

Abstract
Previous studies have suggested that polymorphisms in CASP8 on chromosome 2 are associated with breast cancer risk. To clarify the role of CASP8 in breast cancer susceptibility, we carried out dense genotyping of this region in the Breast Cancer Association Consortium (BCAC). Single-nucleotide polymorphisms (SNPs) spanning a 1 Mb region around CASP8 were genotyped in 46 450 breast cancer cases and 42 600 controls of European origin from 41 studies participating in the BCAC as part of a custom genotyping array experiment (iCOGS). Missing genotypes and SNPs were imputed and, after quality exclusions, 501 typed and 1232 imputed SNPs were included in logistic regression models adjusting for study and ancestry principal components. The SNPs retained in the final model were investigated further in data from nine genome-wide association studies (GWAS) comprising in total 10 052 case and 12 575 control subjects. The most significant association signal observed in European subjects was for the imputed intronic SNP rs1830298 in ALS2CR12 (telomeric to CASP8), with per allele odds ratio and 95% confidence interval [OR (95% confidence interval, CI)] for the minor allele of 1.05 (1.03–1.07), P = 1 × 10−5. Three additional independent signals from intronic SNPs were identified, in CASP8 (rs36043647), ALS2CR11 (rs59278883) and CFLAR (rs7558475). The association with rs1830298 was replicated in the imputed results from the combined GWAS (P = 3 × 10−6), yielding a combined OR (95% CI) of 1.06 (1.04–1.08), P = 1 × 10−9. Analyses of gene expression associations in peripheral blood and normal breast tissue indicate that CASP8 might be the target gene, suggesting a mechanism involving apoptosis.
INTRODUCTION
Breast cancer is a complex disease with high, moderate and low penetrance germ-line variants involved in its etiology (1). In recent years, ∼80 low penetrance breast cancer alleles have been identified, with modest odds ratios, ranging from 1.05 to 1.4, and together accounting for around 15% of familial breast cancer risk (2,3). It is likely that there are many more loci with even smaller effect sizes that remain to be identified, accounting for a further 14–15% of familial risk (2). One of the first low penetrance breast cancer variant associations to be convincingly replicated by large case–control studies was the single-nucleotide polymorphism (SNP) rs1045485 encoding the missense alteration D302H in the caspase 8 apoptosis-related cysteine peptidase (CASP8) gene at chromosome region 2q33 (4,5). This association was first identified by a candidate gene study and replicated in 2007 by the Breast Cancer Association Consortium (BCAC), in a study of >17 000 cases and 16 000 controls (4,5). The minor C allele, common in Europeans and rare in Asians, was found to be associated with a 10% reduction in risk of breast cancer (5). However, further fine-mapping studies have shown that other variants in the region are associated with an increased risk of breast cancer, and in the recent large-scale genotyping study carried out by the BCAC as part of the COGS (Collaborative Oncology Gene-Environment Study), rs1045485 showed only weak evidence of association with breast cancer risk (2,6,7). In addition, in 2010 a UK genome-wide association study (GWAS) of 3659 cases and 4897 controls found suggestive evidence of association [OR (95% confidence interval, CI) 1.14 (1.06–1.22); P = 1.5 × 10−4] with an independent variant in the region; rs10931936, a CASP8 intronic SNP, that is only weakly correlated with rs1045485 (r2 = 0.083) (8).
In order to clarify the breast cancer risk association(s) at this locus, we have analyzed 501 SNPs across a 1 Mb region surrounding CASP8, for 89 050 women, as part of a custom-designed Illumina genotyping chip—the iCOGS array. We present here the results of this fine-mapping analysis, together with a meta-analysis across iCOGS and the combined data from nine breast cancer GWAS, followed by an examination of associations between the key SNPs and RNA expression levels.
RESULTS
Breast cancer risk associations in the CASP8 region on chromosome 2
A summary of the breast cancer risk associations of 1733 typed and imputed SNPs across a 1 Mb region surrounding CASP8, based on the iCOGS European data, is shown in Figure 1. The most significant associations were for SNPs in the CASP8 and ALS2CR12 (amyotrophic lateral sclerosis 2 (juvenile) chromosome region, candidate 12) genes (Fig. 1; Supplementary Material, Table S2). The strongest signals came from imputed SNP rs1830298 in ALS2CR12, with minor allele frequency (MAF) of 0.29 and an estimated OR (95% CI) per copy of the minor allele of 1.05 (1.03–1.07), P = 1.1 × 10−5, and the genotyped SNP rs10197246 (MAF = 0.28), with odds ratio (95% CI) 1.05 (1.02–1.07), P = 2.5 × 10−5. These two SNPs are highly correlated and likely reflect the same signal (r2 = 0.9).
Figure 1.

Breast cancer associations within the 1 Mb region surrounding CASP8. The upper panel plots SNPs based on their chromosomal coordinates on the x-axis and their P-values on the –log10 scale on the y-axis. Circle and diamond symbols represent typed and imputed SNPs, respectively. The colors indicate the pairwise r2 with index SNP for iCHAV1, rs1830298 (highlighted in purple); r2 is calculated based on the European panel in the 1000 genomes project. The ranges of iCHAVs 1–4 are indicated with colored shading. Genes within the region are indicated in the lower panel, with arrows indicating transcript direction, dense blocks for exons and lines for introns. The plot was generated using LocusZoom (9).
Two previously reported susceptibility SNPs, CASP8 D302H (rs1045485) and rs10931936, were weakly replicated in iCOGS European data (Supplementary Material, Table S2), with minor allele OR in the same direction; however, the iCOGS OR estimates were much weaker than those from the original reports (5,8). The minor C allele of rs1045485 (MAF = 0.11) yielded an OR (95% CI) of 0.97 (0.94–1.0), P = 0.03, in contrast to 0.88 (0.84–0.92) reported in Cox et al. (5). Similarly, the rs10931936 minor allele (MAF = 0.28) was associated with a 4% increased breast cancer risk [OR (95% CI) = 1.04 (1.02–1.06), P = 1.9 × 10−4], compared with the 12% increase presented in Turnbull et al. (8). The latter SNP is strongly correlated with the iCOGS best hit rs1830298 (r2 = 0.96), but there is very little correlation between rs1045485 and rs1830298 (r2 = 0.055).
Identification of possible independent signals in iCOGS European data
The SNPs in the main association peak have similar ORs for breast cancer, are strongly correlated with one another (r2 > 0.66) and confined to an 82 kb region spanning the CASP8 and ALS2CR12 genes, and are therefore likely to reflect a single association signal, but this does not preclude the possibility of other signals in the region. To test this hypothesis, we carried out a regression analysis testing the association of individual SNPs adjusted for the top hit rs1830298, in the iCOGS European dataset (Supplementary Material, Table S3). Interestingly, while this resulted in the loss of the signal from the main peak in CASP8/ALS2CR12, residual associations remained (e.g. 43 SNPs with P ≤ 1 × 10−3), suggesting that there may be further signals present in the region, albeit weaker (Supplementary Material, Table S3 and Fig. S1). To investigate this further, we carried out penalized logistic regression analysis of all 1733 SNPs to identify the best subset of SNPs that explain the association, using HyperLasso (10). This identified 59 models containing combinations of 27 SNPs (Supplementary Material, Table S2), but many of these models were equivalent after taking into account linkage disequilibrium between SNPs. To obtain the most parsimonious model, we carried out stepwise forward logistic regression on the 27 SNPs, which resulted in a model containing four SNPs; rs1830298 (ALS2CR12; pconditional = 9.3 × 10−3, MAF = 0.29), rs36043647 (CASP8; pconditional = 1.9 × 10−4, MAF = 0.06), rs59278883 (ALS2CR11; pconditional = 6.1 × 10−4, MAF = 0.07) and rs7558475 (CFLAR; CASP8- and FADD-like apoptosis regulator; pconditional = 9.2 × 10−4, MAF = 0.07). We refer to these four SNPs, marking four independent sets of correlated highly associated variants (iCHAVs), as index SNPs.
Meta-analysis of iCOGS and combined nine GWAS data
We first examined the results for the four index SNPs, together with the previous hits rs1045485 and rs10931936, in the combined nine GWAS meta-analysis, and then carried out a further meta-analysis combining the iCOGS European data with the combined nine GWAS for these SNPs (total sample size 56 502 cases and 55 175 controls; Supplementary Material, Tables S4 and S5). We found that the top index SNP, rs1830298, replicated in the combined GWAS data alone (P = 2.7 × 10−6), and reached genome-wide significance (P = 1.1 × 10−9) in the meta-analysis containing both the iCOGS and combined GWAS data (Supplementary Material, Table S5; Fig. 2). The genotyped proxy rs10197246 also reached genome-wide significance (P = 1.7 × 10−8). When we examined the other three index SNPs in the combined GWAS data, we found a replicated association (P = 1.8 × 10−3) for rs59278883, a null result for rs36043647 (P = 0.58) and borderline evidence for rs7558475 (P = 0.05) (Supplementary Material, Table S5; Fig. 2). However, these three index SNPs all showed some evidence of association in the meta-analysis of iCOGS and combined GWAS (Supplementary Material, Table S5 and Fig. 2), providing some support for the existence of four signals in the region. Consistent with its strong correlation with rs1830298, a similar but slightly weaker signal was found for rs10931936 in the combined analysis (P = 1.0 × 10−7). Weak evidence for association was observed for CASP8 D302H rs1045485 (P = 1.1 × 10−3).
Figure 2.
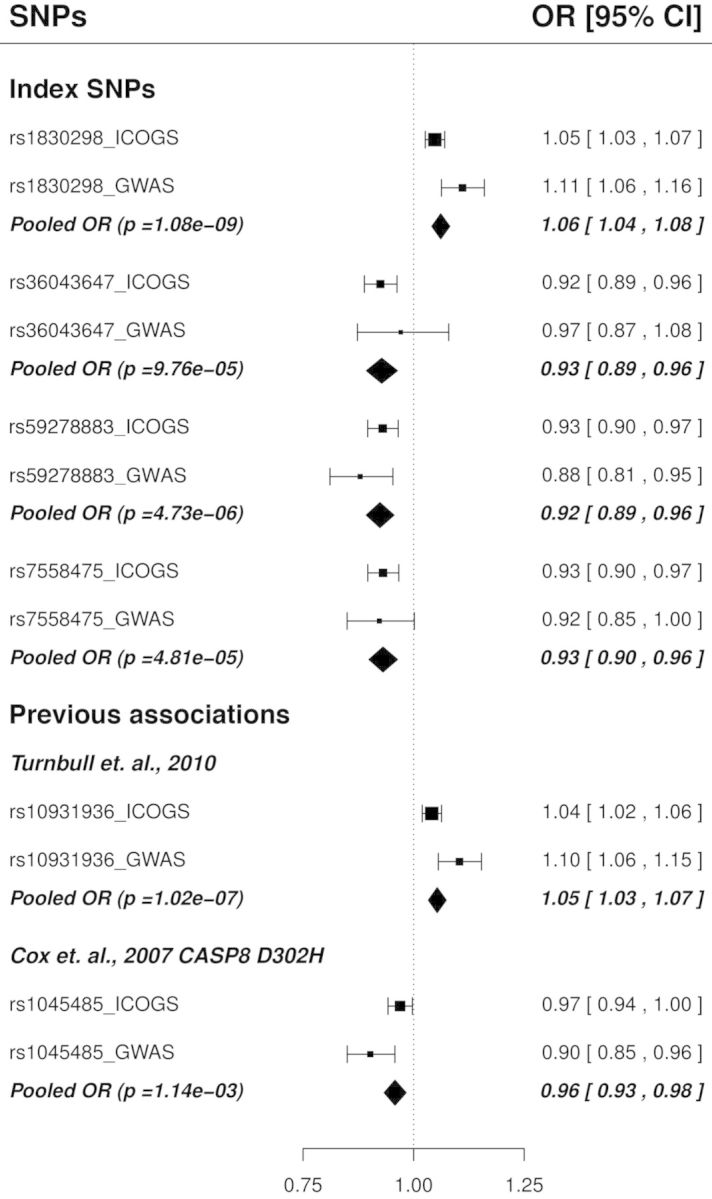
Associations of the four index SNPs corresponding to iCHAVs 1–4, and the two previous associations, in iCOGS European subjects and GWAS data. Squares denote the per-allele OR for the minor allele based on iCOGS and nine GWAS data, with the size of the square proportional to the sample size. Diamonds represent the pooled estimates of ORs under the fixed effect model after exclusion of the 1955 samples from the iCOGS data that were also in the combined GWAS data. Index SNPs correspond to iCHAVs as follows: rs1830298; iCHAV1, rs36043647; iCHAV2, rs59278883; iCHAV3, rs7558475 iCHAV4.
Analysis of index SNPs in different ethnic groups
We next explored these four associations in the available Asian and African-American populations genotyped as part of COGS (Fig. 3; Supplementary Material, Table S6). Figure 3 shows the study-specific OR for rs1830298 by the three ethnic groups. The rs1830298 OR were homogeneous among European studies (phet = 0.54, I2 = 0) and African-American studies (phet = 0.40, I2 = 0), but were more heterogeneous among the nine Asian studies (phet = 0.025, I2 = 54), although the combined effect size in Asians was similar to that seen in Europeans [OR (95% CI) = 1.04 (0.95–1.13); P = 0.44], and slightly stronger in African Americans [OR (95% CI) = 1.12 (0.96–1.30); P = 0.16]. Although estimates in both Asian and African-American populations were not statistically significant, the ORs were consistent with the European data, and the pooled OR (95% CI) was 1.05 (1.03–1.07); P = 4.1 × 10−6 for all populations combined. The MAF of CASP8 rs36043647 was much lower in Asians, in whom the association was in the opposite direction to that in Europeans and African Americans, with an OR (95% CI) of 1.69 (1.13–2.51), P = 0.009, for the minor allele (Supplementary Material, Table S6). We did not observe any association of rs59278883 and rs7558475 in Asian and African-American populations (Supplementary Material, Table S6).
Figure 3.
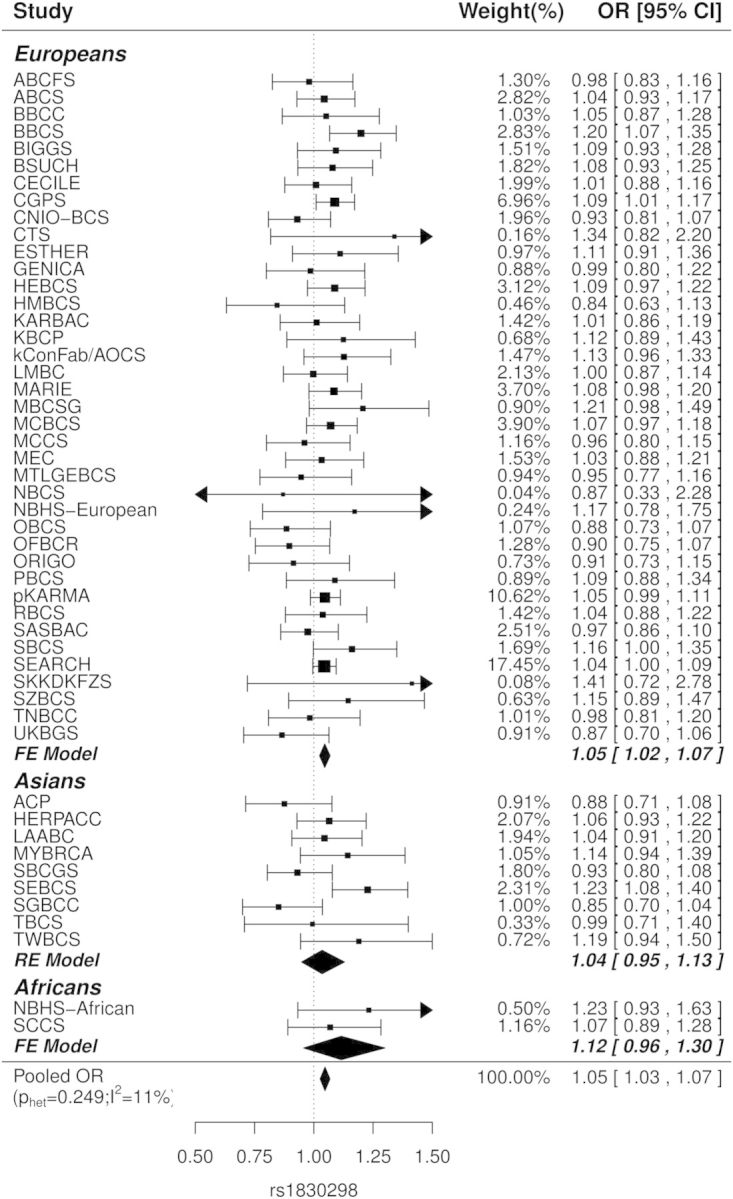
Study-specific OR for the minor allele of rs1830298 in iCOGS European, Asian and African-American subjects. Squares denote the individual study per-allele OR and diamonds indicate the combined effects, with the size of the symbol indicating sample size. Fixed effect models (FE model) were used to combine the study ORs if p for the Cochran's Q test (phet) was >0.05, otherwise random effect models (RE model) were used. Pooled OR across the three populations is shown, with phet and I2 for heterogeneity in parenthesis.
Subtype and survival analysis in iCOGS
To investigate whether these SNP associations vary with clinical subtypes of breast cancer, we explored potential subtype-specific associations by comparing different subtypes to all controls in the iCOGS European data. The OR estimates by tumor estrogen receptor (ER) status, triple negative status and invasiveness of breast cancer were all similar and close to the OR of 1.05 seen in overall breast cancer for rs1830298 (Fig. 4). Similarly, no significant differences in OR were seen when cases were stratified by family history, tumor grade, tumor stage, tumor size and lymph node status (Supplementary Material, Fig. S2). A broadly similar picture was seen for the other index SNPs (Supplementary Material, Figs S2 and S3).
Figure 4.

Associations between rs1830298 and clinical subtypes of breast cancer in iCOGS European subjects. Squares denote the individual study per-allele OR with the size of the symbol indicating sample size. Cases in each subtype group were compared with all controls.
SNP effects were also evaluated for overall survival and breast cancer-specific survival. There were 4191 deaths among 39 140 breast cancer patients with known vital status in the European dataset. Of these deaths, 1979 died from breast cancer. We did not observe any associations between the index SNPs or previous hit SNPs with either overall or breast cancer-specific survival, and all hazard ratios (HR) were close to unity (data not shown).
In silico functional and expression quantitative trait loci annotations
We examined available in silico functional and expression quantitative trait loci (eQTL) data for the four iCHAVs. Of interest in iCHAV1, rs3769823 is a missense alteration encoding K14R in the 4th exon of CASP8, which encodes the N-terminus of protein isoform 9. In addition, this SNP and rs3769821 are both located in a region of deoxyribonuclease I hypersensitivity and histone H3K27 acetylation in breast cell lines (Fig. 5). The minor alleles of both of these SNPs, together with four others in iCHAV1 for which data were available, were associated with a reduction in CASP8 mRNA levels in peripheral blood samples in the eQTL meta-analysis of Westra et al. (P ≤ 9.4 × 10−5; Supplementary Material, Table S7; Fig. 5) (11). The cancer genome atlas (TCGA) dataset only had data available for two SNPs from iCHAV1, and both were associated with a reduction in CASP8 mRNA in normal breast tissue (P ≤ 1 × 10−3; Supplementary Material, Table S7; Fig. 5). No strong eQTL associations were seen for other genes in the region in either the Westra et al. or the TCGA data. Taken together, these data suggest that one or more variants in iCHAV1 may affect levels of CASP8 gene expression. As shown in Figure 5, iCHAVs 3 and 4 overlap enhancer sites identified in Hnisz et al.; a CASP8 enhancer in MCF7 cells and a CFLAR enhancer in human mammary epithelial cells, respectively (12). However, there was limited eQTL data available for these iCHAVs, with no evidence of any significant eQTLs (Supplementary Material, Table S7).
Figure 5.
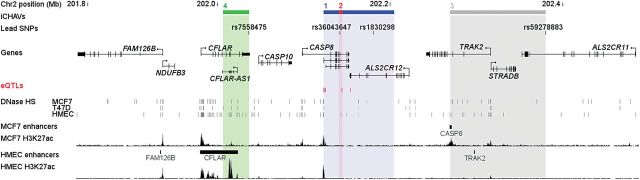
Summary of the CASP8/ALS2CR12 locus. The locations of iCHAVs and lead SNPs are shown relative to genes. eQTL SNPs are displayed as red marks. ENCODE DNaseI hypersensitive sites derived from various mammary cell types are depicted as gray marks. H3K27ac histone modification ChIP-seq data are shown as well as predicted enhancers and target genes from Hnisz et al. (12).
DISCUSSION
In our analysis of the genomic region surrounding CASP8 for association with breast cancer, the strongest signal came from an imputed SNP, rs1830298, in the ASL2CR12 gene (iCHAV1). A strongly correlated genotyped SNP (rs10197246; r2 = 0.9, 23.5 kb telomeric in the same gene), yielded a similar association signal (P = 1.1 × 10−5 and 2.5 × 10−5, respectively). In each case, the rare allele (MAF = 0.28) was associated with an increase in the risk of breast cancer of 5% [OR(95% CI) 1.05 (1.03, 1.07) and 1.05 (1.02, 1.07), respectively]. The odds ratios for both SNPs are consistent in Europeans, Asians and African Americans (although not statistically significant in the smaller non-European cohorts), and were replicated in the combined GWAS data, achieving a genome-wide level of significance when the iCOGS and GWAS data were combined (P = 1.1 × 10−9 and P = 1.7 × 10−8, respectively). This association is consistent between ER-positive and -negative disease, and between invasive and in situ cancers (Fig. 4). The previously published result for rs10931936 in the UK GWAS is consistent with its correlation with rs1830298 (8).
Several of the SNPs in iCHAV1were associated with CASP8 eQTLs. The minor alleles of SNPs in this group, associated with increased risk of breast cancer, are associated with reduced CASP8 mRNA levels in both peripheral blood lymphocytes and normal breast tissue (Supplementary Material, Table S7; Fig. 5). These data suggest that CASP8 may be the target gene of iCHAV1, and are consistent with a hypothesis in which the effect of the risk alleles is via reduced levels of apoptosis, thus promoting tumor initiation. However, further functional studies are required to demonstrate a direct interaction between iCHAV1 and the CASP8 promoter and to investigate the allele-specific functional effects of these SNPs in different tissue types.
Our results also suggest three other independent signals in the region; the most significant SNPs for these three signals are in CASP8 (iCHAV2), ALS2CR11 (iCHAV3) and the anti-apoptotic gene CFLAR (iCHAV4); see Figure 2; Supplementary Material, Table S5. The signals for iCHAVs 3 and 4 were replicated in the combined GWAS, but since they did not achieve genome-wide levels of significance even in the very large datasets analyzed here, they are harder to interpret. However, it is interesting that both these iCHAVs overlap enhancer regions (Fig. 5).
As previously noted, we find only very weak support for an association of rs1045485/D302H in the iCOGS data (P = 0.03) (2), although the odds ratio in the combined GWAS data was more consistent with the original report [OR (95% CI) = 0.90(0.85, 0.96), P = 0.0007] (5). At present, the reasons for the discrepancy with the original report are not clear. D302H is only weakly correlated with any of the four index SNPs identified here (max r2 = 0.06 with rs1830298). However, it is correlated with rs28845859 (r2 = 0.67); the latter SNP is associated with reduced breast cancer risk in the iCOGS data (OR 0.95, P = 1.9 × 10−4; Supplementary Material, Table S2) and combined GWAS (P = 4.0 × 10−5). We found no significant differences between subtypes, although the associated effect for D302H was stronger (and borderline significant) for triple negative disease, despite the smaller sample size (Supplementary Material, Fig. S3). Further investigation with a larger sample of triple negative cases may help clarify this point.
The association for the top CASP8 index SNP, rs1830298, represents one of the smaller effect sizes identified to date for breast cancer. However, it is worth noting that the CASP8 region has recently been reported to be associated with other cancers at genome-wide levels of significance, including melanoma and chronic lymphocytic leukemia (CLL) (13,14). The alleles associated with increased risk in melanoma are correlated with rs1830298, but the signal in CLL appears to be due to uncorrelated SNPs in the region. This difference may reflect the different cell type of origin and it will be interesting to determine the relative importance and function of alleles of the CASP8 gene family in immune cell lineages, compared with that in epithelial cancers.
MATERIALS AND METHODS
Study samples
The iCOGS and nine breast cancer GWAS datasets have been described in detail previously (2). Briefly, the COGS includes a total of 103 991 women from 50 studies participating in the BCAC whose DNA samples were genotyped with the iCOGS array. These were 89 050 Europeans (46 450 cases; 42 600 controls), 12 893 Asians (6269 cases; 6624 controls) and 2048 African Americans (1116 cases and 932 controls). The numbers of subjects by study are detailed in Supplementary Material, Table S1. Approximately 93% of cases had invasive breast cancer (Supplementary Material, Table S1). The combined nine breast cancer GWAS dataset comprised 10 052 cases and 12 575 controls of European ancestry from United States, UK, Australia, Germany, Finland, Sweden and the Netherlands (2).
Ethics statement
Each study was approved by the relevant local/institutional Research Ethics Committee, and all subjects gave written informed consent to take part.
SNP selection for fine-scale mapping on the iCOGS array
The region for analysis on chromosome 2 was defined such that it contained all SNPs correlated (r2 ≥ 0.1) with the SNPs previously reported to be associated with breast cancer, namely CASP8 D302H (rs1045485) and rs10931936 (5,8). This identified a 1 Mb region from 201 566 128 to 202 566 128 (hg19). In March 2010 when the iCOGS array was designed, 2191 SNPs had been catalogued in this region by the 1000 genomes and HapMap3 projects. Of these, 1723 SNPs had an MAF ≥2%, and of these 1723, there were 988 SNPs with Illumina assay design scores of ≥0.8. We selected a total of 280 SNPs correlated at r2 ≥ 0.1 with rs1045485 or rs10931936, plus 288 tagSNPs which tagged the remaining 708 SNPs at r2 ≥ 0.9. Another 45 SNPs in the region, nominated by other consortia members, were included as part of the genotyping array that comprised 211 155 SNPs in total (2).
Genotyping and quality control
Genotyping, allele calling, quality control and principal components analysis for COGS are described in detail in Michailidou et al. (2). Genotyping was carried out at four centers using the Illumina Infinium iCOGS array, including 2% duplicates from each participating study. Final genotype calls were made using Illumina's proprietary GenCall algorithm. SNPs were excluded from analysis if the overall call rate was <95%, duplicate concordance rate was <98%, or if deviation from Hardy–Weinberg equilibrium in controls was significant at P < 1 × 10−7 (2). Subjects were excluded from analysis for the following reasons: genotypically non-female; overall call rate <95%; low or high heterozygosity (P < 1 × 10−6); discordant replicates or cryptic duplicates. Genotype data and ancestry principal components (seven principal components for the European and two each for the Asian and African-American populations) were thus available for 103 991 individuals.
Statistical analysis
The iCOGS CASP8 region genotype data were split into four groups for efficiency of imputation of missing genotypes and untyped SNPs. These comprised 36 793 European ancestry subjects from North American and UK studies in Group 1, with 26 129 and 26 128 of the remaining European subjects in Groups 2 and 3, respectively, and 14 941 Asians and African Americans in Group 4. Imputations were carried out separately by group based on the 1000 genomes phase I reference panel with singleton variants excluded, using IMPUTE2 version 2.3 (15,16). SNPs were included in the subsequent analyses if the mean information score of the European groups was ≥0.9, and untyped imputed SNPs were only included if their MAF was ≥3%; these criteria resulted in inclusion of 501 typed and 1232 imputed SNPs in the final analysis. The imputation accuracy for rs1830298 was verified in whole-genome sequence data from 197 individuals; the correlation between the observed and imputed genotypes was 0.974. The imputation step increases the number of common SNPs captured at r2 > 0.9 from 76% (1198/1583) to 84% (1333/1583).
The main analyses were based on the data for individuals of European ancestry. For each SNP, allelic dosage of the minor allele was estimated, and included in a logistic regression model, to estimate OR and corresponding 95% CI. Covariates for each study plus the seven ancestry principal components were included in the model (2). These analyses were implemented in R. P-values from the Wald test are reported in the text (uncorrected for multiple testing). FDR values in Supplementary Material, Table S2 were calculated according to the Benjamini & Hochberg method, as implemented in the R p.adjust function (17). Penalized logistic regression models (based on the normal exponential gamma probability density) were implemented in HyperLasso (10), including all 501 typed and 1232 imputed SNPs, to identify the best subsets of SNPs to account for the observed association data. Based on the sample size and a type I error of 0.001, a λ of 0.05 and penalty of 491 were specified in HyperLasso, according to equation 7 in Hoggart et al. (10). Candidate SNPs were then compiled from the resulting HyperLasso models and included in a stepwise forward logistic regression procedure with penalty k = 10 in the step function in R to identify the most parsimonious model, as described previously (18). The SNPs retained in the final model are referred to as index SNPs.
Index SNPs were further examined by means of meta-analysis of iCOGS European, Asian and African-American data, and also with individual SNP results from the combined nine breast cancer GWAS (2). Due to an overlap of 1955 samples that exist in both the iCOGS and the combined GWAS data, we removed these samples from the iCOGS data before carrying out the meta-analysis. The meta-analysis was carried out using the MetaFor package in R, with inverse-variance weights and the DerSimonian-Laird estimator for the random effects model (19). We used the threshold of P = 5 × 10−8 to define genome-wide significance (2).
The index SNPs were also examined for associations with breast cancer specific and overall survival in Cox's proportional hazard models, including age at diagnosis, study and seven principal components as covariates, and accounting for the left-censoring time between study entry and diagnosis. Further adjustment was carried out for stage, grade, tumor size and lymph node involvement for SNPs with nominally significant associations with survival (P < 0.05). These analyses were implemented in R.
In silico functional and eQTL annotations
We defined independent sets of iCHAVs with likelihood (determined from the individual-SNP logistic regression analysis) relative to an index SNP of >1/100 and degree of correlation with the index SNP of >0.65. The ENCODE integrated regulation data for each SNP were retrieved from the UCSC Genome Browser by use of ANNOVAR (20). Predicted enhancers and target genes were retrieved from Hnisz et al. (12). Expression QTL data were obtained by interrogation of the GTEx Portal, the online results of the peripheral blood eQTL meta-analysis based on 5311 samples from seven studies by Westra and colleagues (11), and from breast cancer cases in the TCGA Project. For the latter, RNAseq data in the form of fragments per kilobase of transcript per million mapped reads (FPKM) were available for uninvolved breast tissue from 97 TCGA breast cancer cases. Peripheral blood DNA SNP genotypes for these individuals were extracted from the TCGA Level 2 Affymetrix 6.0 array birdseed files. Mean FPKM were compared between individuals homozygous for the common allele and those carrying one or two copies of the rare allele by use of an unpaired, unequal variance t-test in Stata.
SUPPLEMENTARY MATERIAL
FUNDING
Part of this work was supported by the European Community's Seventh Framework Programme under grant agreement number 223175 (grant number HEALTH-F2-2009-223175) (COGS). Funding for the iCOGS infrastructure came from: the European Community's Seventh Framework Programme under grant agreement n° 223175 (HEALTH-F2-2009-223175) (COGS), Cancer Research UK (C1287/A10118, C1287/A 10710, C12292/A11174, C1281/A12014, C5047/A8384, C5047/A15007 and C5047/A10692), the National Institutes of Health (CA128978) and Post-Cancer GWAS initiative (1U19 CA148537, 1U19 CA148065 and 1U19 CA148112—the GAME-ON initiative), the Department of Defence (W81XWH-10-1-0341), the Canadian Institutes of Health Research (CIHR) for the CIHR Team in Familial Risks of Breast Cancer, Komen Foundation for the Cure, the Breast Cancer Research Foundation and the Ovarian Cancer Research Fund. The ABCFS, NC-BCFR and OFBCR work was supported by the United States National Cancer Institute, National Institutes of Health (NIH) under RFA-CA-06-503 and through cooperative agreements with members of the Breast Cancer Family Registry (BCFR) and Principal Investigators, including Cancer Care Ontario (U01 CA69467), Northern California Cancer Center (U01 CA69417) and University of Melbourne (U01 CA69638). Samples from the NC-BCFR were processed and distributed by the Coriell Institute for Medical Research. The content of this manuscript does not necessarily reflect the views or policies of the National Cancer Institute or any of the collaborating centers in the BCFR, nor does mention of trade names, commercial products or organizations imply endorsement by the US Government or the BCFR. The ABCFS was also supported by the National Health and Medical Research Council of Australia, the New South Wales Cancer Council, the Victorian Health Promotion Foundation (Australia) and the Victorian Breast Cancer Research Consortium. J.L.H. is a National Health and Medical Research Council (NHMRC) Australia Fellow and a Victorian Breast Cancer Research Consortium Group Leader. M.C.S. is a NHMRC Senior Research Fellow and a Victorian Breast Cancer Research Consortium Group Leader. The ABCS was supported by the Dutch Cancer Society (grants NKI 2007-3839, 2009 4363); BBMRI-NL, which is a Research Infrastructure financed by the Dutch government (NWO 184.021.007); and the Dutch National Genomics Initiative. The ACP study is funded by the Breast Cancer Research Trust, UK. The work of the BBCC was partly funded by ELAN-Fond of the University Hospital of Erlangen. The BBCS is funded by Cancer Research UK and Breakthrough Breast Cancer and acknowledges NHS funding to the NIHR Biomedical Research Centre, and the National Cancer Research Network (NCRN). The BBCS GWAS received funding from The Institut National de Cancer. E.S. is supported by NIHR Comprehensive Biomedical Research Centre, Guy's & St. Thomas' NHS Foundation Trust in partnership with King's College London, UK. I.T. is supported by the Oxford Biomedical Research Centre. The BSUCH study was supported by the Dietmar-Hopp Foundation, the Helmholtz Society and the German Cancer Research Center (DKFZ). The CECILE study was funded by Fondation de France, Institut National du Cancer (INCa), Ligue Nationale contre le Cancer, Ligue contre le Cancer Grand Ouest, Agence Nationale de Sécurité Sanitaire (ANSES) and Agence Nationale de la Recherche (ANR). The was supported by the Chief Physician Johan Boserup and Lise Boserup Fund, the Danish Medical Research Council and Herlev Hospital. The CNIO-BCS was supported by the Genome Spain Foundation, the Red Temática de Investigación Cooperativa en Cáncer and grants from the Asociación Española Contra el Cáncer and the Fondo de Investigación Sanitario (PI11/00923 and PI081120). The Human Genotyping-CEGEN Unit (CNIO) is supported by the Instituto de Salud Carlos III. The CTS was supported by the California Breast Cancer Act of 1993, National Institutes of Health (grants R01 CA77398 and the Lon V Smith Foundation, LVS39420) and the California Breast Cancer Research Fund (contract 97-10500). Collection of cancer incidence data used in this study was supported by the California Department of Public Health as part of the statewide cancer reporting program mandated by California Health and Safety Code Section 103885. The ESTHER study was supported by a grant from the Baden Württemberg Ministry of Science, Research and Arts. Additional cases were recruited in the context of the VERDI study, which was supported by a grant from the German Cancer Aid (Deutsche Krebshilfe). The German Consortium of Hereditary Breast and Ovarian Cancer (GC-HBOC) is supported by the German Cancer Aid (grant no. 109076; coordinator: R.K.S.). The GENICA was funded by the Federal Ministry of Education and Research (BMBF) Germany grants 01KW9975/5, 01KW9976/8, 01KW9977/0 and 01KW0114, the Robert Bosch Foundation, Stuttgart, Deutsches Krebsforschungszentrum (DKFZ), Heidelberg, Institute for Prevention and Occupational Medicine of the German Social Accident Insurance, Institute of the Ruhr University Bochum (IPA), Bochum, Germany, as well as the Department of Internal Medicine, Evangelische Kliniken Bonn gGmbH, Johanniter Krankenhaus, Bonn, Germany. The HEBCS was financially supported by the Helsinki University Central Hospital Research Fund, Academy of Finland (266528), the Finnish Cancer Society, The Nordic Cancer Union and the Sigrid Juselius Foundation. The GWS population allele and genotype frequencies were obtained from the data source funded by the Nordic Center of Excellence in Disease Genetics based on samples regionally selected from Finland, Sweden and Denmark. The HERPACC was supported by a Grant-in-Aid for Scientific Research on Priority Areas from the Ministry of Education, Science, Sports, Culture and Technology of Japan, by a Grant-in-Aid for the Third Term Comprehensive 10-Year Strategy for Cancer Control from Ministry Health, Labour and Welfare of Japan, by Health and Labour Sciences Research Grants for Research on Applying Health Technology from Ministry Health, Labour and Welfare of Japan and by National Cancer Center Research and Development Fund. The HMBCS was supported by a grant from the Friends of Hannover Medical School and by the Rudolf Bartling Foundation. Financial support for KARBAC was provided through the regional agreement on medical training and clinical research (ALF) between Stockholm County Council and Karolinska Institutet, The Swedish Cancer Society and the Gustav V Jubilee foundation. The KBCP was financially supported by the special Government Funding (EVO) of Kuopio University Hospital grants, Cancer Fund of North Savo, the Finnish Cancer Organizations, the Academy of Finland and by the strategic funding of the University of Eastern Finland. kConFab is supported by grants from the National Breast Cancer Foundation, the NHMRC, the Queensland Cancer Fund, the Cancer Councils of New South Wales, Victoria, Tasmania and South Australia and the Cancer Foundation of Western Australia. The kConFab Clinical Follow Up Study was funded by the NHMRC (145684, 288704 and 454508). Financial support for the AOCS was provided by the United States Army Medical Research and Materiel Command (DAMD17-01-1-0729), the Cancer Council of Tasmania and Cancer Foundation of Western Australia and the NHMRC (199600). G.C.T. and P.W. are supported by the NHMRC. LAABC is supported by grants (1RB-0287, 3PB-0102, 5PB-0018 and 10PB-0098) from the California Breast Cancer Research Program. Incident breast cancer cases were collected by the USC Cancer Surveillance Program (CSP), which is supported under subcontract by the California Department of Health. The CSP is also part of the National Cancer Institute's Division of Cancer Prevention and Control Surveillance, Epidemiology, and End Results Program, under contract number N01CN25403. LMBC is supported by the ‘Stichting tegen Kanker’ (232-2008 and 196-2010). Diether Lambrechts is supported by the FWO and the KULPFV/10/016-SymBioSysII. The MARIE study was supported by the Deutsche Krebshilfe e.V. (70-2892-BR I), the Hamburg Cancer Society, the German Cancer Research Center and the Federal Ministry of Education and Research (BMBF) Germany (01KH0402, 01KH0408 and 01KH0409). MBCSG is supported by grants from the Italian Association for Cancer Research (AIRC) and by funds from the Italian citizens who allocated the 5/1000 share of their tax payment in support of the Fondazione IRCCS Istituto Nazionale Tumori, according to Italian laws (INT-Institutional strategic projects ‘5 × 1000’). The MCBCS was supported by the NIH grant CA128978, an NIH Specialized Program of Research Excellence (SPORE) in Breast Cancer (CA116201), the Breast Cancer Research Foundation, a generous gift from the David F. and Margaret T. Grohne Family Foundation and the Ting Tsung and Wei Fong Chao Foundation. MCCS cohort recruitment was funded by VicHealth and Cancer Council Victoria. The MCCS was further supported by Australian NHMRC grants 209057, 251553 and 504711 and by infrastructure provided by Cancer Council Victoria. The MEC was support by NIH grants CA63464, CA54281, CA098758 and CA132839. The work of MTLGEBCS was supported by the Quebec Breast Cancer Foundation, the Canadian Institutes of Health Research for the ‘CIHR Team in Familial Risks of Breast Cancer’ program—grant # CRN-87521 and the Ministry of Economic Development, Innovation and Export Trade—grant # PSR-SIIRI-701. MYBRCA is funded by research grants from the Malaysian Ministry of Science, Technology and Innovation (MOSTI), Malaysian Ministry of Higher Education (UM.C/HlR/MOHE/06) and Cancer Research Initiatives Foundation (CARIF). Additional controls were recruited by the Singapore Eye Research Institute, which was supported by a grant from the Biomedical Research Council (BMRC08/1/35/19/550), Singapore and the National medical Research Council, Singapore (NMRC/CG/SERI/2010). The NBCS was supported by grants from the Norwegian Research council, 155218/V40, 175240/S10 to ALBD, FUGE-NFR 181600/V11 to VNK and a Swizz Bridge Award to ALBD. The NBCS was supported by grants from the Norwegian Research council, 155218/V40, 175240/S10 to ALBD, FUGE-NFR 181600/V11 to VNK and a Swizz Bridge Award to ALBD. The NBHS was supported by NIH grant R01CA100374. Biological sample preparation was conducted the Survey and Biospecimen Shared Resource, which is supported by P30 CA68485. The OBCS was supported by research grants from the Finnish Cancer Foundation, the Academy of Finland Centre of Excellence grant 251314, the Sigrid Juselius Foundation, the University of Oulu, and the Oulu University Hospital special Govermental EVO Research Funds. The OFBCR work was supported by grant UM1 CA164920 from the National Cancer Institute. The content of this manuscript does not necessarily reflect the views or policies of the National Cancer Institute or any of the collaborating centers in the Breast Cancer Family Registry (BCFR), nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government or the BCFR. The ORIGO study was supported by the Dutch Cancer Society (RUL 1997–1505) and the Biobanking and Biomolecular Resources Research Infrastructure (BBMRI-NL CP16). The PBCS was funded by Intramural Research Funds of the National Cancer Institute, Department of Health and Human Services, USA. The pKARMA study was supported by Märit and Hans Rausings Initiative Against Breast Cancer. The RBCS was funded by the Dutch Cancer Society (DDHK 2004-3124, DDHK 2009-4318). The SASBAC study was supported by funding from the Agency for Science, Technology and Research of Singapore (A*STAR), the US National Institute of Health (NIH) and the Susan G. Komen Breast Cancer Foundation. The SBCGS was supported primarily by NIH grants R01CA64277, R01CA148667 and R37CA70867. Biological sample preparation was conducted the Survey and Biospecimen Shared Resource, which is supported by P30 CA68485. The SBCS was supported by Yorkshire Cancer Research awards S295, S299, S305PA and by the Sheffield Experimental Cancer Medicine Centre Network. N.J.C. was supported by NCI grant R01 CA163353 and The Avon Foundation (02-2009-080). The SCCS is supported by a grant from the National Institutes of Health (R01 CA092447). Data on SCCS cancer cases used in this publication were provided by the Alabama Statewide Cancer Registry; Kentucky Cancer Registry, Lexington, KY; Tennessee Department of Health, Office of Cancer Surveillance; Florida Cancer Data System; North Carolina Central Cancer Registry, North Carolina Division of Public Health; Georgia Comprehensive Cancer Registry; Louisiana Tumor Registry; Mississippi Cancer Registry; South Carolina Central Cancer Registry; Virginia Department of Health, Virginia Cancer Registry; Arkansas Department of Health, Cancer Registry, 4815 W. Markham, Little Rock, AR 72205. The Arkansas Central Cancer Registry is fully funded by a grant from National Program of Cancer Registries, Centers for Disease Control and Prevention (CDC). Data on SCCS cancer cases from Mississippi were collected by the Mississippi Cancer Registry which participates in the National Program of Cancer Registries (NPCR) of the Centers for Disease Control and Prevention (CDC). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of the CDC or the Mississippi Cancer Registry. SEARCH is funded by programme grants from Cancer Research UK (C490/A10124, C490/A16561) and supported by the UK National Institute for Health Research Biomedical Research Centre at the University of Cambridge. The SEBCS was supported by the Korea Health 21 R&D Project [AO30001], Ministry of Health and Welfare, Republic of Korea. SGBCC is funded by the National Medical Research Council start-up Grant and Centre Grant (NMRC/CG/NCIS /2010). Additional controls were recruited by the Singapore Consortium of Cohort Studies-Multi-ethnic cohort (SCCS-MEC), which was funded by the Biomedical Research Council, grant number: 05/1/21/19/425. SKKDKFZS is supported by the DKFZ. The SZBCS was supported by Grant PBZ_KBN_122/P05/2004. The TBCS was funded by The National Cancer Institute Thailand. The TNBCC was supported by: MCBCS (National Institutes of Health Grants CA116167, CA176785 and a Specialized Program of Research Excellence (SPORE) in Breast Cancer (CA116201), a generous gift from the David F. and Margaret T. Grohne Family Foundation and the Ting Tsung and Wei Fong Chao Foundation. This research has been partly financed by the European Union (European Social Fund—ESF) and Greek national funds through the Operational Program “Education and Lifelong Learning” of the National Strategic Reference Framework (NSRF)—Research Funding Program of the General Secretariat for Research & Technology: ARISTEIA. Investing in knowledge society through the European Social Fund; and the Stefanie Spielman Breast Fund and the Ohio State University Comprehensive Cancer Center. The TWBCS is supported by the Taiwan Biobank project of the Institute of Biomedical Sciences, Academia Sinica, Taiwan. The UKBGS is funded by Breakthrough Breast Cancer and the Institute of Cancer Research (ICR). ICR acknowledges NHS funding to the RMH/ICR NIHR Specialist Biomedical Research Centre for Cancer. The Nurses' Health Studies (CGEMS) are supported by NIH grants CA 65725, CA87969, CA49449, CA67262, CA50385 and 5UO1CA098233. The UK2 GWAS was funded by Wellcome Trust and Cancer Research UK. It included samples collected through the BOCS study, which is funded by Cancer Research UK [C8620/A8372]. It included control data obtained through the WTCCC which was funded by the Wellcome Trust. The DFBBCS GWAS was funded by The Netherlands Organisation for Scientific Research (NWO) as part of a ZonMw/VIDI grant number 91756341. Control GWA genotype data from the Rotterdam Study were funded by NWO Groot Investments (project nr. 175.010.2005.011).
Supplementary Material
ACKNOWLEDGEMENTS
We thank all the individuals who took part in these studies and all the researchers, clinicians, technicians and administrative staff who have enabled this work to be carried out. This study would not have been possible without the contributions of the following: Andrew Berchuck (OCAC), Rosalind A. Eeles, Ali Amin Al Olama, Zsofia Kote-Jarai, Sara Benlloch (PRACTICAL), Antonis Antoniou, Lesley McGuffog, Ken Offit (CIMBA), Andrew Lee and Ed Dicks, and the staff of the Centre for Genetic Epidemiology Laboratory, the staff of the CNIO genotyping unit, Sylvie LaBoissière and Frederic Robidoux and the staff of the McGill University and Génome Québec Innovation Centre, the staff of the Copenhagen DNA laboratory and Julie M. Cunningham, Sharon A. Windebank, Christopher A. Hilker, Jeffrey Meyer and the staff of Mayo Clinic Genotyping Core Facility. We also thank Maggie Angelakos, Judi Maskiell, Gillian Dite (ABCFS) and extend our thanks to the many women and their families that generously participated in the Australian Breast Cancer Family Study and consented to us accessing their pathology material. J.L.H. is a National Health and Medical Research Council Australia Fellow. M.C.S. is a National Health and Medical Research Council Senior Research Fellow. JLH and MCS are both group leaders of the Victoria Breast Cancer Research Consortium. We thank Sten Cornelissen, Richard van Hien, Linde Braaf, Frans Hogervorst, Senno Verhoef and Ellen van der Schoot, Femke Atsma (ABCS). The ACP study wishes to thank the participants in the Thai Breast Cancer study. Special thanks also go to the Thai Ministry of Public Health (MOPH), doctors and nurses who helped with the data collection process. Finally, the study thank Dr Prat Boonyawongviroj, the former Permanent Secretary of MOPH and Dr Pornthep Siriwanarungsan, the Department Director-General of Disease Control who have supported the study throughout. We thank Eileen Williams, Elaine Ryder-Mills, Kara Sargus (BBCS), Niall McInerney, Gabrielle Colleran, Andrew Rowan, Angela Jones (BIGGS), Peter Bugert and Medical Faculty Mannheim (BSUCH). We thank the staff and participants of the Copenhagen General Population Study, and for excellent technical assistance: Dorthe Uldall Andersen, Maria Birna Arnadottir, Anne Bank, and Dorthe Kjeldgård Hansen. The Danish Breast Cancer Group (DBCG) is acknowledged for the tumor information. We thank Guillermo Pita, Charo Alonso, Daniel Herrero, Nuria Álvarez, Pilar Zamora, Primitiva Menendez, the Human Genotyping-CEGEN Unit (CNIO), Hartwig Ziegler, Sonja Wolf, and Volker Hermann (ESTHER). GC-HBOC would like to thank the following persons for providing additional informations and samples: Prof. Dr Norbert Arnold, Dr Sabine Preissler-Adams, Dr Raymonda Varon-Mateeva, Dr Dieter Niederacher, Prof. Dr Brigitte Schlegelberger, Heide Hellebrand, and Stefanie Engert. The GENICA Network: Dr Margarete Fischer-Bosch-Institute of Clinical Pharmacology, Stuttgart, and University of Tübingen, Germany; (H.B., Wing-Yee Lo, Christina Justenhoven), German Cancer Consortium (DKTK) and German Cancer Research Center (DKFZ) (H.B.), Department of Internal Medicine, Evangelische Kliniken Bonn gGmbH, Johanniter Krankenhaus, Bonn, Germany (Y.D.K., Christian Baisch), Institute of Pathology, University of Bonn, Germany (Hans-Peter Fischer), Molecular Genetics of Breast Cancer, Deutsches Krebsforschungszentrum (DKFZ), Heidelberg, Germany (Ute Hamann) and Institute for Prevention and Occupational Medicine of the German Social Accident Insurance, Institute of the Ruhr University Bochum (IPA), Bochum, Germany (T.B., Beate Pesch, Sylvia Rabstein, Anne Lotz); Institute of Occupational Medicine and Maritime Medicine, University Medical Center Hamburg-Eppendorf, Germany (Volker Harth). HEBCS thanks Kirsimari Aaltonen, Tuomas Heikkinen, and Dr Karl von Smitten and RN Irja Erkkilä for their help with the HEBCS data and samples. We thank Peter Hillemanns, Hans Christiansen and Johann H. Karstens (HMBCS), Eija Myöhänen and Helena Kemiläinen (KBCP). kConFab thanks Heather Thorne, Eveline Niedermayr, the AOCS Management Group (D. Bowtell, G. Chenevix-Trench, A. deFazio, D. Gertig, A. Green and P. Webb), the ACS Management Group (A. Green, P. Parsons, N. Hayward, P. Webb and D. Whiteman). LAABC thanks all the study participants and the entire data collection team, especially Annie Fung and June Yashiki. We thank Gilian Peuteman, Dominiek Smeets, Thomas Van Brussel and Kathleen Corthouts (LMBC). MARIE thank Alina Vrieling, Katharina Buck, Muhabbet Celik, Ursula Eilber and Sabine Behrens for their valuable contributions to data management and assessment, and Lars Beckmann, Thomas Illig, Kirsten Mittelstraß for their valuable contributions to analysis and generation of the GWAS data. MBCSG thank Bernard Peissel, Giulietta Scuvera and Daniela Zaffaroni of the Fondazione IRCCS Istituto Nazionale dei Tumori (INT); Monica Barile and Irene Feroce of the Istituto Europeo di Oncologia (IEO) and the personnel of the Cogentech Cancer Genetic Test Laboratory. We thank Martine Tranchant (CHU de Québec Research Center), Marie-France Valois, Annie Turgeon and Lea Heguy (McGill University Health Center, Royal Victoria Hospital; McGill University) for DNA extraction, sample management and skillful technical assistance (MTLGEBCS). J.S. is Chairholder of the Canada Research Chair in Oncogenetics. We thank Phuah Sze Yee, Peter Kang, Kang In Nee, Kavitta Sivanandan, Shivaani Mariapun, Yoon Sook-Yee, Daphne Lee, Teh Yew Ching and Nur Aishah Mohd Taib for DNA Extraction and patient recruitment (MYBRCA). We thank Mervi Grip, Meeri Otsukka, Kari Mononen (OBCS) and Teresa Selander, Nayana Weerasooriya (OFBCR). ORIGO thanks E. Krol-Warmerdam, and J. Blom for patient accrual, administering questionnaires and managing clinical information. The LUMC survival data were retrieved from the Leiden hospital-based cancer registry system (ONCDOC) with the help of Dr J. Molenaar. We thank Mark Sherman, Stephen Chanock, Neonila Szeszenia-Dabrowska, Beata Peplonska, Witold Zatonski, Pei Chao, Michael Stagner (PBCS). SASBAC and pKARMA thank The Swedish Medical Research Counsel. We thank Petra Bos, Jannet Blom, Ellen Crepin, Anja Nieuwlaat, Annette Heemskerk, the Erasmus MC Family Cancer Clinic (RBCS), and Sue Higham, Helen Cramp, Ian Brock, Sabapathy Balasubramanian, Jenny Barrett, Mark Iles, John Taylor and Dan Connley (SBCS). We thank the SEARCH and EPIC teams, and Breakthrough Breast Cancer and the Institute of Cancer Research for support and funding of the Breakthrough Generations Study. We thank Muriel Adank for selecting the samples and Margreet Ausems, Christi van Asperen, Senno Verhoef and Rogier van Oldenburg for providing samples from their Clinical Genetic centers (DFBBCS). The SGBCC thank the participants and research coordinator Kimberley Chua. The results published here are in part based upon data generated by The Cancer Genome Atlas pilot project established by the NCI and NHGRI (dbGAP phs000178.v8.p7, accessed 15 November 2013). Information about TCGA and the investigators and institutions who constitute the TCGA research network can be found at http://cancergenome.nih.gov. We also thank Westra et al. for the provision of eQTL data at http://genenetwork.nl/bloodeqtlbrowser/. The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health (commonfund.nih.gov/GTEx). Additional funds were provided by the NCI, NHGRI, NHLBI, NIDA, NIMH and NINDS. Donors were enrolled at Biospecimen Source Sites funded by NCI\SAIC-Frederick, Inc. (SAIC-F) subcontracts to the National Disease Research Interchange (10XS170), Roswell Park Cancer Institute (10XS171) and Science Care, Inc. (X10S172). The Laboratory, Data Analysis and Coordinating Center (LDACC) was funded through a contract (HHSN268201000029C) to the The Broad Institute, Inc. Biorepository operations were funded through an SAIC-F subcontract to Van Andel Institute (10ST1035). Additional data repository and project management were provided by SAIC-F (HHSN261200800001E). Statistical Methods development grants were made to the University of Geneva (MH090941), the University of Chicago (MH090951 and MH090937), the University of North Carolina—Chapel Hill (MH090936) and to Harvard University (MH090948). The datasets used for the analyses described in this manuscript were obtained from the GTEx Portal on 01/16/2014.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Stratton M.R., Rahman N. The emerging landscape of breast cancer susceptibility. Nat. Genet. 2008;40:17–22. doi: 10.1038/ng.2007.53. [DOI] [PubMed] [Google Scholar]
- 2.Michailidou K., Hall P., Gonzalez-Neira A., Ghoussaini M., Dennis J., Milne R.L., Schmidt M.K., Chang-Claude J., Bojesen S.E., Bolla M.K., et al. Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nat. Genet. 2013;45:353–361. doi: 10.1038/ng.2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Welter D., MacArthur J., Morales J., Burdett T., Hall P., Junkins H., Klemm A., Flicek P., Manolio T., Hindorff L., et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014;42:D1001–D1006. doi: 10.1093/nar/gkt1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.MacPherson G., Healey C.S., Teare M.D., Balasubramanian S.P., Reed M.W.R., Pharoah P.D.P., Ponder B.A.J., Meath M., Bhattacharyya N.P., Cox A. Association of a common variant of the CASP8 gene with reduced risk of breast cancer. J. Natl. Cancer Inst. 2004;96:1866–1869. doi: 10.1093/jnci/dji001. [DOI] [PubMed] [Google Scholar]
- 5.Cox A., Dunning A.M., Garcia-Closas M., Balasubramanian S., Reed M.W.R., Pooley K.A., Scollen S., Baynes C., Ponder B.A.J., Chanock S., et al. A common coding variant in CASP8 is associated with breast cancer risk. Nat. Genet. 2007;39:352–358. doi: 10.1038/ng1981. [DOI] [PubMed] [Google Scholar]
- 6.Shephard N.D., Abo R., Rigas S.H., Frank B., Lin W.Y., Brock I.W., Shippen A., Balasubramanian S.P., Reed M.W.R., Bartram C.R., et al. A breast cancer risk haplotype in the caspase-8 gene. Cancer Res. 2009;69:2724–2728. doi: 10.1158/0008-5472.CAN-08-4266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Camp N.J., Parry M., Knight S., Abo R., Elliott G., Rigas S.H., Balasubramanian S.P., Reed M.W., McBurney H., Latif A., et al. Fine-mapping CASP8 risk variants in breast cancer. Cancer Epidemiol. Biomarkers Prev. 2012;21:176–181. doi: 10.1158/1055-9965.EPI-11-0845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Turnbull C., Ahmed S., Morrison J., Pernet D., Renwick A., Maranian M., Seal S., Ghoussaini M., Hines S., Healey C.S., et al. Genome-wide association study identifies five new breast cancer susceptibility loci. Nat. Genet. 2010;42:504–U547. doi: 10.1038/ng.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pruim R.J., Welch R.P., Sanna S., Teslovich T.M., Chines P.S., Gliedt T.P., Boehnke M., Abecasis G.R., Willer C.J. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics. 2010;26:2336–2337. doi: 10.1093/bioinformatics/btq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hoggart C.J., Whittaker J.C., De Iorio M., Balding D.J. Simultaneous analysis of All SNPs in genome-wide and re-sequencing association studies. PLoS Genet. 2008;4:e1000130. doi: 10.1371/journal.pgen.1000130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Westra H.J., Peters M.J., Esko T., Yaghootkar H., Schurmann C., Kettunen J., Christiansen M.W., Fairfax B.P., Schramm K., Powell J.E., et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat. Genet. 2013;45:1238–1243. doi: 10.1038/ng.2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hnisz D., Abraham B.J., Lee T.I., Lau A., Saint-André V., Sigova A.A., Hoke H.A., Young R.A. Super-enhancers in the control of cell identity and disease. Cell. 2013;155:934–947. doi: 10.1016/j.cell.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barrett J.H., Iles M.M., Harland M., Taylor J.C., Aitken J.F., Andresen P.A., Akslen L.A., Armstrong B.K., Avril M.F., Azizi E., et al. Genome-wide association study identifies three new melanoma susceptibility loci. Nat. Genet. 2011;43:1108–1113. doi: 10.1038/ng.959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berndt S.I., Skibola C.F., Joseph V., Camp N.J., Nieters A., Wang Z., Cozen W., Monnereau A., Wang S.S., Kelly R.S., et al. Genome-wide association study identifies multiple risk loci for chronic lymphocytic leukemia. Nat. Genet. 2013;45:868–876. doi: 10.1038/ng.2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Howie B.N., Donnelly P., Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009;5:e1000529. doi: 10.1371/journal.pgen.1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Howie B., Marchini J., Stephens M. Genotype imputation with thousands of genomes. G3 (Bethesda, MD) 2011;1:457–470. doi: 10.1534/g3.111.001198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Benjamini Y., Hochberg Y. Controlling the false discovery rate – a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Methodol. 1995;57:289–300. [Google Scholar]
- 18.French J.D., Ghoussaini M., Edwards S.L., Meyer K.B., Michailidou K., Ahmed S., Khan S., Maranian M.J., O'Reilly M., Hillman K.M., et al. Functional variants at the 11q13 risk locus for breast cancer regulate cyclin D1 expression through long-range enhancers. Am. J. Hum. Genet. 2013;92:489–503. doi: 10.1016/j.ajhg.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Viechtbauer W. Conducting meta-analyses in R with the metafor package. J. Stat. Softw. 2010;36:1–48. [Google Scholar]
- 20.Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.