;47(1):78–86. [DOI] [PubMed] [Google Scholar]
- 3. O’Donnell PH, Ratain MJ. Germline pharmacogenomics in oncology: decoding the patient for targeting therapy. Mol Oncol. 2012;6(2):251–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Simon R, Roychowdhury S. Implementing personalized cancer genomics in clinical trials. Nat Rev Drug Discov. 2013;12(5):358–369. [DOI] [PubMed] [Google Scholar]
- 5. Dienstmann R, Rodon J, Barretina J, et al. Genomic Medicine Frontier in Human Solid Tumors: Prospects and Challenges. J Clin Oncol. 2013;31(15):1874–1884. [DOI] [PubMed] [Google Scholar]
- 6. Jang S, Atkins MB. Which drug, and when, for patients with BRAF-mutant melanoma? Lancet Oncol. 2013;14(2):e60–e69. [DOI] [PubMed] [Google Scholar]
- 7. Arnedos M, Vielh P, Soria J-C, et al. The genetic complexity of common cancers and the promise of personalized medicine: is there any hope? J Pathol. 2014;232(2):274–282. [DOI] [PubMed] [Google Scholar]
- 8. Evans JP, Meslin EM, Marteau TM, et al. Deflating the Genomic Bubble. Science. 2011;331(6019):861–862. [DOI] [PubMed] [Google Scholar]
- 9. Gerlinger M, Rowan AJ, Horswell S, et al. Intratumor Heterogeneity and Branched Evolution Revealed by Multiregion Sequencing. N Engl J Med. 2012;366(10):883–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Miamen AG, Gustafson MP, Roberts LR. Rethinking cancer immunotherapy: Using advanced cancer genetics in immune-mediated eradication of gastrointestinal cancers. Hepatology. 2014;60(6):2121–2124. [DOI] [PubMed] [Google Scholar]
- 11. Bedard PL, Hansen AR, Ratain MJ, et al. Tumour heterogeneity in the clinic. Nature. 2013;501(7467):355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hyman DM, Blay J-Y, Chau I, et al. VE-BASKET, a first-in-kind, phase II, histology-independent “basket” study of vemurafenib (VEM) in nonmelanoma solid tumors harboring BRAF V600 mutations (V600m). J Clin Oncol. 2014;32(suppl 5):abstr 2533. [Google Scholar]
- 13. Kim ES, Herbst RS, Wistuba II, et al. The BATTLE trial: personalizing therapy for lung cancer. Cancer Discov. 2011;1(1):44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Von Hoff DD, Stephenson JJ, Rosen P, et al. Pilot Study Using Molecular Profiling of Patients’ Tumors to Find Potential Targets and Select Treatments for Their Refractory Cancers. J Clin Oncol. 2010;28(33):4877–4883. [DOI] [PubMed] [Google Scholar]
- 15. Iyer G, Hanrahan AJ, Milowsky MI, et al. Genome Sequencing Identifies a Basis for Everolimus Sensitivity. Science. 2012;338(6104):221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lung-MAP: S1400 Biomarker-Targeted Second-Line Therapy in Treating Patients With Recurrent Stage IIIB-IV Squamous Cell Lung Cancer (NCT02154490). http://www.lung-map.org/. Accessed August 5, 2014.
- 17. Tsimberidou AM, Iskander NG, Hong DS, et al. Personalized medicine in a phase I clinical trials program: the MD Anderson Cancer Center initiative. Clin Cancer Res. 2012;18(22):6373–6383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Doroshow JH. Selecting Systemic Cancer Therapy One Patient at a Time: Is There a Role for Molecular Profiling of Individual Patients With Advanced Solid Tumors? J Clin Oncol. 2010;28(33):4869–4871. [DOI] [PubMed] [Google Scholar]
- 19. Dal-Ré R, Katsanis N, Katsanis S, et al. Managing Incidental Genomic Findings in Clinical Trials: Fulfillment of the Principle of Justice. PLoS Med. 2014;11(1):e1001584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ignatiadis M, Dawson SJ. Circulating Tumor Cells and Circulating tumor DNA for precision medicine: Dream or reality? Ann Oncol. 2014;25(12):2304–2313. [DOI] [PubMed] [Google Scholar]