

Abstract
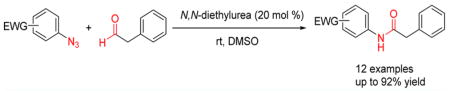
Urea structures, of which N,N-diethylurea (DEU) proved to be the most efficient, were discovered to catalyze amidation reactions between electron-defficient aryl azides and phenylacetaldehydes. Experimental data support 1,3-dipolar cycloaddition between DEU-activated enols and electrophilic phenyl azides, especially perfluoroaryl azides, followed by rearrangement of the triazoline intermediate. The activation of the aldehyde under near-neutral conditions was of special importance in inhibiting dehydration/aromatization of the triazoline intermediate, thus promoting the rearrangement to form aryl amides.
Amide bonds are frequently present as functional and structural units in natural products and synthetic polymers, and as linkers in constructing functional nanomaterials and surfaces. Many reaction types lead to this transformation, of which carboxyl-amine coupling reactions are arguably the most popular.1 Azides, which can be considered as a protected form of amines, have recently been shown to perform well in certain amidation reactions, for example, Staudinger ligation,2 thioacid–azide amidation,3 alkyne–sulfonyl azide coupling,4 alcohol–azide amide synthesis,5 and azide–aldehyde amidations.6 Of these, the amidation reaction between aldehydes and azides is redox-neutral, involving only the release of nitrogen as the side product and is therefore an attractive process.
The first reported azide–aldehyde amidation reaction dates back to the 1950s and is known as the Boyer reaction.7 While intramolecular Boyer amidation proceeds well, intermolecular reactions between aldehydes and aliphatic azides require strongly acidic conditions (>1 equiv of TiCl4, TFA, or TsOH) to give aliphatic amides in very limited yields.8,9 An alternative approach used a strong base (1–2 equiv of tert-butoxide salts) to activate special azides to give aryl amides in moderate to good yields.6a,b Transition-metal-catalyzed aldehyde–azide amidation was also recently reported; however, it had limited substrate scope and slow kinetics.6c,d Herein, we report a new catalytic azide–aldehyde amidation reaction which could be carried out under mild conditions to give aryl amides in good yields.
Perfluoroaryl azides (PFAAs) were initially selected to evaluate the reaction with aldehydes. PFAAs exhibit unique reactivities, owing to the presence of highly electronegative fluorine atoms.10 They have been utilized in photoaffinity labeling,11 and in surface and nanomaterial functionalization.12 PFAAs also show higher reactivities than phenyl azide in 1,3-dipolar cycloadditions toward activated dipolarophiles. For example, PFAAs react with enamines at room temperature without any catalysts, with cycloaddition rate constants in the range of 0.01–1.2 M−1 s−1.13
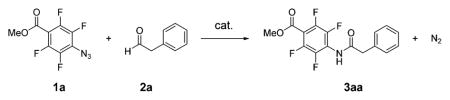
When methyl 4-azido-2,3,5,6-tetrafluorobenzoate (1a) and phenylacetaldehyde (2a) were mixed in an amide solvent or DMSO, aryl amide (3aa) was formed (Table 1, Figure S1). After various amide and urea structures were screened, it was found that N,N-diethylurea (DEU) was the most efficient (Table S1) and was thus chosen for further studies. The best conditions included 20 mol % DEU in DMSO, under which the reaction was completed at room temperature within 4 h to give the aryl amide in 91% isolated yield (entry 2, Table 1). The solvent proved important, and amide or urea solvents such as DMF or DMPU (1,3-dimethyltetrahydropyrimidin-2(1H)-one) reduced the catalytic effect resulting in lower conversion (entries 3, 4). When other solvents such as THF, MeOH, or acetone were used, no product was formed (entry 5). Reducing the DEU catalyst to 10 mol % gave slower conversion (entries 6, 7). The catalytic effect of other amides/ureas was more sluggish. For example, 73% amide formation was observed when the reaction was carried out in the presence of 1.3 equiv of DMPU for 16 h (entry 8). Without any catalyst, only 7% of the product was formed after 64 h (entry 9).
Table 1.
Model Reaction and Optimizationa
| ||||
|---|---|---|---|---|
| entry | cat., mol % | solvent | time/temp | yield (%)b |
| 1 | DEU, 20 | DMSO | 2 h/rt | 86 |
| 2 | DEU, 20 | DMSO | 4 h/rt | >99 (91c) |
| 3 | DEU, 20 | DMF | 2 h/rt | 63 |
| 4 | DEU, 20 | DMPU | 2 h/rt | 49 |
| 5 | DEU, 30 | otherd | 16 h/60 °C | 0 |
| 6 | DEU, 10 | DMSO | 2 h/rt | 75 |
| 7 | DEU, 10 | DMSO | 24 h/rt | >99 |
| 8 | DMPU, 130 | DMSO | 16 h/rt | 73 |
| 9 | – | DMSO | 64 h/rt | 7 |
Conditions: 1a (0.5 M, 1 equiv), 2a (0.63 M). rt (≈22 °C).
Determined by 19F NMR.
Isolated yield.
THF, MeOH, acetone.
Substrate screening was then performed for both the azide and the aldehyde (Figure 1). Perfluorophenyl azides worked efficiently, and these azides were generally converted to the corresponding aryl amides (3aa, 3ab, 3ba, 3ca, 3cb) in over 80% isolated yield at room temperature within 4 h. For 4-nitro-2,3,5,6-tetrafluorophenyl azide, heating at 80 °C for 2 days (3da) was required, whereas the reaction with pentafluoropyridinyl azide efficiently resulted in amide 3ea in 90% yield. Other electrophilically activated phenyl azides were also tested. o-Nitrophenyl- and p-nitrophenyl azides gave aryl amides in slightly lower but still good yields (3fa, 3fb, 3gb). o-Bromophenyl azide gave aryl amide 3hb as the major product in 32% yield together with a large amount of the starting material. Phenyl azide did not give any amide product even after extensive optimization of the reaction conditions. Interestingly, tosyl azide did not lead to any amide product under these conditions.
Figure 1.

Substrate scope. Conditions: 1 (0.5 mmol), 2 (0.63 mmol), DEU (0.1 mmol), DMSO (1 mL), rt (≈22 °C), 4 h, isolated yields. a80 °C, 2 d. bNeat DMPU, 80 °C, 2 d. c60 °C, 4 h.
α-Substituted phenylacetaldehydes, e.g., 2-phenylpropanal, generally gave higher yields than phenylacetaldehyde (3aa vs 3ab, 3ca vs 3cb, 3fa vs 3fb). Other aliphatic aldehydes such as 3-phenylpropanal resulted in lower yields where, for example, 20% of product 3ac was isolated after heating the reaction at 80 °C for 2 days. However, when DMPU was used as the solvent, even without the addition of DEU, the yield of 3ac increased to 70%.
We next conducted a series of experiments to investigate the mechanism of this transformation. Benzaldehydes did not give any aryl amide products. When phenylacetaldehyde (2a) was dissolved in DMPU, a clear transformation was observed for the aldehyde proton in the 1H NMR spectrum (Figures S2, S3). Small amounts (15%) of the typical aldol product 4 could also be isolated from the mixture (Scheme 1a). This conversion was however absent when azide 1a was added. Simply mixing the azide with DMPU or DEU did not result in any changes in the NMR spectrum, indicating that the azide was not activated by the urea, whereas the aldehyde was. When the reaction was carried out using 20 mol % DEU as the catalyst in D2O/DMSO (20:80 v/v), one deuterium atom was incorporated at the benzyl position in the product (Scheme 1b; supported by the NMR spectrum of the isolated compound 3aa-D). This suggests that the benzylic proton in the aldehyde was activated and could exchange with D2O. For 2-phenylpropanal, on the other hand, no deuteration was observed in the product (Scheme 1c). These results suggest that one of the benzylic protons in the amide product emanates from the aldehyde, accomplished via a hydride shift process.
Scheme 1. Control Experimentsa.
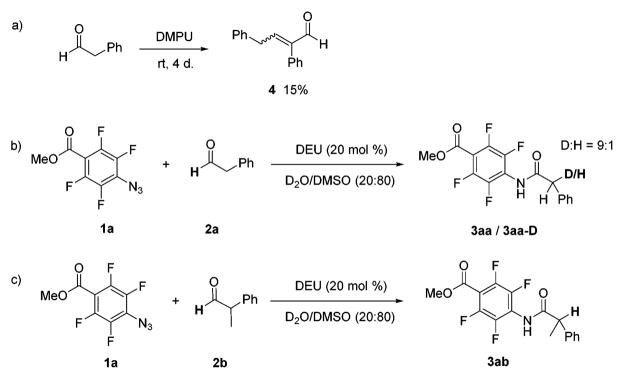
a(a) Reaction of phenylacetaldehyde in DMPU in the absence of the azide. (b) Model reaction carried out in 20:80 D2O/DMSO. (c) DEU-catalyzed reaction of PFPA 1a and 2-phenylpropanal in 20:80 D2O/DMSO.
From the above results, a plausible mechanism can be envisioned that involves activated enol formation followed by (3 + 2) cycloaddition with the azide to give the triazoline intermediate, which subsequently undergoes nitrogen extrusion and a hydride shift to give the amide product. When methyl vinyl ether was tested with azide 1a under identical conditions, the conversion was sluggish (30% after 16 h), indicating that the unactivated enol was unlikely to be the active species in the cycloaddition step. This was further supported by the fact that addition of catalytic amounts of TsOH completely impaired amide formation. On the other hand, when a base such as DMAP or DBU was added, the azide conversion was accelerated and a large amount of triazole was formed together with the amide (Scheme 2). In the case of DBU, triazole was formed exclusively.14 The isolation of a small amount of triazoles (3fa-t, 3ca-t) as byproducts in the DEU-catalyzed reactions also supports the formation of the triazoline as the intermediate. However, ureas, including DEU and DMPU, are generally considered to be superweak Brønsted bases15 and are unlikely to deprotonate the enol form of phenylacetaldehyde (pKa = 9.5–9.8).16
Scheme 2.
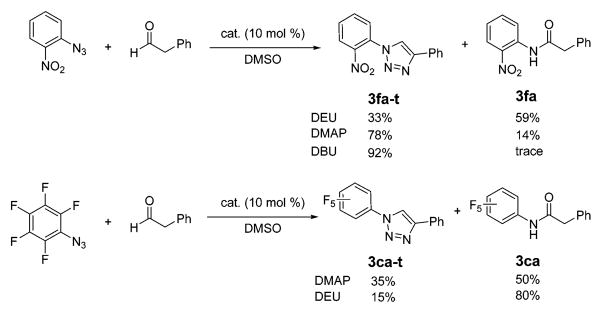
Catalysis by DEU or Base
Figure 2 summarizes the catalytic effect of various ureas/amides on the model reaction (Table 1). The fact that thioureas completely inhibited the reaction ruled out the possibility of hydrogen bond donation-assisted electrophilic activation of the carbonyl group as the main mechanism of the reaction.17 Relative weak catalysis was also observed for N,N′-disubstituted ureas. Ureas have fair hydrogen bond basicities18 and are therefore likely to interact with, and activate, the enol form of the aldehyde. The catalytic effect is however also affected by the urea structure. For example, DMEU displayed a significantly lower catalytic effect than all other ureas tested, including TMU and DMPU (Figure S4), despite their similar properties.19
Figure 2.

Catalytic effects of ureas/amides on the model reaction (Table 1) in DMSO.
An activated enol-mediated mechanism can be proposed (Scheme 3), where ureas such as DEU and DMPU stabilize the enol form and activate the phenylacetaldehyde structure through hydrogen bonding interactions. DMSO, a strong hydrogen bond acceptor,18 could also work synergistically with DEU. The exact nature of the interactions is however complex, but evidence points to a cooperative interaction mode where both the electronic effect and the geometry of the urea play important roles. Similar phenomena were observed by Seebach and co-workers where DMPU was reported to cause increased nucleophilicity of the reagents and was especially useful for reactions involving polyanionic species.20
Scheme 3.
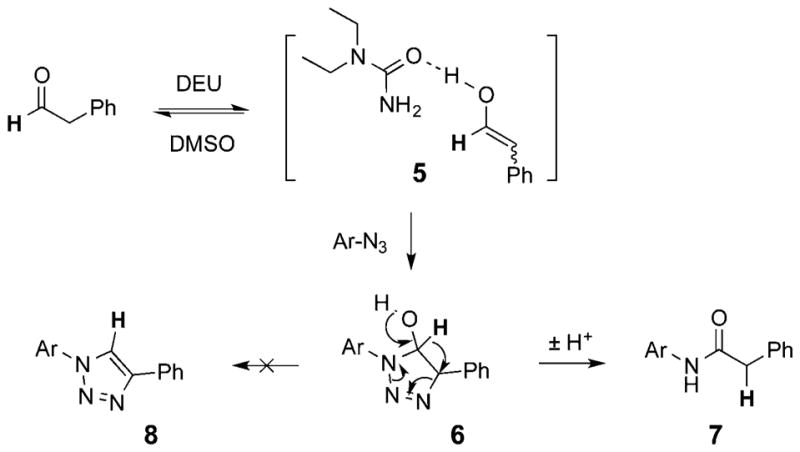
Proposed Mechanism
Following the formation of the activated enol species (5), (3 + 2) cycloaddition with the highly electrophilic azide gives the triazoline intermediate (6). The near-neutral reaction conditions facilitate synchronous rearrangement of the unstable triazoline intermediate, involving extrusion of nitrogen and a 1,5-hydride shift to give the aryl amide (7) as the final product. This rearrangement is similar to the cycloaddition reaction of PFAA with enamines to yield amidines, where the electron-deficient aryl group promoted the transformation at room temperature.13 In the presence of a stronger base, the triazoline instead underwent dehydration to form the 1,4-triazole (8), which was also reported recently by Ramachary et al.14
In summary, we have demonstrated that DEU in DMSO acted as an efficient catalyst for the reaction of phenyl-acetaldehydes with electron-deficient phenyl azides, especially perfluoroaryl azides. Unlike other aldehyde–azide amidations that use strong acids, bases, or transition metals, this reaction uses catalytic amounts of DEU and proceeds under very mild conditions at room temperature to give the products in high yield. The ability to form amide structures without using harsh reagents at elevated temperatures is highly attractive to a wide range of applications including bioconjugation, surface functionalization, and nanomaterials synthesis.
Supplementary Material
Acknowledgments
The study was in part supported by the Royal Institute of Technology, the National Institutes of Health (R01GM080295), and the National Science Foundation (CHE-1112436). S.X. thanks the China Scholarship Council for a special scholarship award.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Experimental procedures and spectroscopic data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Allen CL, Williams JM. Chem Soc Rev. 2011;40:3405. doi: 10.1039/c0cs00196a. [DOI] [PubMed] [Google Scholar]; (b) Pattabiraman VR, Bode JW. Nature. 2011;480:471. doi: 10.1038/nature10702. [DOI] [PubMed] [Google Scholar]; (c) Lundberg H, Tinnis F, Selander N, Adolfsson H. Chem Soc Rev. 2014;43:2714. doi: 10.1039/c3cs60345h. [DOI] [PubMed] [Google Scholar]
- 2.Saxon E, Armstrong JI, Bertozzi CR. Org Lett. 2000;2:2141. doi: 10.1021/ol006054v. [DOI] [PubMed] [Google Scholar]
- 3.Shangguan N, Katukojvala S, Greenberg R, Williams LJ. J Am Chem Soc. 2003;125:7754. doi: 10.1021/ja0294919. [DOI] [PubMed] [Google Scholar]
- 4.Cassidy MP, Raushel J, Fokin VV. Angew Chem, Int Ed. 2006;45:3154. doi: 10.1002/anie.200503805. [DOI] [PubMed] [Google Scholar]
- 5.Fu Z, Lee J, Kang B, Hong SH. Org Lett. 2012;14:6028. doi: 10.1021/ol302915g. [DOI] [PubMed] [Google Scholar]
- 6.(a) Kulkarni SS, Hu X, Manetsch R. Chem Commun. 2013;49:1193. doi: 10.1039/c2cc37289d. [DOI] [PubMed] [Google Scholar]; (b) Carbone G, Burnley J, Moses JE. Chem Commun. 2013;49:2759. doi: 10.1039/c3cc40452h. [DOI] [PubMed] [Google Scholar]; (c) Zhou B, Yang Y, Shi J, Feng H, Li Y. Chem—Eur J. 2013;19:10511. doi: 10.1002/chem.201301168. [DOI] [PubMed] [Google Scholar]; (d) Jin LM, Lu H, Cui Y, Lizardi CL, Arzua TN, Wojtas L, Cui X, Zhang XP. Chem Sci. 2014;5:2422. doi: 10.1039/C4SC00697F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Boyer JH, Hamer J. J Am Chem Soc. 1955;77:951. [Google Scholar]; (b) Boyer JH, Canter FC, Hamer J, Putney RK. J Am Chem Soc. 1956;78:325. [Google Scholar]; (c) Boyer J, Morgan JL. J Org Chem. 1959;24:561. [Google Scholar]
- 8.Review: Scott G, Aubé J. In: Organic Azides: Syntheses and Applications. 1. Bräse S, Banert K, editors. John Wiley & Son; Chichester, U.K: 2010. pp. 191–237.
- 9.Selected examples: Milligan GL, Mossman CJ, Aubé J. J Am Chem Soc. 1995;117:10449.Lee HL, Aubé J. Tetrahedron. 2007;63:9007. doi: 10.1016/j.tet.2007.05.079.Spangenberg T, Breit B, Mann A. Org Lett. 2009;11:261. doi: 10.1021/ol802314g.Chen H, Li R, Gao F, Li X. Tetrahedron Lett. 2012;53:7147.Gu P, Sun J, Kang XY, Yi M, Li XQ, Xue P, Li R. Org Lett. 2013;15:1124. doi: 10.1021/ol400213f.Su B, Chen F, Wang Q. J Org Chem. 2013;78:2775. doi: 10.1021/jo302725q.Yi M, Gu P, Kang XY, Sun J, Li R, Li XQ. Tetrahedron Lett. 2014;55:105.
- 10.(a) Banks RE, Prakash A. J Chem Soc, Perkin Trans. 1974;1:1365. [Google Scholar]; (b) Poe R, Schnapp K, Young MJT, Grayzar J, Platz MS. J Am Chem Soc. 1992;114:5054. [Google Scholar]; (c) Gritsan NP, Gudmundsdottir AD, Tigelaar D, Zhu Z, Karney WL, Hadad CM, Platz MS. J Am Chem Soc. 2001;123:1951. doi: 10.1021/ja9944305. [DOI] [PubMed] [Google Scholar]
- 11.(a) Schnapp KA, Poe R, Leyva E, Soundararajan N, Platz MS. Bioconjugate Chem. 1993;4:172. doi: 10.1021/bc00020a010. [DOI] [PubMed] [Google Scholar]; (b) Kapfer I, Jacques P, Toubal H, Goeldner MP. Bioconjugate Chem. 1995;6:109. doi: 10.1021/bc00031a013. [DOI] [PubMed] [Google Scholar]; (c) Baruah H, Puthenveetil S, Choi YA, Shah S, Ting AY. Angew Chem, Int Ed. 2008;47:7018. doi: 10.1002/anie.200802088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Liu LH, Yan M. Acc Chem Res. 2010;43:1434. doi: 10.1021/ar100066t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Park J, Yan M. Acc Chem Res. 2013;46:181. doi: 10.1021/ar300172h. [DOI] [PubMed] [Google Scholar]; (c) Chen X, Ramström O, Yan M. Nano Res. 2014;7:1381. doi: 10.1007/s12274-014-0507-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xie S, Lopez SA, Ramström O, Yan M, Houk KN. J Am Chem Soc. 2014 doi: 10.1021/ja511457g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ramachary DB, Shashank AB, Karthik S. Angew Chem, Int Ed. 2014;53:10420. doi: 10.1002/anie.201406721. [DOI] [PubMed] [Google Scholar]
- 15.(a) Grant HM, Mctigue P, Ward DG. Aust J Chem. 1983;36:2211. [Google Scholar]; (b) Fawcett WR. J Phys Chem. 1993;97:9540. [Google Scholar]; (c) Wang F, Ma SG, Zhang DX, Cooks RG. J Phys Chem A. 1998;102:2988. [Google Scholar]; (d) Notario R, Castano O, Herreros M, Abboud JLM. THEOCHEM. 1996;371:21. [Google Scholar]
- 16.(a) Chiang Y, Kresge AJ, Krogh ET. J Am Chem Soc. 1988;110:2600. [Google Scholar]; (b) Chiang Y, Kresge AJ, Walsh PA, Yin Y. J Chem Soc, Chem Commun. 1989:869. [Google Scholar]
- 17.(a) Doyle AG, Jacobsen EN. Chem Rev. 2007;107:5713. doi: 10.1021/cr068373r. [DOI] [PubMed] [Google Scholar]; (b) MacMillan DW. Nature. 2008;455:304. doi: 10.1038/nature07367. [DOI] [PubMed] [Google Scholar]
- 18.(a) Le Questel J-Y, Laurence C, Lachkar A, Helbert M, Berthelot M. J Chem Soc, Perkin Trans. 1992;2:2091. [Google Scholar]; (b) Laurence C, Berthelot M. Perspect Drug Discovery Des. 2000;18:39. [Google Scholar]
- 19.Barker BJ, Rosenfarb J, Caruso JA. Angew Chem, Int Ed. 1979;18:503. [Google Scholar]
- 20.(a) Mukhopadhyay T, Seebach D. Helv Chim Acta. 1982;65:385. [Google Scholar]; (b) Beck AK, Seebach D, Nikonov G. e-EROS. 1995 doi: 10.1002/047084289X.rd366.pub2. [DOI] [Google Scholar]; (c) Volz N, Clayden J. Angew Chem, Int Ed. 2011;50:12148. doi: 10.1002/anie.201104037. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.