

Abstract
Sequence-programmable transcription activator-like effector (TALE) proteins have emerged as a highly efficient tool for genome engineering. Recent crystal structures depict a transition between an open unbound solenoid and more compact DNA-bound solenoid formed by the 34 amino acid repeats. How TALEs switch conformation between these two forms without substantial energetic compensation, and how the repeat-variable di-residues (RVDs) discriminate between the cognate base and other bases still remain unclear. Computational analysis on these two aspects of TALE-DNA interaction mechanism has been conducted in order to achieve a better understanding of the energetics. High elasticity was observed in the molecular dynamics simulations of DNA-free TALE structure that started from the bound conformation where it sampled a wide range of conformations including the experimentally determined apo- and bound- conformations. This elastic feature was also observed in the simulations starting from the apo form which suggests low free energy barrier between the two conformations and small compensation required upon binding. To analyze binding specificity, we performed free energy calculations of various combinations of RVDs and bases using Poisson-Boltzmann/surface area (PBSA) and other approaches. The PBSA calculations indicated that the native RVD-base structures had lower binding free energy than mismatched structures for most of the RVDs examined. Our theoretical analyses provided new insight on the dynamics and energetics of TALE-DNA binding mechanism.
Keywords: TALE, specificity, elasticity, bound, unbound
1. INTRODUCTION
TALEs are sequence-programmable transcription factors derived from bacterial plant pathogens. They have garnished wide attention in recent years due to their modular design consisting of highly similar repeats. Each repeat can recognize one base by the repeat-variable di-residues (RVDs) with well-documented specificity, including NI (Asn-Ile) to A, HD (His-Asp) to C, NH (Asn-His) to G and NG (Asn-Gly) to T[1,2]. This simple recognition code as well as the low toxicity has led to its fast-developing applications in diverse fields[3–5]. For instance, the precise targeting of genomic loci in numerous species has been demonstrated using engineered TALE nucleases (TALENs)[6]. Engineered TALE transcription factors and recombinases have also been described[1,7].
However, our understanding of the mechanism by which TALE proteins interact with DNA and achieve such high specificity lags far behind our ability to use them as successful tools. For example, the recent structures of several TALE-DNA complexes have been determined by X-ray crystallography[8,9]. Based on these structures, each repeat consists of two helical segments connected by a short loop that contains the RVD sequences. Surprisingly, only the second residue of the RVD contributes directly to the base recognition, while the first residue mainly contributes to the C-terminal capping of the first helix[8,9]. Although specific hydrogen bonding and other interactions have been observed from the X-ray structures, the available data do not provide a quantitative explanation for the apparent high specificity imparted by the RVDs. Structural data of mismatched RVD-base pairings is presently lacking. Another interesting finding is that although the apo and DNA-bound forms share the same helical architecture, the bound TALE is much more compact[8]. Specifically, while both contain 11 repeats per turn, the pitch changes from 60Å to 35Å per turn upon binding, accompanied by subtle repacking at the repeat interfaces. These two distinct conformations have been observed in independent X-ray structures[9,10]. However, the mechanism by which the ligand-free TALE switches from the apo form to the bound form upon DNA binding is not yet revealed., This is a critical issue because it would require large compensation upon binding if there is a significant free energy barrier separating these two conformations.
Semi-quantitative experiments have investigated the binding specificity of RVDs. Using a reporter assay, Cong et al. interrogated the binding specificity of 23 RVDs which confirmed the specific recognition of NI to A, HD to C, NN to G/A and NG to T and discovered highly specific recognition of NH to G[11]. They further evaluated the binding free energy and found that NH-G binding was 0.86 kcal/mol more favorable than NN-G binding. Streubel et al. examined the specificity and efficiency of 14 RVDs also using a reporter assay and various TALE constructs[12]. HD and NN were identified as strong RVDs, while NG, NI, NK, and N* were scored as weak RVDs (* indicates the absence of the second RVD residue). In addition, NH displayed higher specificity to G than did NN, while NS, NT and HN displayed recognition to both A and G. Our more recent quantitative study, using DNA electrophoretic mobility-shift assays with highly purified TALE proteins showed the relative RVD affinity in the order NG > HD ~ NN ≫ NI > NK, with each repeat contributing an average of 1–4 kJ/mol to binding free energy[13]. The discrepancies with the cellular measurements underscore the need for more quantitative measurements in vitro and in silico in order to probe the physical basis and mechanisms of TALE-DNA binding. Despite the great importance of TALEs, a comprehensive investigation of the binding specificity by free energy calculation has yet to be reported, partly due to the challenge of evaluating protein-DNA interaction energies.
In this work, we investigated the dynamics and energetics of TALE-DNA binding mechanism through computational analyses. First, we conducted molecular dynamics (MD) simulations to investigate the conformational elasticity of TALE. Our MD simulations started with both bound and free forms where consistent features were observed. Second, we applied Poisson-Boltzmann Surface Area (PBSA) [14] calculations to evaluate binding free energies between RVDs and bases. This physics-based approach was compared with two empirical approaches, namely Rosetta[15] and DDNA3[16]. Here we report insights gained from our computational analyses.
2. METHODS
2.1. Molecular dynamics simulations of TALE
The AMBER (version 12) software package[17] and FF03 force field[18] were used for the MD simulations. The initial TALE coordinates for our MD simulations were extracted from X-ray crystallographic structures of the free apo- (PDB code 3V6P) and bound- (PDB code 3V6T) forms of dHAX3 [8]. In order to allow room for substantial movement, large water boxes were used with minimum 27 Å from the protein or complex surface to the solvent wall, resulting in 128774 atoms for the bound system and 127978 atoms for the free system. The systems were neutralized by adding Na+ and Cl− to the systems using the tleap program in AMBERTOOLS. Short minimization (500 steps, steepest decent) and equilibration (500 ps, NPT, constant pressure and temperature) with positional restraints on TALE were performed to bring the solvated systems to normal pressure (1 atm) and room temperature (300 °K). For the bound system, standard production run was performed for 50 ns with triple replicates using different random seeds at the beginning of the replicate simulations. For the free system extracted from the bound structure after the removal of the DNA, a standard production run was performed for 250 ns with triple replicates, again using different random seeds at the beginning of the replicate simulations. Briefly, the production simulations were conducted at NVT mode (constant volume and temperature, T=300 K). No positional restraints were applied in the production run. Temperature was controlled by using Berendsen’s thermostat with a coupling constant of 2.0 ps. SHAKE was applied to constrain all bonds connecting hydrogen atoms. The particle-mesh Ewald method was used to treat long range electrostatic interaction under periodic boundary condition. The cutoff distance for short range non-bonded interaction was 10 Å, while the long range van der Waals interaction was treated by a uniform density approximation. To reduce computation, non-bonded forces were calculated using the two-step RESPA approach. To eliminate the “block of ice” problem, we reset the translation and rotation of the center of mass every 500 steps. Coordinates were saved every 10 ps, resulting in 5000 snapshots for each bound system and 25000 snapshots for each free system. In addition, simulations were also performed on free system starting from the apo structure with a set of triple simulations conducted for 50 ns each using the same protocol. The simulations were performed on NVIDIA GPU using the GPU version of pmemd[19]. Each 10 ns of the simulations required about 50 hours.
2.2. Evaluation of binding free energy for different RVDs
Our template system for binding energy calculation was extracted from the DNA-bound dHAX3 X-ray structure (PDB code: 3V6T)[8]. There are 11.5 repeats in the original structure (“0.5” refers to the most C-terminal DNA-binding repeat). We extracted repeats 7–11 as the template for permutation and kept the original DNA intact. The RVDs for repeats 7–11 were NS-NG-HD-NG-HD in the X-ray structure. We fixed all the RVDs except for the central repeat 9, which was mutated to 15 other RVDs, NG, NN, NH, NK, NI, NS, NT, NP, ND, N*(* indicates the absence of the second RVD residue), NA, HG, HN, HS and HT. For each of these 16 RVDs (including the original HD), all four possible base pairs at the recognition site were also constructed by the tleap program in AMBERTOOLS, resulting in a total of 64 systems. During the in silico mutagenesis, only the backbone atoms of the mutated amino acid or base were kept and the side chain or base atoms were automatically generated by tleap. In order to relax the mutation site, a short MD run (500 ps, NVT) with explicit solvent was performed, preceded by energy minimization (500 steps, steepest decent) and equilibration (500 ps, NPT) according to the standard AMBER protocol. During the entire process of the simulations, all atoms were fixed by positional restraints with harmonic forces (5 kcal/mol/Å2) except for the central RVD loop and the central base pair at the mutation site. From each MD simulation, 100 snapshots were saved for energy evaluation. We conducted three types of binding free energy calculations. PBSA was performed with the AMBER package following the standard protocol. The solvation free energies were calculated for the structures obtained from the simulations. The average binding free energy of the 100 snapshots was reported directly by the PBSA calculation. Rosetta (version 3.4) software (www.rosettacommons.org) was installed following the instruction. The 100 snapshots were converted to individual PDB files by the ptraj program in AMBERTOOLS and submitted to Rosetta for energy calculation and the free energies were averaged for each of the 64 complexes. The scoring option for the RosettaDNA module was used to calculate the binding energy. No further structural optimization was applied prior to energy calculation. The binary code of DDNA3 was downloaded (sparks.informatics.iupui.edu/yueyang/DFIRE/ddna3-service). No options for structure optimization were available in DDNA3. Similar to the Rosetta calculation, DDNA3 binding free energies of the individual 100 snapshots were evaluated and then averaged.
3. RESULTS
3.1. Elastic motion of the ligand-free TALE
One of the main goals of this study was to examine the elasticity of ligand-free TALE and how it may contribute to DNA binding. As a reference and a validation of the simulation protocals, we first conducted simulations on the bound TALE-DNA complex. Within the 50-ns MD simulations with explicit solvent, the three independent trajectories displayed similar stable features with fluctuations around a 3-Å backbone RMSD compared to the starting structure (Figure 1). This fluctuation was considerably smaller when compared to the simulations with the free TALE which will be described later. The results indicated that the bound TALE conformation in the presence of the DNA is in a free energy minimum as would be expected from the experimental X-ray structure. According to previous experimental studies[8], two major forces contribute to the favorable interaction between TALE and DNA. These include the non-specific contribution from the interaction between Lys16/Gln17 near the RVD loop and the DNA backbone phosphate group, and the specific contribution from the RVD-base interactions. The combination of the two favorable forces led to the free energy minimum observed in our simulation. In addition, this stability test provided a validation for the suitability of the simulation protocol used in this study.
Figure 1.
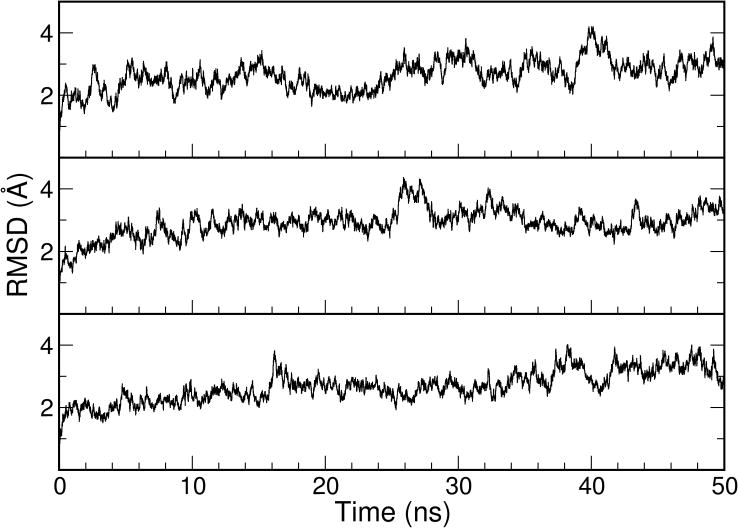
RMSD profiles from the three 50-ns MD simulations with the TALE-DNA complex (PDB code: 3V6T, the complete system).
In contrast to the simulations in the presence of the DNA, the simulations with ligand-free TALE all displayed high elasticity. Two sets of MD simulations were performed in the absence of DNA; one started from the apo form (PDB code: 3V6P) and the other from the bound form (PDB code: 3V6T) with the DNA removed. All simulations were performed with explicit solvent. Large water boxes were used in anticipation of substantial elastic movement. The backbone RMSDs from these two simulations are shown in Figure 2 and Figure 3, respectively. The RMSDs were calculated relative to both the apo (black trace) and bound (green trace) forms in both cases.
Figure 2.
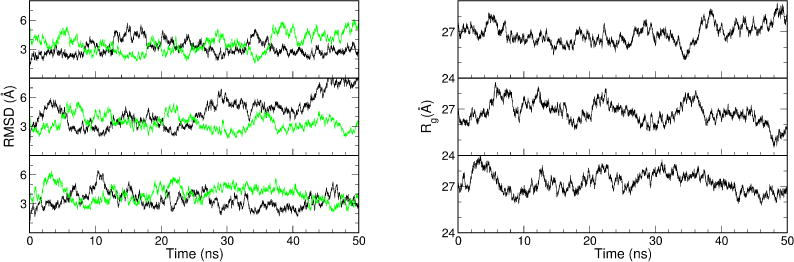
The profiles of RMSD (left) and radius of gyration (Rg, right) from the three 50-ns MD simulations with the ligand-free TALE starting from the apo structure (PDB code: 3V6P). In the RMSD profiles, the RMSDs against the apo structure are shown in black, the RMSDs against the bound structure are shown in green.
Figure 3.
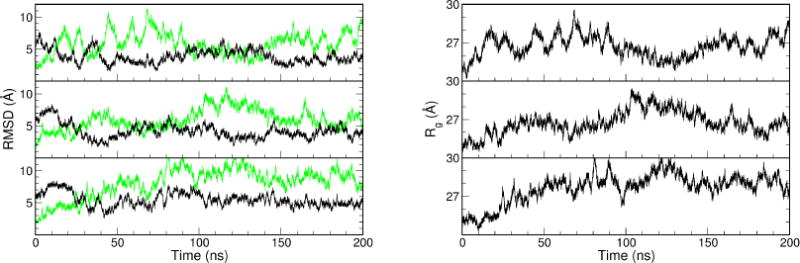
The profiles of RMSD (left) and Rg (right) from the three 200-ns MD simulations with the ligand-free TALE starting from the bound structure (PDB code: 3V6T, DNA removed). In the RMSD profiles, the RMSDs against the bound structure are shown in green, the RMSDs against the apo structure are shown in black.
A consistent picture emerged from these simulations was the constant oscillation of TALE although these simulations started from two different conformations. This is clearly illustrated by the fact that RMSDs relative to apo (black trace) and bound (green trace) forms both exhibit large degree of fluctuations. These two RMSD profiles also moved generally in the opposite directions. For example, when the RMSD relative to the apo structure went up to 6 Å, the RMSD relative to the bound structure decreased to below 3 Å and vice versa. Thus, TALE is quite elastic in the absence of DNA. More importantly, TALE oscillates in a wide range encompassing the apo and bound forms.
Since the apo-form TALE is an experimentally determined structure, we expected it to be reasonably stable in the simulation. Indeed, the backbone RMSD was fluctuating around 3 Å in two of the three 50-ns simulation trajectories started from the apo-form (Figure 2, left, black trace). However, we still observed considerable fluctuations as shown by RMSD and radius of gyration (Rγ) (Figure 2, RMSD, left, and Rg, right). In one of these three trajectories (Figure 2, top panel), although the apo basin around 3 Å backbone RMSD was the predominant conformation, it moved away to 5–6 Å a few times, and for several short periods it was close to the bound form (green trace). In another trajectory (Figure 2, bottom panel), it displayed high fluctuation in the first half, but the apo basin was heavily sampled in the second half. Transient sampling close to the bound form was also observed in this trajectory. In the third trajectory (Figure 2, middle panel), it moved away from the apo basin near 25 ns and stayed away during the second half of the 50-ns simulation. Sampling close to the bound form was also observed in the second half of the simulation. The substantial elastic motion could also be seen from the Rγ profiles. The Rγ of the apo form was near 27 Å according to the initial values at the beginning of the three simulations. It fluctuated between 24.5 and 29 Å during the simulations, with the lower boundary close to the bound form and the upper boundary more extended than the apo form.
With the removal of the DNA, the bound conformation was expected to be less stable in the simulation. Indeed, in all three trajectories that started from the bound form, TALE moved away from the bound conformation within 50 ns (Figure 3, left, green trace). Another interesting observation was the transient sampling back to the bound form in two trajectories (Figure 3, top and middle). This elastic motion can also be seen from the Rγ profiles that fluctuated between 24 and 32 Å during the simulations (Figure 3, right). It should be noted that these Rγ values are not directly comparable to the Rγ values in Figure 2 because the bound structure (PDB code 3V6T) had a longer chain than the apo structure (PDB code 3V6P). Nonetheless, we still observed the similar features that the TALE repeat structures sampled a wide range of conformations including the apo and bound forms.
To further illustrate the substantial elastic motion, we selected three representative snapshots based on the closeness to the apo or bound forms (Figure 4, red color) from the trajectory shown in the top panel of Figure 3 and compared them against the starting bound form (Figure 4, green color). At 68.25 ns (Figure 4, left), it adopted a rather extended conformation with significant deviation from the starting bound structure (11.08 Å backbone RMSD to the bound form, Rg=29.4 Å). At 73.95 ns (Figure 4, middle), it reached a conformation very close to the apo form (backbone RMSD=1.56 Å to the apo form, Rg=27.1 Å). At 130.29-ns (Figure 4, right), however, it transiently moved back to the compact conformation very close to the starting structure (2.08 Å backbone RMSD to the bound form, Rg=24.9 Å).
Figure 4.
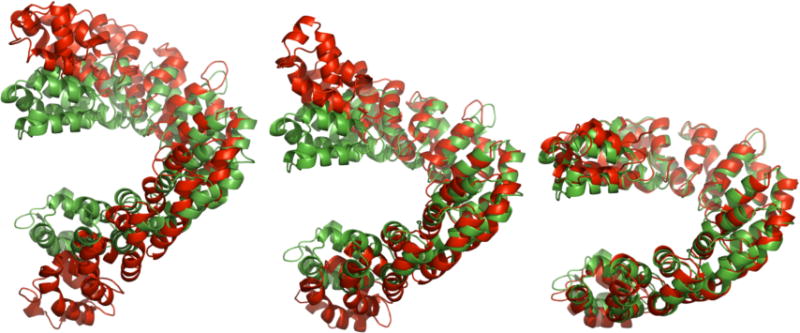
Three representative snapshots from the MD simulation trajectory shown in the top panel of Figure 3(left, 68.25 ns, highly extended; middle, 73.95 ns, close to the apo form; and right, 130.29 ns, close to the bound form). The structures from the simulation are show in red, the reference bound structure is shown in green.
In summary, we observed consistent features in the DNA-free simulations started from both the apo and bound forms: 1) the DNA-free TALE is highly dynamic with constant elastic movement; 2) the apo form is closer to the energy minima than the bound form as demonstrated by the RMSD profiles; 3) the bound form can be transiently reached during the elastic motions.
The conformational sampling can be further illustrated using a three dimensional contour map (Figure 5). To construct this map, sampling data from the three trajectories shown in Figure 3 (started with the bound form, DNA removed) were merged. Sampling over a large conformational space is evident from this map. The RMSD to the bound TALE varied mostly from 2 to 12 Å while the RMSD to the apo TALE varied mostly from 1.5 to 8 Å. The most heavily sampled region was within RMSD 3–4.5 Å to the apo form and 5–7 Å to the bound form. We have also performed clustering analysis for all the conformations on the map. The top 5 cluster comprise ~75% of the conformations, and the representative structures for the top 5 clusters are shown on the map. In brief, clusters #1 and #4 are close to the apo form (RMSD_apo=3.51 Å and 2.32 Å, population=33.9% and 8.0%, respectively) for a combined ~42% population. Cluster #5 is close to the bound form (RMSD_bound=3.01 Å, population=5.2%). The other two clusters have intermediate conformations (RMSD_apo=4.76 Å, RMSD_bound=8.72 Å, and population=17.5% for cluster #2, RMSD_apo=4.77 Å, RMSD_bound=3.95 Å, and population=10.5% for cluster #3).
Figure 5.
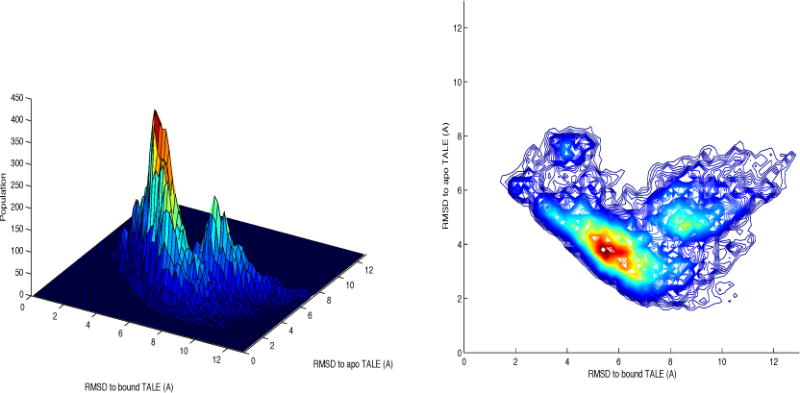
Conformational sampling of the ligand-free TALE from the three MD simulations shown in Figure 3.
Overall, the sampling was biased toward the apo form in these simulations, even though they all started from the bound structure. Since the RMSD to the bound form also reflected the compactness of the TALE, the map illustrated the broad conformational sampling with large variation in the compactness. The DNA-free TALE constantly underwent oscillation movement with center close to the apo form while the bound form can be reached during the oscillation process.
3.2. Evaluation of the binding free energy between RVDs and bases
Another critical component of the binding mechanism is the specific recognition of RVDs and bases. In order to understand the energetic contribution to the binding specificity, we evaluated the binding free energy between RVDs and bases using three different methods, PBSA, Rosetta and DDNA3. A minimal local environment was included in the calculations in which a five-repeat segment from the high resolution X-ray structure (PDB code: 3V6T, repeats 7–11) and performed in silico mutations on the central repeat (repeat #9). Such a minimal environment helps to reduce the uncertainty associated with inevitable fluctuation due to the remaining parts. Furthermore, during the relaxation of the structure, only the central RVD loop and the central base pair were allowed full flexibility whereas all other atoms were restrained by harmonic forces. We attempted other protocols and found that DNA has tendency to untwist when simulations exceeds 10 ns. Thus, to reduce the influence of the inevitable approximation in simulations including both parameterization and limited sampling, it was necessary to keep the TALE-DNA close to the experimental structures. We evaluated 16 RVDs (Table 1) for which observed experimental binding preferences are described in the literature[1,11,12]. All 64 possible RVD-base pair combinations were evaluated and, for each, the average binding free energy of 100 relaxed complex structures was calculated by the three methods.
Table 1.
An overall comparison of the performance for binding free energy evaluation by PBSA, VDW, Rosetta and DDNA3. Note: For RVDs with single preference, the energy ranking of the base is shown as 1 (lowest energy), 2 (second lowest energy), or X (others). For RVDs which recognize multiple bases, the base with lowest energy is shown (“x” stands for wrong energy ranking). The second preferred base for NN is A. NS also recognizes other bases. For comparison, the performance by VDW (van der Waals) is also shown.
| RVD – base | PBSA | VDW | DDNA3 | Rosetta |
|---|---|---|---|---|
| NI – A | 1 | 2 | X | X |
| NS – A | 1 | 2 | 2 | X |
| NK – G | 1 | X | X | 1 |
| NH – G | X | 2 | X | 1 |
| NN – G | 2 | 1 | X | 1 |
| NG – T | 2 | X | X | X |
| HG – T | 2 | X | 1 | 1 |
| HD – C | 1 | X | X | X |
| HN – AG | A | A | T(x) | G |
| NT – AG | A | A | C(x) | C(x) |
| NP – ACT | T | C | A | G(x) |
| N* – CT | T | T | C | T |
| HT – AG | C(x) | G | A | G |
| NA – CT | C | C | A(x) | T |
| ND – C | 1 | 2 | 1 | 2 |
| HS – A | 1 | 2 | 2 | X |
A summary of the energy evaluation by the three methods is shown in Table 1 (more detailed free energy values can be found in Supplementary Table 1). Overall, the PBSA energies exhibited better correlations with experimental observations. For the ten RVDs with single base preferences, PBSA had six ranked at No.1 and three ranked at No.2, while Rosetta had four ranked at No.1 and one ranked at No.2, and DDNA3 only had two ranked at No.1 and two ranked at No.2. For the six RVDs which recognize multiple bases, PBSA had incorrect ranking for only one RVD, while Rosetta had two RVDs incorrectly ranked, and DDNA3 had three RVDs incorrectly ranked. A potential problem with Rosetta was that it showed preference over either G or T for all but two of the sixteen RVDs examined.
The results of the PBSA energy evaluation are summarized in Figure 6. For HT, NG, HG and NN, the native recognition was only 0.2–0.6 kcal/mol away from the lowest binding free energy. The consistency of PBSA with experimental findings prompted us to further dissect the energy components of PBSA. The ranking performance by van der Waals (VDW) is shown in Table 1. It is evident that the ranking by VDW is much less satisfactory then the total PBSA energy. Similarly, none of the other energy components demonstrated better correlation with experimental observation than the total PBSA energy (data not shown). Therefore, the specific recognition of TALE arises from the combination of VDW, electrostatics and solvation free energy, not dominated by any of the individual terms.
Figure 6.
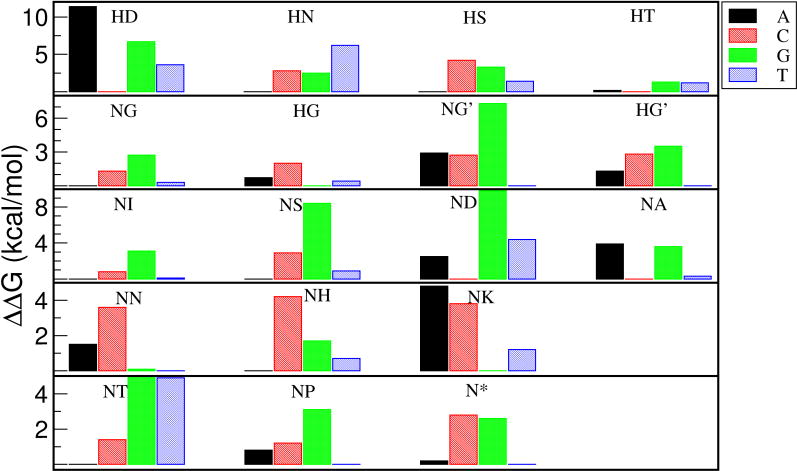
Binding free energy evaluation of 16 RVDs and all four possible bases for each RVD by PBSA. For each RVD, the lowest binding free energy was set to zero while others were assigned to positive energy based on the energy difference. For NG and HG, a second template with NG at the central RVD of the original structure was used for energy evaluation (shown as NG’ and HG’).
In order to examine the effect of the native conformation on energy evaluation, we re-conducted energy evaluations for RVDs NG and HG using another template that consisted of repeats 6–10 with NG as the central RVD (repeat #8) in the original structure (PDB code 3V6T). It is evident that both NG and HG had clear preference over T using this template (NG’ and HG’ in Figure 6). This suggests that the NG-T interaction was more optimized in the X-ray structure than our constructed structure by mutation. Another interesting insight regarding NG-T interaction can be gained from the energy evaluation. Based on the X-ray structure, it has been hypothesized that specific recognition of NG to T was likely due to the exclusion mechanism, i.e. that NG can accommodate the thymine 5-methyl group and other RVD side chains would be expected to clash with the group. However, in our in silico constructed TALE systems, the RVD interactions with T at the recognition site were all well tolerated, there were no visible clashes in any of structures even with restraints on most of the atoms, and the T recognition was not the least favorable interaction for most of the RVDs examined (Figure 6), suggesting that NG-T recognition may not due to the exclusion mechanism.
4. DISCUSSION
4.1. Low free energy barrier implied from the high elasticity
The observation of two distinctive TALE conformations at the apo and bound states from crystallography data prompted us to conduct a computational analysis of the conformational space for the DNA-free TALE. The MD simulations starting from both the apo and bound forms demonstrated consistent features. Although the apo conformation was more favorable, the DNA-free TALE was highly elastic. A wide range of conformations were sampled in the simulations and some were significantly more extended than the apo form while others were more compact. The bound conformation was also transiently sampled in the DNA-free simulations and the overall feature was the constant oscillation with center close to the apo conformation. Together with the more favorable binding free energy for the specific RVD-base recognition, the high elasticity may help us to dissect the energetics in TALE-DNA interactions. The cylindrical TALE-DNA complex structure requires the wrapping of TALE around DNA major groove, which can be difficult without the high elasticity observed in our simulation. The ability of apo TALE to reach the bound conformation implies a low free energy barrier separating the bound and unbound conformations. Favorable interactions with the DNA backbone as described earlier can help TALE to overcome this small free energy barrier. Since TALE does not bind to a random DNA sequence, this non-specific TALE-DNA interaction is likely in the similar scale as the free energy barrier between the two TALE states. The overall favorable binding free energy likely comes entirely from the specific RVD-base interaction including the neighbor effect. Therefore, it is critical to quantitatively determine the binding energies of RVD-base interactions. We have also attempted ab initio TALE-DNA binding simulation. However, the preliminary test showed that the time scale for binding is far beyond our reach. Therefore, more details regarding the initial binding process can not be revealed from the simulation.
4.2. Technical considerations for the binding free energy evaluation
Due to the errors in parameterization and difficulty in evaluating entropy, accurate and quantitative free energy calculation has been a major challenge in the field of computational biology. Not surprisingly, the evaluation of binding free energy for RVD-base recognition in this study turned out to be technically challenging. We tested several alternative strategies to perform the analysis. Since extended simulations can provide extensive conformation sampling, we first attempted longer simulations to allow the structures to relax to their bound states. However, extended relaxation of the central repeat or the whole five-repeat segment without restraints led to significantly distorted DNA structure with the DNA clearly untwisted. Calculations using those simulated structures had notably worse correlation with experimental RVD specificities, and in many cases, yielded values close to random ranking for all three energy evaluation methods (data not shown). This implies inherent problems in the underlying simulation parameters, in particular the parameter set representing DNA because notable distortion of DNA conformation was observed consistently in the simulations without restraints. Good correlation with experimental observation was obtained only when stringent restraints were applied (Table 1 and Figure 6). The limited conformational sampling during the short MD simulations (500 ps) ensured that the simulation sampled the local minimum only and retained the critical features of experimental structures. Clearly, much work is needed to improve the simulation parameters. Nevertheless, it is encouraging that such difficulty can be partially circumvented.
The selection of template was also critical in this study. We have conducted full analyses on these 16 RVDs using two different templates, one with repeats 7–11 (template #1) and the other with repeats 6–10 (template #2). Although template #2 gave better results for NG and HG (NG’ and HG’ in Figure 6), the overall performance was less satisfactory for all three methods (data not shown). One of the potential problems with template #2 was the side chain interaction among neighboring RVDs which was weak with template #1 because the flanking RVDs were NG on both sides of the central repeat. Again, this suggests deficiency in the force field.
Since the RVD side chain orientations are critical for the favorable RVD-base interaction, we also used the side chain orientations from the X-ray structures of the same or similar RVDs whenever possible, whereas the direct assignment of side chain orientation by AMBER tleap led to less satisfactory correlations (data not shown).
The empirical methods Rosetta and DDNA3 had less satisfactory performance compared to PBSA even though the experimental protein-DNA interactions were not included in the parameterization process of PBSA. Although PBSA has not been extensively tested for protein-DNA interactions, the results from this study suggest that PBSA might be the better choice for understanding the energetics of TALE-DNA interactions. The lessons learned from this study shall be carefully considered in future computational studies on TALE-DNA binding mechanism. However, we note that this result does not necessarily diminish the usefulness of Rosetta, DDNA3 or other empirical methods. For example, structure refinement may lead to better ranking in Rosetta which was not tested in this work. Given the increasing availability of DNA-protein complex structures, these methods are expected to improve over time.
4.3. Concluding remarks
In this work, we conducted computational analyses on the conformational elasticity and specific recognition of TALEs. Novel insights regarding the binding mechanism were gained from the molecular dynamics simulations of the DNA-free TALE. While the DNA-bound TALE structure was relatively stable, the DNA-free TALE underwent significant and reversible conformational transition in the simulations irrespective of the starting conformation. This spring-like motion may be a critical part of the binding mechanism for TALE-DNA interactions. The PBSA binding free energy calculation was validated by the result that the native pairing of RVD and base was favored compared to the mismatched pairings, and showed better consistency than the empirical approaches including Rosetta and DDNA3. An additional insight from the free energy evaluation is the proposition that NG to T recognition is not due to exclusion of other larger side chains by the base as suggested by many, since all the substitutions examined were well tolerated in the simulations. Based on the computational analyses on these two aspects, we proposethat the high elasticity of DNA-free TALE leads to low free energy barrier between the apo and bound states which requires only small compensation from the non-specific TALE–DNA interaction upon binding. Therefore, the binding affinity may come entirely from the specific RVD-base interaction.
Supplementary Material
Acknowledgments
We would like to thank Dr. Yuedong Yang for helping us solving some technical issues with DDNA3. This work was supported by research grants from NIH (GM79383 to YD; GM097073 to DJS) and MOST (Grant 2014CB964901 to HL).
Footnotes
The authors have no conflicts of interest to declare.
References
- 1.Boch J, Scholze H, Schornack S, Landgraf A, Hahn S, Kay S, et al. Breaking the code of DNA binding specificity of TAL-type III effectors. Science. 2009;326:1509–1512. doi: 10.1126/science.1178811. [DOI] [PubMed] [Google Scholar]
- 2.Moscou MJ, Bogdanove AJ. A simple cipher governs DNA recognition by TAL effectors. Science. 2009;326:1501. doi: 10.1126/science.1178817. [DOI] [PubMed] [Google Scholar]
- 3.Bedell VM, Wang Y, Campbell JM, Poshusta TL, Starker CG, Krug RG, 2nd, et al. In vivo genome editing using a high-efficiency TALEN system. Nature. 2012;491:114–118. doi: 10.1038/nature11537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tremblay JP, Chapdelaine P, Coulombe Z, Rousseau J. Transcription activator-like effector proteins induce the expression of the frataxin gene. Hum Gene Ther. 2012;23:883–890. doi: 10.1089/hum.2012.034. [DOI] [PubMed] [Google Scholar]
- 5.Sanjana NE, Cong L, Zhou Y, Cunniff MM, Feng G, Zhang F. A transcription activator-like effector toolbox for genome engineering. Nat Protoc. 2012;7:171–192. doi: 10.1038/nprot.2011.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Perez-Pinera P, Ousterout DG, Gersbach CA. Advances in targeted genome editing. Curr Opin Chem Biol. 2012;16:268–277. doi: 10.1016/j.cbpa.2012.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mercer AC, Gaj T, Fuller RP, Barbas CF., 3rd Chimeric TALE recombinases with programmable DNA sequence specificity. Nucleic Acids Res. 2012;40:11163–11172. doi: 10.1093/nar/gks875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deng D, Yan C, Pan X, Mahfouz M, Wang J, Zhu JK, et al. Structural basis for sequence-specific recognition of DNA by TAL effectors. Science. 2012;335:720–723. doi: 10.1126/science.1215670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mak AN, Bradley P, Cernadas RA, Bogdanove AJ, Stoddard BL. The crystal structure of TAL effector PthXo1 bound to its DNA target. Science. 2012;335:716–719. doi: 10.1126/science.1216211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gao H, Wu X, Chai J, Han Z. Crystal structure of a TALE protein reveals an extended N-terminal DNA binding region. Cell Res. 2012;22:1716–1720. doi: 10.1038/cr.2012.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cong L, Zhou R, Kuo YC, Cunniff M, Zhang F. Comprehensive interrogation of natural TALE DNA-binding modules and transcriptional repressor domains. Nat Commun. 2012;3:968. doi: 10.1038/ncomms1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Streubel J, Blucher C, Landgraf A, Boch J. TAL effector RVD specificities and efficiencies. Nat Biotechnol. 2012;30:593–595. doi: 10.1038/nbt.2304. [DOI] [PubMed] [Google Scholar]
- 13.Meckler JF, Bhakta MS, Kim MS, Ovadia R, Habrian CH, Zykovich A, Yu A, Lockwood SH, Morbitzer R, Elsäesser J, Lahaye T, Segal DJ, Baldwin EP. Quantitative Analysis of TALE-DNA Interactions Suggests Polarity Effects. Nucleic Acids Res. 2013 doi: 10.1093/nar/gkt085. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong L, et al. Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models. Acc Chem Res. 2000;33:889–897. doi: 10.1021/ar000033j. [DOI] [PubMed] [Google Scholar]
- 15.Leaver-Fay A, Tyka M, Lewis SM, Lange OF, Thompson J, Jacak R, et al. ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 2011;487:545–574. doi: 10.1016/B978-0-12-381270-4.00019-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao H, Yang Y, Zhou Y. Structure-based prediction of DNA-binding proteins by structural alignment and a volume-fraction corrected DFIRE-based energy function. Bioinformatics. 2010;26:1857–1863. doi: 10.1093/bioinformatics/btq295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DA Case TAD, Cheatham TE, III, Simmerling CL, Wang J, Duke RE, Luo R, Walker RC, Zhang W, Merz KM, Roberts B, Hayik S, Roitberg A, Seabra G, Swails J, Goetz AW, Kolossváry I, Wong KF, Paesani F, Vanicek J, Wolf RM, Liu J, Wu X, Brozell SR, Steinbrecher T, Gohlke H, Cai Q, Ye X, Wang J, Hsieh M-J, Cui G, Roe DR, Mathews DH, Seetin MG, Salomon-Ferrer R, Sagui C, Babin V, Luchko T, Gusarov S, Kovalenko A, Kollman PA. AMBER 12. University of California; San Francisco: 2012. [Google Scholar]
- 18.Duan Y, Wu C, Chowdhury S, Lee MC, Xiong G, Zhang W, et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J Comput Chem. 2003;24:1999–2012. doi: 10.1002/jcc.10349. [DOI] [PubMed] [Google Scholar]
- 19.Gotz AW, Williamson MJ, Xu D, Poole D, Le Grand S, Walker RC. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. J Chem Theory Comput. 2012;8:1542–1555. doi: 10.1021/ct200909j. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.