

Abstract
Microbial infections can induce aberrant responses in cellular stress pathways, leading to translational attenuation, metabolic restriction, and activation of oxidative stress, with detrimental effects on cell survival. Here we show that infection of human airway epithelial cells with Streptococcus pneumoniae leads to induction of endoplasmic reticulum (ER) and oxidative stress, activation of mitogen-associated protein kinase (MAPK) signaling pathways, and regulation of their respective target genes. We identify pneumococcal H2O2 as the causative agent for these responses, as both catalase-treated and pyruvate oxidase-deficient bacteria lacked these activities. Pneumococcal H2O2 induced nuclear NF-κB translocation and transcription of proinflammatory cytokines. Inhibition of translational arrest and ER stress by salubrinal or of MAPK signaling pathways attenuate cytokine transcription. These results provide strong evidence for the notion that inhibition of translation is an important host pathway in monitoring harmful pathogen-associated activities, thereby enabling differentiation between pathogenic and nonpathogenic bacteria.
Keywords: Streptococcus pneumonia, hydrogen peroxide, ER stress, MAPK, immune response
Streptococcus pneumoniae is a frequent cause of community-acquired pneumonia (CAP), meningitis, sinusitis, and otitis media. It is estimated that annually >1 million children worldwide die from pneumococcal infections, which, moreover, account for up to 25% of all preventable deaths in children aged <2 years. S. pneumoniae primarily resides on the mucosal surface of the nasopharynx but can cause severe disease following translocation to the lower respiratory tract, lungs, blood, and brain [1]. Colonization requires production of the capsular polysaccharide (CPS), phosphorylcholine (ChoP), and an array of bacterial surface proteins, including pneumococcal adhesion and virulence A (PavA) protein, pneumococcal surface protein A (PspA) and PspC, that bind extracellular matrix components as well as the plasmonigen-binding enolase [2]. Other surface molecules, including hyaluronidase (Hyl), serine protease (PrtA), and the neuraminidases NanA, BgaA, and StrH, enable the spread of the bacteria [2]. An essential virulence factor produced by S. pneumoniae is the cholesterol-dependent cytolysin pneumolysin (PLY). Its role and contribution to S. pneumoniae virulence is well established and has been documented in many studies comparing wild-type S. pneumoniae to isogenic PLY-deficient strains [3–5]. Curiously, although a role for pneumococcal H2O2 in the pathogenesis of S. pneumoniae infections has been known for a long time [6–8], it is not featured in recent reviews on this bacterium.
Infection with S. pneumoniae has been shown to have many effects on host physiology and immune defense. It can promote activation of host complement, potentiate neutrophil activity, and enhance production of proinflammatory cytokines in macrophages and monocytes. Studies have revealed the activation of p38 mitogen-activated protein kinase (MAPK), as well as the activation of nuclear factor of activated T cells (NFAT). In addition pneumococcal infection has been reported to induce calcium influx, damage of mitochondria, and activation of cytoskeletal rearrangements [9]. Many of these effects have been attributed to the membrane permeable properties of PLY [10].
We recently showed that the cholesterol-dependent cytolysin toxin of Listeria monocytogenes listeriolysin O (LLO) induces an endoplasmic reticulum (ER) stress response in cells even before bacterial infection [11]. ER stress is mitigated by several complementary mechanisms collectively known as the unfolded protein response (UPR). The UPR is a complex signal transduction pathway that leads to translational attenuation permitting selective expression of proteins involved in folding, quality control, and transport into the ER and organelle biogenesis. Several sensors located at the ER membrane, such as the inositol-requiring enzyme 1 (IRE1), protein kinase RNA (PKR)–like ER kinase (PERK), and activating transcription factor 6 (ATF6), control the UPR. These sensors transduce information about protein folding status at the ER lumen to the nucleus and cytosol by controlling the expression of specific transcription factors [12]. We demonstrated that all of the 3 ER sensor pathways are activated during infection with L. monocytogenes by extracellular LLO and that induction of the UPR restricts L. monocytogenes growth in infected cells [11].
In this study, we examined the ability of S. pneumoniae to induce ER stress in club cell–like H441 lung epithelial cells following infection. We reasoned that, because PLY and LLO are both members of the family of cholesterol-dependent cytolysin toxins, PLY would be a major pneumococcal factor inducing ER stress. Unexpectedly, our results demonstrated that a single bacterially produced molecule, pneumococcal H2O2, rather than PLY, is responsible for the induction of ER stress and further perturbations of cellular activity.
MATERIALS AND METHODS
Cell Culture
Human club cell–like H441 lung adenocarcinoma cells were obtained from ATCC (Wesel, Germany) and were grown in Roswell Park Memorial Institute (RPMI) 1640 medium (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS; PAA, Pasching, Austria) in a humidified atmosphere of 5% CO2 at 37°C.
The infection was done in medium containing 0.5% FBS. Bovine catalase at 1000 U/mL (Sigma-Aldrich, St. Louis, MO) was added directly before infection. Cells were pretreated with JNK and ERK inhibitor (SP600125 10 µM, U0126 50 µM; Sigma), as well as p38 inhibitor (SB203580 10 µM; Tocris, Bristol, United Kingdom) and salubrinal (50 µM; Sigma) 30 minutes before infection. H2O2 was obtained from Sigma.
Bacterial Strains
All S. pneumoniae strains used in this study are listed in the Supplementary Materials. Bacteria were cultured in Todd-Hewitt broth plus 0.5% yeast extract or on blood-agar plates in 5% CO2. Unless otherwise indicated, infections were performed at a multiplicity of infection (MOI) of 25–45.
Preparation of Bacterial Culture Supernatant
Bacteria were inoculated in RPMI 1640 medium plus 0.5% FBS. After 5 hours of growth at 37°C, the bacterial solution was centrifuged for 10 minutes at 6000 × g at 4°C. The sterile filtered (0.22-µm-pore membrane) supernatants were then stored protected from light at 4°C.
Purification of PLY
PLY was purified as described before [48].
Immunoblotting
Cells were prepared as described before [11]. GAPDH was used as a loading control. Antibodies are listed in the Supplementary Materials.
NanoPro Technology
After infection, cells were pelleted, washed twice with Cell Wash buffer (ProteinSimple, Santa Clara, CA), and lysed in CHAPS buffer containing dimethyl sulfoxide and aqueous inhibitors (ProteinSimple). Preparation of cell lysates for nanofluidic isoelectric focusing, using the PEGGY system, occurred as described in the manufacturer's protocol (ProteinSimple). Antibodies are listed in the Supplementary Materials.
Quantitative Polymerase Chain Reaction (qPCR) Analysis
Protocols of RNA isolation and qPCR analysis were described before [11]. All primers used in this study are listed in the Supplementary Materials. hprt1, tbp, and b2m were used as internal controls.
Immunofluorescence
Infected cells were fixed with 4% paraformaldehyde, permeabilized with methanol, and stained with anti-NFκB antibody (sc-8008, Santa Cruz Biotechnology, Santa Cruz, CA). Cells were observed under confocal fluorescence microscopy (Leica Mikrosysteme Vertrieb, Wetzlar, Germany). Quantification of nuclear NFkB translocation was performed as previously described [49].
Immunoelectron Microscopy of Ultrathin Cryosections
H441 cells were infected at a MOI of 65–80. Preparation of cells for transmission electron microscopy and staining with anti-PDI antibody (sc-59640, Santa Cruz Biotechnology) was performed as previously described [11].
Measurement of Reactive Oxygen Species (ROS) Production
Production of mitochondrial ROS was measured using the MitoSox dye from Life Technologies.
Statistics
Data are expressed as means ± standard deviation. Differences between 2 groups were assessed using the Student t test.
RESULTS
S. pneumoniae Induces the Unfolded Protein Response via the Signal Transducer PERK
We used transmission electron microscopy to monitor for changes in ER morphology following S. pneumoniae infection of H441, using gold-labeled antibody against the ER resident protein disulfide-isomerase (PDI). We observed strong widening of ER lumen in infected cells that is indicative of ER stress induction (Figure 1A and Supplementary Figure 1) [13].
Figure 1.
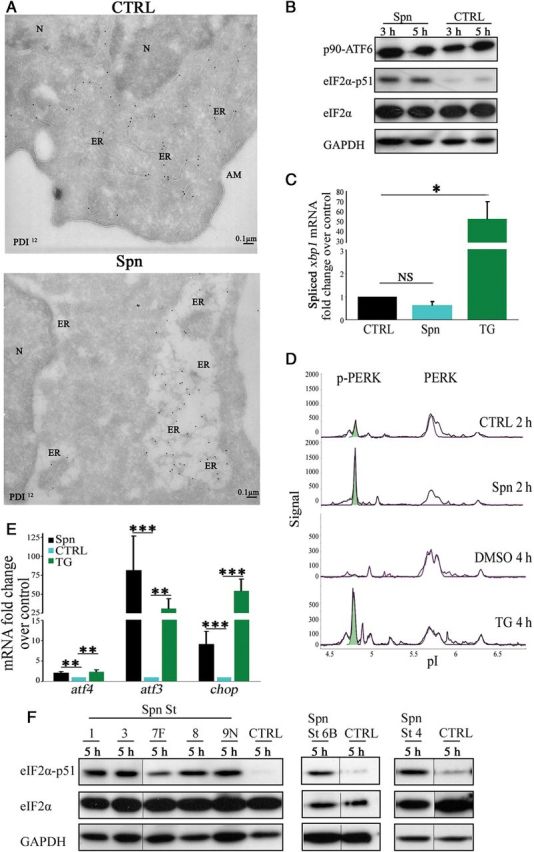
Streptococcus pneumoniae induces the protein kinase RNA (PKR)–like endoplasmic reticulum (ER) kinase (PERK) branch of unfolded protein response (UPR) in H441 cells. A, Cells were infected, fixed after 5 hours, sectioned, stained for ER marker protein disulfide-isomerase (PDI), and analyzed by transmission electron microscopy. B, Representative immunoblot of unprocessed activating transcription factor 6, phosphorylated eukaryotic initiation factor 2α (eIF2α), and total eIF2α in cells infected with S. pneumoniae. C and E, Quantitative polymerase chain reaction analysis of spliced xbp1, atf4, atf3, and chop in cells infected with S. pneumoniae or treated with 20 µM thapsigargin (TG) for 5 hours, normalized to noninfected/dimethyl sulfoxide–treated cells. D, Representative nanofluidic isoelectric focusing of PERK in lysates of cells infected with S. pneumoniae or treated with 20 µM TG. F, Representative immunoblot of phosphorylated and total eIF2α in lysates of cells infected with S. pneumoniae serotype 1, 3, 4, 6B, 7F, 8, and 9. Bar graphs show mean + standard deviation from 4 independent experiments. *P < .05, **P < .01, and ***P < .001, by the Student t test. Abbreviations: AM, apical membrane; CTRL, control; N, nucleus; NS, not significant; Spn, Streptococcus pneumoniae; St, serotype.
Analysis of infected and noninfected cells for activation of the 3 UPR signaling pathways revealed phosphorylation of the eukaryotic initiation factor 2α (eIF2α), indicating an arrest of protein synthesis in infected cells (Figure 1B). By contrast, no activation of the ATF6 and IRE1 pathway, as determined by monitoring ATF6 cleavage and splicing of xbp1 messenger RNA (mRNA), respectively, was detected upon S. pneumoniae infection (Figure 1B and 1C). In eukaryotic cells, a family of kinases that respond to stress induces eIF2α phosphorylation [14]. To rule out the involvement of any kinases independent from ER stress induction, we confirmed the activation of PERK, PKR, and general control non-derepressible-2 (GCN2) by nanofluidic isoelectric focusing (NanoPro), which allows antibody-mediated detection of protein expression and posttranslational modifications in a highly sensitive manner. Phosphorylation increases the acidic properties of the protein that can be monitored by changes in the isoelectric point (pI), resulting in the appearance of additional peaks of lower pI following protein separation. Noninfected cells showed major peaks at a pI of around 5.6 (nonphosphorylated PERK) and a small peak at 4.75 (phosphorylated PERK). Infection of the cells with S. pneumoniae led to a significant increase of the peak at a pI of 4.75 (Figure 1D), indicating increased phosphorylation of PERK. Treatment of cells with the well-established ER stress inductor thapsigargin (TG) also resulted in an increase of the peak at a pI of 4.75 (Figure 1D). We found no changes in phosphorylation levels in PKR and GCN2 (Supplementary Figure 2). However, while phosphorylation of PKR was clearly inducible by addition of poly I:C, we were unable to find conditions to independently verify changes in phosphorylation of GCN2 in H441 cells through amino acid starvation or the addition of urea. To further confirm selective induction of eIF2α by PERK, we examined downstream signals emanating from its activation. Expression of ATF4 and ATF3 (activating transcription factors 4 and 3, respectively), as well as the proapoptotic factor CHOP/GADD153 (growth arrest and DNA damage induced gene-153), are increased upon activation of PERK along with phosphorylation of eIF2α [12]. The transcription of all 3 downstream factors was increased following S. pneumoniae infection and was comparable to that in cells treated with TG (Figure 1E). The phosphorylation of PERK and eIF2α, as well as the increased expression of atf4, atf3, and chop, suggests that an accumulation of unfolded proteins leads to the selective activation of the PERK pathway of the UPR following S. pneumoniae infection of H441 cells.
We wondered whether activation of the UPR is a commonly occurring response to S. pneumoniae infection and examined different clinically relevant S. pneumoniae serotypes (1, 3, 4, 6B, 7F, 8, and 9N) [15] for their ability to induce this stress response. All of the representative serotype strains examined showed phosphorylation of eIF2α, suggesting that activation of the UPR is a common feature of the S. pneumoniae serotypes tested (Figure 1F).
Pneumococcal H2O2 and Not Pneumolysin Is Required for UPR Induction
We were puzzled by the observation that the S. pneumoniae serotype 1 and 8 strains, both of which produce a nonhemolytic form of PLY [16], led to phosphorylation of eIF2α (Figure 1F). Induction of UPR previously has been associated with pore-forming toxins and led us to assume that PLY is involved in PERK activation [11, 17]. To investigate whether PLY is involved, we performed 2 sets of experiments. First, we infected cells with a PLY-negative S. pneumoniae strain (Δply) and found that the strain was still capable of inducing phosphorylation of eIF2α and the transcription of atf4, atf3, and chop mRNA (Figure 2A). Second, we detected no phosphorylation of eIF2α in cells treated with increasing concentrations of purified PLY (Figure 2B). Thus, these results suggest that PLY is not required for the induction of the UPR in H441 cells.
Figure 2.

Pneumococcal H2O2 is required for unfolded protein response (UPR) induction. A, Representative immunoblot of phosphorylated and total eukaryotic initiation factor 2α (eIF2α) and quantitative polymerase chain reaction (qPCR) of atf4, atf3, and chop of cells infected with Streptococcus pneumoniae Δply for 5 hours. B–D and F, Representative immunoblot of phosphorylated and total eIF2α of cells treated with purified pneumolysin (PLY; B), cells treated with bacterial supernatant (SN) and H2O2 with and without catalase (Cat; C), and cells infected with wild-type (WT) S. pneumoniae with and without Cat (D) and S. pneumoniae ΔspxB (F). E, qPCR analysis of atf4, atf3, and chop of cells infected with WT S. pneumoniae with and without Cat and S. pneumoniae ΔspxB for 5 hours. Bar graphs show mean + standard deviation from 3 to 4 independent experiments. *P < .05, **P < .01, and ***P < .001, by the Student t test. Abbreviations: CTRL, control; NS, not significant; Spn, Streptococcus pneumoniae.
To examine whether a secreted factor is responsible for activation of the UPR, we exposed H441 cells to S. pneumoniae culture supernatants. Indeed, cells treated with the supernatants showed phosphorylation of eIF2α (Figure 2C). Treatment of the supernatants with catalase abolished the phosphorylation of eIF2α, suggesting a role for H2O2. When H2O2 was added directly to H441 cells in culture, eIF2α phosphorylation was also detected, indicating that H2O2 is indeed the active molecule involved (Figure 2C).
S. pneumoniae, like many lactobacilli, accumulates H2O2 in the supernatant because of a lack of catalase [18]. To determine the impact of pneumococcal H2O2 on the UPR induction, we treated cells with catalase during the entire course of infection. The reduction of H2O2 levels by catalase abolished the phosphorylation of eIF2α and significantly diminished mRNA levels of atf4, atf3, and chop during infection (Figure 2D and 2E). We used a mutant strain lacking spxB encoding pyruvate oxidase, which is responsible for most of the H2O2 produced by S. pneumoniae to definitively confirm that bacteria were the source of the H2O2 seen in our study. Infection of cells with S. pneumoniae strain ΔspxB did not result in the phosphorylation of eIF2α or in an increase of mRNA levels of atf4, atf3, and chop contrary to wild-type S. pneumoniae–infected cells (Figure 2E and 2F). To exclude the involvement of other pneumococcal virulence factors, we infected cells with mutants lacking the capsule, the autolysins, or the adhesion factor PavA. Infection with all tested mutants still increased the eIF2α phosphorylation (Supplementary Figure 3). Taken together, these results show that the activation of the PERK UPR pathway in H441 cells is due to pneumococcal H2O2 upon S. pneumoniae infection.
Activation of Global Stress Responses by Pneumococcal H2O2
Because accumulation of ROS, including H2O2, induces oxidative stress, we wondered whether this was also provoked by infection with S. pneumoniae. To test this, we investigated the production of mitochondrial ROS (mtROS). Infection with either wild-type S. pneumoniae or S. pneumoniae Δply but not with S. pneumoniae ΔspxB led to an increased production of mtROS, compared with observations for noninfected cells, indicating induction of oxidative stress by pneumococcal H2O2 accumulation (Figure 3A). To prevent oxidative injuries, cells have evolved a stress response that enables the increased expression of antioxidants. The transcription factor NRF2 (nuclear factor erythroid 2-related factor 2) plays a major role in transcriptional activation of antioxidant enzymes and ROS scavengers, including heme oxygenase 1 (HO-1), NADH dehydrogenase, and superoxide dismutase [19]. To determine whether an antioxidant response is induced by S. pneumoniae infection, we examined the transcription level of nrf2 and ho-1. Cells infected with wild-type S. pneumoniae showed increased levels of both mRNAs. However, infection with S. pneumoniae ΔspxB abolished the transcription levels of nrf2 and ho-1, indicating that the antioxidant response following S. pneumoniae infection is also dependent on the production of pneumococcal H2O2 (Figure 3B).
Figure 3.

Pneumococcal H2O2 induces oxidative stress and mitogen-associated protein kinase (MAPK) signaling. A, Representative mitochondrial reactive oxygen species measurement by flow cytometry in cells infected with either wild-type (WT) Streptococcus pneumoniae and S. pneumoniae Δply or S. pneumoniae ΔspxB. B, Quantitative polymerase chain reaction analysis of nrf2 and ho-1 in lysates of cells infected with WT S. pneumoniae, S. pneumoniae Δply, and S. pneumoniae ΔspxB for 5 hours. C, Example nanofluidic isoelectric focusing of either “phospho-JNK, phospho-p38 and ERK” or “phospho c-Jun N-terminal kinase (JNK), phospho-p38 and extracellular-signal regulated kinase (ERK) of cells infected with WT S. pneumoniae, S. pneumoniae Δply, and S. pneumoniae ΔspxB. Bar graphs show mean + standard deviation from 3–4 independent experiments. *P < .05, **P < .01, and ***P < .001, by the Student t test. Abbreviations: CTRL, control; NS, not significant; Spn, Streptococcus pneumoniae.
A variety of stimuli, such as growth factors and cellular stresses, activate pathways of members of the MAPK family, which exerts an important role in signal transduction [20–21]. To examine the role of pneumococcal H2O2 on MAPK signaling during S. pneumoniae infection, we analyzed the activation of these kinases by using specific antibodies to detect their phosphorylation status following nanofluidic isoelectric focusing. Cells infected with wild-type S. pneumoniae and S. pneumoniae Δply showed peaks corresponding to phosphorylated p38 and JNK at 1 hour and 3 hours after infection that were not visible after 5 hours. These peaks were not detected in cells infected with S. pneumoniae ΔspxB or in noninfected cells (Figure 3C). Infection with wild-type S. pneumoniae and S. pneumoniae Δply also led to an increase in peaks representing the single and dually phosphorylated forms of ERK1 and ERK2, which were not detected in S. pneumoniae ΔspxB–infected or noninfected cells (Figure 3C). Thus, infection with S. pneumoniae induces activation of all 3 MAPK subfamilies, which depends on the production of pneumococcal H2O2.
Induction of an Immune Response by Pneumococcal H2O2
Lung parenchymal cells have previously been shown to contribute to cytokine production in response to different pathogens, including Legionella pneumophila, Klebsiella pneumonia, Haemophilus influenza, and Pseudomonas aeruginosa [22–25]. We wondered whether pneumococcal H2O2 directly induced an immune response in the H441 cells. First, we analyzed nuclear translocation of the proinflammatory activator NFκB. Infection with wild-type S. pneumoniae and S. pneumoniae Δply led to an increase in nuclear localization of NFκB, as compared to noninfected cells. By contrast, cells infected with S. pneumoniae ΔspxB showed no NFκB translocation. Furthermore, addition of catalase during infection with wild-type S. pneumoniae diminished NFκB activation (Figure 4A). Next, we analyzed the transcription of il8 and il23a, which encode a proinflammatory chemokine and cytokine, respectively, and are important in mucosal immunity. Infection with S. pneumoniae indeed resulted in an increased transcription of both proinflammatory mediators. The addition of catalase reduced the S. pneumoniae–induced transcription of il8 and il23a. Cells exposed to S. pneumoniae ΔspxB had a significantly diminished interleukin response, compared with the wild-type strain. However, cells treated with S. pneumoniae Δply exhibited no significant differences in interleukin expression, compared with the wild-type strain (Figure 4B). Taken together, these results show an H2O2-dependent immune response induction following infection of H441 cells with S. pneumoniae.
Figure 4.

Activation of innate immune response by pneumococcal H2O2. A, Immunofluorescence staining of NFκB and quantification of nuclear:cytoplasmic ratios of NFκB staining in cells infected with wild-type (WT) Streptococcus pneumoniae with and without catalase (Cat), as well as S. pneumoniae strains Δply and ΔspxB, for 3 hours. Arrows indicate NFκB fluorescence in the nucleus. B, Quantitative polymerase chain reaction (qPCR) analysis of il8 and il23a in cells infected with WT S. pneumoniae with and without catalase (Cat), as well as S. pneumoniae strains Δply and ΔspxB, for 5 hours. Bar graphs show mean ± standard deviation from 3 independent experiments. *P < .05 and **P < .01, by the Student t test. Abbreviations: CTRL, control; NS, not significant; Spn, Streptococcus pneumoniae.
Inhibition of ER Stress and MAPK Signaling Attenuate S. pneumoniae–Mediated Cytokine Transcription
Examination of ER stress and the UPR has demonstrated many links to inflammatory signaling. Induction of ER stress is involved in regulation of interleukin 8, interleukin 6, and monocyte chemoattractant protein 1 in human endothelial cells [26]. We used the ER stress inhibitor salubrinal [27, 28] to investigate the role of ER stress induction during S. pneumoniae–mediated transcriptional activation of cytokines. Treatment of cells with salubrinal prior to infection diminished S. pneumoniae–mediated transcription of il8 and il23a, as well as that of the UPR target genes atf3 and chop (Figure 5A). Previous studies of infection of lung epithelial cells have shown that S. pneumoniae–mediated production of IL8 is regulated by p38 and JNK [29, 30]. Using specific inhibitors for p38, JNK, and ERK, we tested the involvement of MAPK signaling in S. pneumoniae–mediated cytokine expression in H441 cells. Transcription of il8 and il23a was reduced by inhibiting JNK and ERK, while inhibition of p38 had no effect on il8 transcription and even increased il23a transcription following S. pneumoniae infection. In addition, inhibition of JNK and ERK also reduced transcription of atf3 and chop (Figure 5B). Thus, S. pneumoniae–mediated transcriptional activation of proinflammatory interleukins is regulated by ER stress induction, as well as by JNK and ERK pathways, in H441 cells.
Figure 5.

Transcription of proinflammatory cytokines is regulated by endoplasmic reticulum (ER) stress induction, as well as by JNK and ERK signaling. A, Quantitative polymerase chain reaction (qPCR) analysis of il8 and il23a, as well as atf3 and chop, in cells treated with salubrinal (Sal) or dimethyl sulfoxide (DMSO) vehicle prior to infection with wild-type (WT) Streptococcus pneumoniae for 5 hours. B, qPCR analysis of il8 and il23a, as well as atf3 and chop, in cells treated with inhibitors of JNK (SP600125), ERK (U0126), p38 (SB203580), or DMSO vehicle prior to infection with S. pneumoniae WT for 5 hours. Bar graphs show mean + standard deviation from 3 independent experiments. *P < .05, **P < .01, and ***P < .001, by the Student t test. Abbreviations: NS, not significant; Spn, Streptococcus pneumoniae.
DISCUSSION
In this study, we demonstrated that S. pneumoniae infection of lung epithelial cells has profound effects on cellular stress response pathways, including activation of the UPR and the MAPK pathways, as well as on the induction of the oxidative stress response. Exposure of cells to S. pneumoniae also resulted in the activation of the proinflammatory transcription factor NFκB, accompanied by increased transcription of the interleukin-encoding genes il8 and il23a. Induction of the UPR and the MAPK pathways is involved in regulation of interleukin transcription. These cellular activities do not involve the recognition of well-known pathogen associated molecular patterns (PAMPs) of S. pneumoniae and are dependent on the production of a single bacterial molecule, pneumococcal H2O2 (Figure 6). Indeed, our data suggest an explanation for the virulent properties of S. pneumoniae isolates that lack pneumolysin.
Figure 6.

Pneumococcal H2O2 targets cellular stress responses accompanied by induction of the innate immune response. Infection of H441 cells with Streptococcus pneumoniae results in induction of endoplasmic reticulum (ER) and oxidative stress responses, as well as in the triggering of mitogen-associated protein kinase (MAPK) signaling pathways. Pneumococcal H2O2 is the causative agent responsible for these reactions. Infection with S. pneumoniae moreover leads to H2O2-dependent activation of an innate immune response. This is in part regulated by ER stress induction, as well as by JNK and ERK MAPKs. Abbreviations: IL-8, interleukin 8; IL-23a, interleukin 23a; Spn, Streptococcus pneumoniae.
S. pneumoniae produces large amounts of H2O2 (1–3 mM) [31], and its role in inducing cellular damage and promoting virulence has been previously documented [6, 8]. For S. pneumoniae, the major enzyme involved in the production of H2O2 is the pyruvate oxidase SpxB. Apart from its role in central metabolism, SpxB has an important role in colonization and the virulence properties of this bacterium [8, 32]. Levels of H2O2 produced by S. pneumoniae are also variable and are often associated with mutations within the spxB gene. Thus, for example, a variant spxB allele of R36A and R6 is associated with increased cellular pyruvate oxidase activity relative to that of the ancestral strain D39 [33]. The gene is absent in other species of streptococci, except for some streptococcal species that colonize the oropharynx, such as Streptococcus gordonii, Streptococcus oralis, and Streptococcus sanguinis [34].
These data suggest that surveillance and detection of pathogen-associated activities—here, pneumococcal production of H2O2—represent complementary modes of innate immune recognition. In epithelial cells, PAMP recognition may be more limited than the activity of innate immune cells, to avoid recurrent inflammatory responses. A role for detection of pathogen-associated disruption of the integrity of cellular signaling pathways may have particular relevance for epithelial cells, which are exposed to a variety of microorganisms but do not possess the full repertoire of pattern-recognition receptors (PRRs), by distinguishing between properties exhibited by pathogenic and nonpathogenic bacteria [35]. As demonstrated both here and previously, MAPKs activate both stress- and immune-related genes, particularly those encoding proinflammatory cytokines, and are part of the response to pathogen-induced H2O2 stress [36]. Hence, screening of pneumococcal H2O2 levels produced by various S. pneumoniae represents a mechanism by which enhanced pathogenic potential is discerned by the host.
The detection of pathogen-associated disruption of the integrity of cellular signaling pathways and its association to a protective response was first recognized as an important component of the innate immune response in plants [35]. These observations have recently been extended to infections of vertebrates and invertebrates and constitute a novel mode of immune surveillance that relies not on the recognition of pathogen-conserved molecular patterns by PRRs, but rather on the detection of pathogen-associated disruption of cellular processes. Thus, toxins and effector proteins that target the host translational machinery, such as exotoxin A of P. aeruginosa or secreted glucosyltransferases of L. pneumophila, can trigger an immune response, independent of signaling via PRR pathways [37, 38].
In addition, several toxins have been shown to induce ER stress in a wide variety of cell types [11, 17, 39, 40]. A prime example of bacterial effectors are the family of pore-forming toxins, in which where diverse but specific cell-dependent response pathways are triggered in cells exposed to sublytic concentrations of toxins. Toxin-dependent disruption of plasma-membrane integrity activates signaling pathways such as the p38 MAPK pathway, induces ER stress, and modulates mitochondrial dynamics [11, 17, 41]. Curiously, it is just these processes that are targeted by pneumococcal H2O2, as shown in this study.
Triggering of the UPR and MAPK pathways can lead to transcriptional activation of proinflammatory genes [26, 42, 43]. Apart from reduced proinflammatory interleukin transcription, inhibition of ER stress and of JNK and ERK pathways result in diminished chop transcription upon S. pneumoniae infection. Since it was shown that CHOP acts as transcriptional activator for il8 and il23a [44, 45], our results suggest the regulation of proinflammatory cytokines and chemokines via stress-induced transcription factors, including CHOP, upon S. pneumoniae infection (Figure 5). As demonstrated here, salubrinal, a small-molecule inhibitor of ER stress, can significantly dampen host cell inflammatory responses and has a potential for use in adjuvant therapy of pneumococcal infections.
Our data presented here have important implications for innate immune surveillance of pneumococcal infections. Here we demonstrate that pneumococcal H2O2 is a novel member of the growing list of bacterial virulence factors that are sensed through activation of cellular stress responses. In previous studies on the role of nonhematopoietic cells in infection with L. pneumophila, it was demonstrated that airway epithelial cells are key players in the control of bacterial airway infection [46]. The expression and secretion of the chemokine interleukin 8 by epithelial cells leads to attraction and activation of neutrophils and dendritic cells [47]. This promotes defense against invasive pathogens. Studies on the role of cell stress responses and the contribution of airway epithelial cells to immune recognition and bacterial clearance during infection with S. pneumoniae are now warranted.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online (http://jid.oxfordjournals.org). Supplementary materials consist of data provided by the author that are published to benefit the reader. The posted materials are not copyedited. The contents of all supplementary data are the sole responsibility of the authors. Questions or messages regarding errors should be addressed to the author.
Notes
Acknowledgments. We thank Dr Bastian Opitz and Dr Kathrin Mühlemann, for kindly providing different S. pneumoniae serotypes and the spxB mutant; Dr Sanjeev Kumar, for discussions; and Mrs Bettina Goritzka and Mrs Larissa Thiessen, for technical support. CIBER de Enfermedades Respiratorias (CIBERES) is an initiative of ISCIII.
Financial support. This work was supported by the Deutsche Forschungsgemeinschaft (SFB/TR84 project A04 to T. C.), the Bundesministerium für Bildung und Forschung (PROGRESS project A1 and A2 to T. C.), and the National Heart, Lung, and Blood Institute, National Institutes of Health (grant RO1HL094609 to R. L.).
Potential conflicts of interest. All authors: No potential conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Prevention of pneumococcal disease: recommendations of the Advisory Committee on Immunization Practices (ACIP) MMWR Recomm Rep. 1997;46:1–24. [PubMed] [Google Scholar]
- 2.Mitchell AM, Mitchell TJ. Streptococcus pneumoniae. Virulence factors and variation. Clin Microbiol Infect. 2010;16:411–8. doi: 10.1111/j.1469-0691.2010.03183.x. [DOI] [PubMed] [Google Scholar]
- 3.Berry AM, Yother J, Briles DE, Hansman D, Paton JC. Reduced virulence of a defined pneumolysin-negative mutant of Streptococcus pneumoniae. Infect Immun. 1989;57:2037–42. doi: 10.1128/iai.57.7.2037-2042.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Canvin JR, Marvin AP, Sivakumaran M, et al. The role of pneumolysin and autolysin in the pathology of pneumonia and septicemia in mice infected with a type 2 pneumococcus. J Infect Dis. 1995;172:119–23. doi: 10.1093/infdis/172.1.119. [DOI] [PubMed] [Google Scholar]
- 5.Hirst RA, Gosai B, Rutman A, et al. Streptococcus pneumoniae deficient in pneumolysin or autolysin has reduced virulence in meningitis. J Infect Dis. 2008;197:744–51. doi: 10.1086/527322. [DOI] [PubMed] [Google Scholar]
- 6.Bermpohl D, Halle A, Freyer D, et al. Bacterial programmed cell death of cerebral endothelial cells involves dual death pathways. J Clin Invest. 2005;115:1607–15. doi: 10.1172/JCI23223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.N'Guessan PD, Schmeck B, Ayim A, et al. Streptococcus pneumoniae R6x induced p38 MAPK and JNK-mediated caspase-dependent apoptosis in human endothelial cells. Thromb Haemost. 2005;94:295–303. doi: 10.1160/TH04-12-0822. [DOI] [PubMed] [Google Scholar]
- 8.Spellerberg B, Cundell DR, Sandros J, et al. Pyruvate oxidase, as a determinant of virulence in Streptococcus pneumoniae. Mol Microbiol. 1996;19:803–13. doi: 10.1046/j.1365-2958.1996.425954.x. [DOI] [PubMed] [Google Scholar]
- 9.Lucas R, Yang G, Gorshkov BA, et al. Protein kinase C-α and arginase I mediate pneumolysin-induced pulmonary endothelial hyperpermeability. Am J Respir Cell Mol Biol. 2012;47:445–53. doi: 10.1165/rcmb.2011-0332OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marriott HM, Mitchell TJ, Dockrell DH. Pneumolysin: A double-edged sword during the host-pathogen interaction. Curr Mol Med. 2008;8:497–509. doi: 10.2174/156652408785747924. [DOI] [PubMed] [Google Scholar]
- 11.Pillich H, Loose M, Zimmer K, Chakraborty T. Activation of the unfolded protein response by Listeria monocytogenes. Cell Microbiol. 2012;14:949–64. doi: 10.1111/j.1462-5822.2012.01769.x. [DOI] [PubMed] [Google Scholar]
- 12.Schröder M. Endoplasmic reticulum stress responses. Cell Mol Life Sci. 2008;65:862–94. doi: 10.1007/s00018-007-7383-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sriburi R. XBP1: a link between the unfolded protein response, lipid biosynthesis, and biogenesis of the endoplasmic reticulum. J Cell Biol. 2004;167:35–41. doi: 10.1083/jcb.200406136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wek R, Jiang H, Anthony T. Coping with stress: eIF2 kinases and translational control. Biochem Soc Trans. 2006;34:7. doi: 10.1042/BST20060007. [DOI] [PubMed] [Google Scholar]
- 15.Scott JAG, Hall AJ, Dagan R, et al. Serogroup-Specific epidemiology of Streptococcus pneumoniae: Associations with age, sex, and geography in 7,000 episodes of invasive disease. Clin Infect Dis. 1996;22:973–81. doi: 10.1093/clinids/22.6.973. [DOI] [PubMed] [Google Scholar]
- 16.Jefferies JMC, Johnston CHG, Kirkham LS, et al. Presence of nonhemolytic pneumolysin in serotypes of Streptococcus pneumoniae associated with disease outbreaks. J Infect Dis. 2007;196:936–44. doi: 10.1086/520091. [DOI] [PubMed] [Google Scholar]
- 17.Bischof LJ, Kao C, Los FCO, et al. Activation of the unfolded protein response is required for defenses against bacterial pore-forming toxin in vivo. PLoS Pathog. 2008;4:e1000176. doi: 10.1371/journal.ppat.1000176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Avery OT, Morgan HJ. The occurrence of peroxide in cultures of pneumococcus. J Exp Med. 1924;39:275–87. doi: 10.1084/jem.39.2.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ma Q. Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol. 2013;53:401–26. doi: 10.1146/annurev-pharmtox-011112-140320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gehart H, Kumpf S, Ittner A, Ricci R. MAPK signalling in cellular metabolism: stress or wellness? EMBO Rep. 2010;11:834–40. doi: 10.1038/embor.2010.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Son Y, Cheong Y, Kim N, Chung H, Kang DG, Pae H. Mitogen-activated protein kinases and reactive oxygen species: How can ROS activate MAPK pathways? J Signal Transduct. 2011;2011:792639. doi: 10.1155/2011/792639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schmeck B, N'Guessan PD, Ollomang M, et al. Legionella pneumophila-induced NF- B- and MAPK-dependent cytokine release by lung epithelial cells. Eur Respir J. 2006;29:25–33. doi: 10.1183/09031936.00141005. [DOI] [PubMed] [Google Scholar]
- 23.Kube D, Sontich U, Fletcher D, Davis PB. Proinflammatory cytokine responses to P. aeruginosa infection in human airway epithelial cell lines. Cell Mol Phyiol. 2001;280:L493–502. doi: 10.1152/ajplung.2001.280.3.L493. [DOI] [PubMed] [Google Scholar]
- 24.Clemans DL, Bauer RJ, Hanson JA, et al. Induction of proinflammatory cytokines from human respiratory epithelial cells after stimulation by nontypeable Haemophilus influenzae. Infect Immun. 2000;68:4430–40. doi: 10.1128/iai.68.8.4430-4440.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Raju S, Painter R, Bagby G, Nelson S, Wang G. Response of differentiated human airway epithelia to alcohol exposure and Klebsiella pneumoniae challenge. Med Sci. 2013;1:2–19. doi: 10.3390/medsci1010002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gargalovic PS, Gharavi NM, Clark MJ, et al. The unfolded protein response is an important regulator of inflammatory genes in endothelial cells. Arterioscler Thromb Vasc Biol. 2006;26:2490–6. doi: 10.1161/01.ATV.0000242903.41158.a1. [DOI] [PubMed] [Google Scholar]
- 27.Boyce M. A Selective Inhibitor of eIF2 dephosphorylation protects cells from ER stress. Science. 2005;307:935–9. doi: 10.1126/science.1101902. [DOI] [PubMed] [Google Scholar]
- 28.Gong T, Wang Q, Lin Z, Chen M, Sun G. Endoplasmic reticulum (ER) stress inhibitor salubrinal protects against ceramide-induced SH-SY5Y cell death. Biochem Biophys Res Commun. 2012;427:461–5. doi: 10.1016/j.bbrc.2012.08.068. [DOI] [PubMed] [Google Scholar]
- 29.Schmeck B, Zahlten J, Moog K, et al. Streptococcus pneumoniae-induced p38 MAPK-dependent phosphorylation of RelA at the interleukin-8 promotor. J Biol Chem. 2004;279:53241–7. doi: 10.1074/jbc.M313702200. [DOI] [PubMed] [Google Scholar]
- 30.Schmeck B, Moog K, Zahlten J, et al. Streptococcus pneumoniae induced c-Jun-N-terminal kinase- and AP-1 -dependent IL-8 release by lung epithelial BEAS-2B cells. Respir Res. 2006;7:98. doi: 10.1186/1465-9921-7-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Duane P, Rubins JB, Weisel H, Janoff E. Identification of hydrogen peroxide as a Streptococcus pneumoniae toxin for rat alveolar epithelial cells. Infect Immun. 1993;61:4392–7. doi: 10.1128/iai.61.10.4392-4397.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Regev-Yochay G, Trzcinski K, Thompson CM, Lipsitch M, Malley R. SpxB is a suicide gene of Streptococcus pneumoniae and confers a selective advantage in an in vivo competitive colonization model. J Bacteriol. 2007;189:6532–9. doi: 10.1128/JB.00813-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Belanger AE, Clague MJ, Glass JI, Leblanc DJ. Pyruvate oxidase is a determinant of Avery's rough morphology. J Bacteriol. 2004;186:8164–71. doi: 10.1128/JB.186.24.8164-8171.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Okahashi N, Nakata M, Sumitomo T, Terao Y, Kawabata S, Vadivelu J. Hydrogen peroxide produced by oral Streptococci induces macrophage cell death. PLoS One. 2013;8:e62563. doi: 10.1371/journal.pone.0062563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stuart LM, Paquette N, Boyer L. Effector-triggered versus pattern-triggered immunity: how animals sense pathogens. Nat Rev Immunol. 2013;13:199–206. doi: 10.1038/nri3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pelaia G, Cuda G, Vatrella A, et al. Effects of hydrogen peroxide on MAPK activation, IL-8 production and cell viability in primary cultures of human bronchial epithelial cells. J Cell Biochem. 2004;93:142–52. doi: 10.1002/jcb.20124. [DOI] [PubMed] [Google Scholar]
- 37.McEwan DL, Kirienko NV, Ausubel FM. Host translational inhibition by Pseudomonas aeruginosa exotoxin A triggers an immune response in Caenorhabditis elegans. Cell Host Microbe. 2012;11:364–74. doi: 10.1016/j.chom.2012.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fontana MF, Banga S, Barry KC, et al. Secreted bacterial effectors that inhibit host protein synthesis are critical for induction of the innate immune response to virulent Legionella pneumophila. PLoS Pathog. 2011;7:e1001289. doi: 10.1371/journal.ppat.1001289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wolfson JJ, May KL, Thorpe CM, Jandhyala DM, Paton JC, Paton AW. Subtilase cytotoxin activates PERK, IRE1 and ATF6 endoplasmic reticulum stress-signalling pathways. Cell Microbiol. 2008;10:1775–86. doi: 10.1111/j.1462-5822.2008.01164.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee S, Lee M, Cherla RP, Tesh VL. Shiga toxin 1 induces apoptosis through the endoplasmic reticulum stress response in human monocytic cells. Cell Microbiol. 2008;10:770–80. doi: 10.1111/j.1462-5822.2007.01083.x. [DOI] [PubMed] [Google Scholar]
- 41.Braun JS, Sublett JE, Freyer D, et al. Pneumococcal pneumolysin and H2O2 mediate brain cell apoptosis during meningitis. J Clin Invest. 2002;109:19–27. doi: 10.1172/JCI12035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hoffmann E, Dittrich-Breiholz O, Holtmann H, Kracht M. Multiple control of interleukin-8 gene expression. J Leukoc Biol. 2002;72:847–55. [PubMed] [Google Scholar]
- 43.Brereton CF, Sutton CE, Lalor SJ, Lavelle EC, Mills KHG. Inhibition of ERK MAPK suppresses IL-23- and IL-1-driven IL-17 production and attenuates autoimmune disease. J Immunol. 2009;183:1715–23. doi: 10.4049/jimmunol.0803851. [DOI] [PubMed] [Google Scholar]
- 44.Cucinotta M, Visalli M, Aguennouz M, et al. Regulation of interleukin-8 gene at a distinct site of its promoter by CCAAT enhancer-binding protein homologous protein in prostaglandin E2-treated human T cells. J Biol Chem. 2008;283:29760–9. doi: 10.1074/jbc.M803145200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Goodall JC, Wu C, Zhang Y, et al. Endoplasmic reticulum stress-induced transcription factor, CHOP, is crucial for dendritic cell IL-23 expression. Proc Natl Acad Sci U S A. 2010;107:17698–703. doi: 10.1073/pnas.1011736107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.LeibundGut-Landmann S, Weidner K, Hilbi H, Oxenius A. Nonhematopoietic cells are key players in innate control of bacterial airway infection. J Immunol. 2011;186:3130–7. doi: 10.4049/jimmunol.1003565. [DOI] [PubMed] [Google Scholar]
- 47.Eckmann L, Kagnoff MF, Fierer J. Epithelial cells secrete the chemokine interleukin-8 in response to bacterial entry. Infect Immun. 1993;61:4569–74. doi: 10.1128/iai.61.11.4569-4574.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lucas R, Sridhar S, Rick FG, et al. Agonist of growth hormone-releasing hormone reduces pneumolysin-induced pulmonary permeability edema. Proc Natl Acad Sci U S A. 2012;109:2084–9. doi: 10.1073/pnas.1121075109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Noursadeghi M, Tsang J, Haustein T, Miller RF, Chain BM, Katz DR. Quatitative imaging assay for NF-κB nuclear translocation in primary human macrophages. J Immunol Methods. 2008;329:194–200. doi: 10.1016/j.jim.2007.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.