

Abstract
The divergent TGF-β superfamily member, macrophage inhibitory cytokine-1 (MIC-1/GDF15), is overexpressed by most cancers, including prostate cancer (PCa). Whilst its circulating levels are linked to cancer outcome, the role MIC-1/GDF15 plays in cancer development and progression is incompletely understood. To investigate its effect on PCa development and spread, we have used TRAMP prostate cancer prone mice bearing a germline deletion of MIC-1/GDF15 (TRAMPMIC-/-). On average TRAMPMIC-/- mice died about 5 weeks earlier and had larger prostatic tumors compared with TRAMP mice that were wild type for MIC-1/GDF15 (TRAMPMIC+/+). Additionally, at the time of death or ethical end point, even when adjusted for lifespan, there were no significant differences in the number of mice with metastases between the TRAMPMIC+/+ and TRAMPMIC-/- groups. However, consistent with our previous data, more than twice as many TRAMP mice overexpressing MIC-1/GDF15 (TRAMPfmsmic-1) had metastases than TRAMPMIC+/+ mice (p<0.0001). We conclude that germ line gene deletion of MIC-1/GDF15 leads to increased local tumor growth resulting in decreased survival consistent with an overall protective role for MIC-1/GDF15 in early primary tumor development. However, in advancing disease, as we have previously noted, MIC-1/GDF15 overexpression may promote local invasion and metastatic spread.
Introduction
Prostate cancer (PCa) is one of the most frequently diagnosed cancers in men. It caused an estimated 29,480 deaths in the USA in 2014 and is the second leading cause of cancer deaths in men [1]. Despite its clinical importance, our understanding of its biology is incomplete and aside from surgery for early stage disease, its therapy is palliative. Like most, if not all tumors, PCa displays altered expression of many gene products, including cytokines and growth factors. One cytokine commonly overexpressed in many cancers, including PCa, is MIC-1/GDF15, a divergent member of the transforming growth factor-β superfamily[2]. Expression of this cytokine is also induced by most cancer therapies and its serum levels are clearly linked to cancer outcome [3–9].
MIC-1/GDF15 is detectible in the blood of all individuals [10]. Its expression by cancers is frequently reflected by rises in its blood levels, usually in proportion to the stage and extent of tumor [5,11–16]. For example, there is a continuing rise in MIC-1/GDF15 serum levels with progression to colonic polyps, high grade dysplastic polyps, localized colorectal cancer (CRC) and then disseminated CRC [13]. Further, patients with CRC with elevated serum MIC-1/GDF15 levels at presentation, have a worse overall prognosis and earlier disease relapse [13,17]. For PCa, MIC-1/GDF15 serum levels are an independent predictor of the presence of cancer [14] and in more advanced disease they predict overall survival and bone metastasis [12,16]. High MIC-1/GDF15 serum levels also predict diagnosis and/or outcome for a wide range of malignancies including melanoma [18,19], cancers of the pancreas [15,20,21], thyroid [22,23], ovary [24] and endometrium [25].
In patients with advanced cancers, serum MIC-1/GDF15 levels commonly rise from a normal mean of about 450pg/ml [13] to 10,000–100,000 pg/ml or more [5] and may cause cancer anorexia/cachexia [26,27]. This common cancer complication is mediated by actions of MIC-1/GDF15 on feeding centres in the brain and can be reversed by neutralising antibodies [26,27]. MIC-1/GDF15 serum levels in cancer are influenced not only by its over-expression, but also depend on how it is processed by the tumor. Intracellular processing leads to removal of the MIC-1/GDF15 propeptide and diffusion into the blood stream after secretion. However, as the propeptide interacts with tumor stroma, unprocessed secreted protein remains bound to the extracellular matrix proximate to the producing tumor [21,28]. In PCa, increased stromal MIC-1/GDF15 is associated with better patient outcomes, especially in those with low-grade localized prostate tumors (Gleason sum score of 6 or less) [28], suggesting that its increased local availability is beneficial. By contrast, high circulating concentrations of MIC-1/GDF15 are associated with a poor outcome [28]. However, whether MIC-1/GDF15 overexpression in cancer has a beneficial, harmful or mixed effect on disease outcome is difficult to determine from epidemiological studies alone.
The in vivo cancer related activity of MIC-1/GDF15, has been examined in a number of tumor xenograft studies with mixed results. For example, enforced MIC-1/GDF15 overexpression in HCT-116 colon cancer cells [29] or in the DU145 [30] PCa cell line, xenografted into immunodeficient mice, reduced tumor size. A tumorigenic glioblastoma cell line, that remained unaffected by MIC-1/GDF15 in vitro, on transfection with MIC-1/GDF15, failed to develop tumors in nude mice [31]. The authors suggested that MIC-1/GDF15 may have acted on the local tumor microenvironment to inhibit tumor growth. By contrast, knock down of MIC-1/GDF15 in a human melanoma [18] and a mouse glioblastoma [32] cell line significantly decreased the growth of engrafted tumors. Further, the xenografts of PC3 PCa cell line engineered to overexpress MIC-1/GDF15 grew faster [33] and when orthotopically implanted, led to more metastases [34].
Unlike the xenograft models in immunodeficient mice, carcinogen induced and spontaneously developing cancer models are performed in immune competent mice, which more closely mimic the pathogenesis of cancers. In chemically induced cancer models, transgenic overexpression of MIC-1/GDF15 leads to resistance to urethane induced lung cancer [35] and azoxymethane induced colon cancer [36]. However, whilst transgenic overexpression led to protection in these two instances, gene deletion did not modify the development of diethylnitrosamine induced hepatocellular carcinoma [37].
Spontaneously developing cancers in transgenic mice often most closely conform to human cancers and all studies based on their use suggest that MIC-1/GDF15 is largely protective in early disease. Development of large bowel polyps and cancer in Apcmin mice is reduced by transgenic overexpression of MIC-1/GDF15 [36]. Further, germline deletion of MIC-1/GDF15 in Apcmin mice abolished the protection afforded from the COX inhibitor sulindac [38]. As this class of drug is known to induce expression of MIC-1/GDF15 in both mice and men [38], this data suggests that tumor suppression may be dependent on the expression of MIC-1/GDF15 [38]. Further supporting this view is a study utilising samples from the Polyp Prevention Trial [39]. This demonstrated that non-steroidal anti inflammatory drug (NSAID) users had a higher serum MIC-1/GDF15 level than non-users and only NSAID users with an elevated serum MIC-1/GDF15 level were protected from colonic adenoma recurrence [39,40].
More recently we have assessed the effect of MIC-1/GDF15 overexpression on the course of cancer in Transgenic Adenocarcinoma of Mouse Prostate (TRAMP) prostate cancer prone transgenic mice. TRAMP mice express SV40 early genes (T and t; Tag) under the control of rat probasin (rPB) promoter [41], which targets its expression to prostatic epithelium. Heterozygous TRAMP male mice develop progressive prostate cancer exhibiting the same spectrum of disease as found in men. Over the course of 6–12 months these mice progressively develop localized then invasive cancer that exhibits metastatic spread to distant sites, primarily the pelvic lymph nodes, liver, kidney and lungs [42]. Our data indicates that TRAMP mice, overexpressing MIC-1/GDF15, have substantially increased survival due to decreased growth and histological grade of the primary tumor [43], further supporting a beneficial role for MIC-1/GDF15 in early cancer. However, as the tumor advanced, these mice also developed more metastases [43], suggesting that MIC-1/GDF15 overexpression may have deleterious actions late in the course of cancer. There are no other data from transgenic cancer models where the effect of MIC-1/GDF15 on advanced cancers has been investigated.
It is important to understand the effect that MIC-1/GDF15 has on the biology of cancers as it is highly overexpressed by many cancers and its expression is induced by cancer therapies. Thus any effect it has on the biology of cancer is likely to be of clinical significance. To further advance our understanding of this cytokine in cancer, we have determined how MIC-1/GDF15 deficiency influenced the evolution of PCa. We have utilised TRAMP prostate cancer prone mice that also bear a germline deletion of the MIC-1/GDF15 gene (TRAMPMIC-/-) or wild type MIC-1/GDF15 (TRAMPMIC+/+), to compare survival rate, pattern of PCa growth and metastatic spread. TRAMPMIC-/- mice had significantly larger prostate tumors and shorter survival than TRAMPMIC+/+ mice, but there was no significant difference in the incidence and rate of metastasis in the two mouse lines suggesting that different mechanisms mediate the effects of MIC-1/GDF-15 on local and metastatic PCa development. These data are consistent with earlier studies, identifying a largely protective role for MIC-1/GDF15 in the local growth of early cancers.
Materials and Methods
Ethics Statement
All research and animal care procedures were approved by the Garvan Institute/St Vincent’s Hospital Animal Experimentation Ethics Committee (Ethics No: 07/05, 10/05 and 13/08) and were in agreement with the Australian Code of Practice for the Care and Use of Animals for Scientific Purpose.
Transgenic mice
Heterozygous male TRAMP mice (TRAMP+/-) [41] were generated by mating TRAMP+/- females (C57BL/6 background) with non-transgenic C57BL/6 males. Mice with a germline deletion of the MIC-1/GDF15 gene (MIC-1-/-) [44], also on a C57BL/6 background were bred with TRAMP mice to generate MIC-1-/- mice also bearing the TRAMP transgene (TRAMPMIC-/-). The PB-SV40 T transgene was identified using DNA extracted from tail samples and PCR primers directed at the PB-SV40 T-antigen sequence: Pb-forward: 5’-CCGGTCGACCGGAAGCTTCCACAAGTGCATTTA-3’ and SV40Tag-reverse: 5’-CTCCTTTCAAGACCTAGAAGGTCCA-3’. MIC-1/GDF15 gene deletion was identified using primers MIC1Exon2for: 5’-GGCGGCGCACAGCTGGAACTGC-3’ with MIC1Exon2Rev: 5’-CAGCCCCGGGCCACCAGGTCAT-3’ (Wild type pair) and MIC-1/GDF15KOfor: 5’-GAGAGGACTCGAACTCAGAACCA-3’ with MIC-1/GDF15KORev: 5’-GAAGTTATATTAAGGGTTCCGCAAGC-3’ (Knock-out pair). Syngeneic mice overexpressing MIC-1/GDF15 under control of the myeloid cell specific c-fms promoter (MIC-1fms) were used to breed TRAMP mice that also overexpress MIC-1/GDF15 (TRAMPfmsmic-1). The double transgenic TRAMPfmsmic-1 mice were generated by crossing TRAMP+/- females with homozygous MIC-1fms males. The MIC-1/GDF15 transgene in TRAMPfmsmic-1 mice was identified by PCR using primers, Flag-forward: 5’-GACTACAAGGACGACGATGACAAG-3’ and MS8-reverse: 5’-CGAAGCCTACCGCGTGCACCGAG-3’. The reaction conditions used were: denaturation at 95°C for 10 s, annealing at 60°C for 20 s, and extension at 72°C for 30 s.
Survival study
Based on a statistical power analysis for sample size, (Alpha error = 0.05, statistical power = 0.95), 35 TRAMPMIC+/+ and 35 TRAMPMIC-/- mice were allocated at 4–6 weeks of age, for a survival study. From that time, mice were weighed once a week and monitored twice a week for tumor size and extent by palpating the abdomen. Mice either died or were culled when they reached ethical end points of tumor size larger than 11mm X 11mm X 11mm, more than 20% weight loss or meeting any other ethical end point criteria for euthanasia. The overall survival of individual mice was calculated from birth to ethical end point or death from the tumor. Survival distribution was estimated using the method of Kaplan-Meier. At necropsy the genitourinary complex (GU) consisting of prostate (including dorsal, lateral, ventral, and anterior lobes), urethra, ampullary gland, seminal vesicle (SV) and urinary bladder was taken out and weighed. Prostate was excised from GU and weighed separately. Weight of the GU and prostate of each mouse was normalized by its body weight (organ wt/body wt).
Primary tumor size
In a separate cohort to that above, prostate tumor growth was compared in TRAMPMIC+/+ and TRAMPMIC-/- mice. At the start of the study 88 TRAMP and 88 TRAMPMIC-/- mice, 22 of each for each stage, were pre-allocated to be sacrificed at different time points from early to advanced tumor stages (8, 17, 25 and 33 weeks of age). For each of the 88 mice necropsied, the GU was excised and prostate was separated from GU. Total GU and prostate weight were recorded and normalized for the donor mouse total body weight (organ wt/body wt).
Identification of tumor metastases
To estimate the occurrence of metastasis at the time of death or culling in TRAMPMIC+/+ and TRAMPMIC-/- mice, examined a different cohort of TRAMPMIC+/+ (n = 63) and TRAMPMIC-/- (n = 63). For comparison, we also examined a similar number of MIC-1/GDF15 overexpressing TRAMPfmsmic-1 mice (n = 63), whose PCa was known to be associated with increased metastases [43]. Mice were looked after and euthanized using the same criteria as mentioned above in the survival study. At the necropsy pelvic lymph nodes, kidney, and liver tumors (if present) were harvested and fixed in 10% neutral buffered formalin. Lungs were excised, weighed and fixed in Bouin’s fixative (Sigma-Aldrich) to visualize and count lung tumor colonies. Metastatic lesions on all the organs were counted under a dissecting microscope. Some of the lesions were confirmed by H&E staining and further by immunostaining of frozen tissue sections with anti Tag antibody (Santa Cruz) to confirm the prostatic origin of the tumor. The number of mice having distant organ metastasis was compared in all the three mouse lines.
Statistical analysis
All graphs and statistical evaluation of all the experiments were performed with GraphPad Prism software version 6 for Mac OS X, (GraphPad Software, San Diego, CA, USA). Kaplan–Meier analysis and log-rank statistic were used to compare survival curves. The data for GU and prostate tumor sizes between groups were compared using 2-way ANOVA or t-test. The Chi-square test was used for categorical analysis. Statistical power analysis for sample size was done with the online power and sample size analysis tool (http://www.stat.ubc.ca/) using an Alpha Error of 0.05 and statistical power level of 0.95. Multivariate logistic regression analysis was used to examine the relationship of metastasis with survival time. A p value less than 0.05 considered statistically significant.
Results
MIC-1/GDF15 gene deleted TRAMP mice die earlier of PCa
In order to assess the effects of MIC-1/GDF15 gene deletion on the overall survival of TRAMP mice, we monitored a cohort (n = 35) of TRAMPMIC+/+ and TRAMPMIC-/- mice (Survival Group) till death or ethical end point. Kaplan-Meier survival analysis showed that TRAMPMIC-/- mice had significantly shorter survival than TRAMPMIC+/+ mice (Fig. 1A, p = 0.0416, log-rank test). The mean survival of 39.5 weeks in TRAMPMIC+/+ mice was reduced by about 5 weeks (34.4 weeks) in the TRAMPMIC-/- group. Further, while only 20% of TRAMPMIC-/- mice survived at week 40, 42.85% of TRAMPMIC+/+ mice were still alive (Fig. 1A). These data indicate that germline gene deletion of MIC-1/GDF15 reduced PCa related survival in TRAMP mice.
Fig 1. TRAMPMIC-/- mice have shorter survival and larger prostate tumors than TRAMPMIC+/+ mice.

(A) Survival data for TRAMPMIC+/+ and TRAMPMIC-/- mice (n = 35). Overall survival of individual mice from birth to death was plotted using the Kaplan-Meier method. The log-rank statistic for median survival time is shown. (B) The genitourinary complex (GU) and prostate tumor weights (C), in TRAMPMIC+/+ and TRAMPMIC-/- mice, at the necropsy, are corrected for body weight and presented as mean ± SEM. Differences are analyzed using an unpaired 2-tailed t test. (D) The number of TRAMPMIC+/+ and TRAMPMIC-/- mice having large prostate tumor (corrected tumor wt>10mg/g), was compared using a Chi-square test. p values are shown as *, p< 0.05; **, p< 0.01.
TRAMPMIC-/- mice have larger prostate tumors at necropsy
At the necropsy of the above-mentioned survival group of TRAMPMIC+/+ and TRAMPMIC-/- mice, GU and prostate were isolated and their weights, corrected for body weight, were recorded. Despite dying earlier than TRAMPMIC+/+ mice, TRAMPMIC-/- mice on average had significantly heavier prostate tumors at the time of death (Fig. 1C, p = 0.0240). Further, the TRAMPMIC-/- group had far more mice with very large prostate tumors (normalized prostate weight >10mg/g) than the TRAMPMIC+/+ group (Fig. 1D, p = 0.0027, Chi-square test). There was no significant difference in total GU wt (Fig. 1B) between two mouse lines because TRAMPMIC+/+ had significantly larger SV tumors than TRAMPMIC-/- mice (data not shown). This data suggests that deletion of MIC-1/GDF15 gene was associated with increased local prostate tumor growth in TRAMP mice, perhaps with reduced seminal vesicle invasion.
MIC-1/GDF15 deletion enhances PCa growth in TRAMP mice
To further assess the impact of MIC-1/GDF15 on prostate cancer growth, at 4–6 weeks of age, we pre-assigned another cohort of 88 TRAMPMIC+/+ and 88 TRAMPMIC-/- mice to be culled progressively at four predefined time point up to 33 weeks of age. Consistent with the data from the survival study group mice, discussed above, there was no significant difference in the normalized GU weights between two mouse lines at any time point observed (Fig. 2A). When we looked at the prostate size, there were no detectible differences in the average corrected prostate weights between TRAMPMIC+/+ and TRAMPMIC-/- mouse lines at week 8 and 17. At week 25 and 33, TRAMPMIC-/- mice had a 6.9 and 8 fold increase in corrected average prostate tumor weight respectively, as compared to TRAMPMIC+/+ mice (Fig. 2B) and this difference was statistically significant at week 33 (p = 0.0098, 2 way ANOVA).
Fig 2. TRAMPMIC-/- mice have comparatively larger prostate tumor than TRAMPMIC+/+ mice.

The corrected tumor weights of (A) GU and (B) prostates were compared in TRAMPMIC+/+ and TRAMPMIC-/- mice (n = 22/group/time point) sacrificed at 8, 17, 25 and 33 weeks of age. Results were analyzed using a 2 way ANOVA and are presented as mean ± SEM. p values are shown as *, p< 0.05.
MIC-1/GDF15 gene deletion has no effect on metastases
Since metastasis is the major cause of death in patients with human PCa, we evaluated the effect of MIC-1/GDF15 gene deletion on the incidence and extent of metastasis in TRAMP mice. We examined a separate cohort of 63 TRAMPMIC+/+, 63 TRAMPMIC-/- and 63 MIC-1/GDF15 overexpressing TRAMP (TRAMPfmsmic-1) mice, which were followed until death or ethical end point. The latter group was included as a positive control, as our previous study had indicated TRAMPfmsmic-1 mice have more metastases but survive longer [43]. Kaplan-Meier survival analysis reconfirmed that TRAMPMIC-/- mice die significantly earlier than TRAMPMIC+/+ mice (Fig. 3A, p = 0.0267, log-rank test), consistent with data in the survival group. Further, TRAMPfmsmic-1 mice died at significantly slower rate than TRAMPMIC+/+ mice (Fig. 3A, p<0.0001, log-rank test) confirming data from our previous publication [43]. In this cohort 19% of the TRAMPMIC+/+ mice developed macroscopically detectible distant organ metastasis in the surveyed organs, which was not significantly different to that of 14.2% in TRAMPMIC-/- mice (Fig. 3B). The incidence of metastases in these two mouse lines was significantly less that in TRAMPfmsmic-1 mice, 59% of which developed metastases (Fig. 3B). These data show that although TRAMPMIC-/- mice die significantly earlier than TRAMPMIC+/+ mice (Fig. 3A) there were no significant differences in the incidence of distant organ metastasis between the two mouse lines (Fig. 3B). In contrast, as previously reported, a significantly higher proportion of TRAMPfmsmic-1 mice showed distant organ metastasis compared with TRAMPMIC+/+ or TRAMPMIC-/- mice (Fig. 3B, p<0.0001, Chi-square test). Multivariate logistic regression analysis confirmed that the increased proportion of TRAMPfmsmic-1 mice with metastases was independent of their longer survival times (p>0.5) and only dependent on genotype (p<0.0001). Further, using a similar approach, the lack of difference in the proportion of TRAMPMIC-/- compared with TRAMPMIC+/+ mice with metastases, could not be accounted for by their shorter survival (p>0.5).
Fig 3. Effect of MIC-1/GDF15 gene modification on metastases.
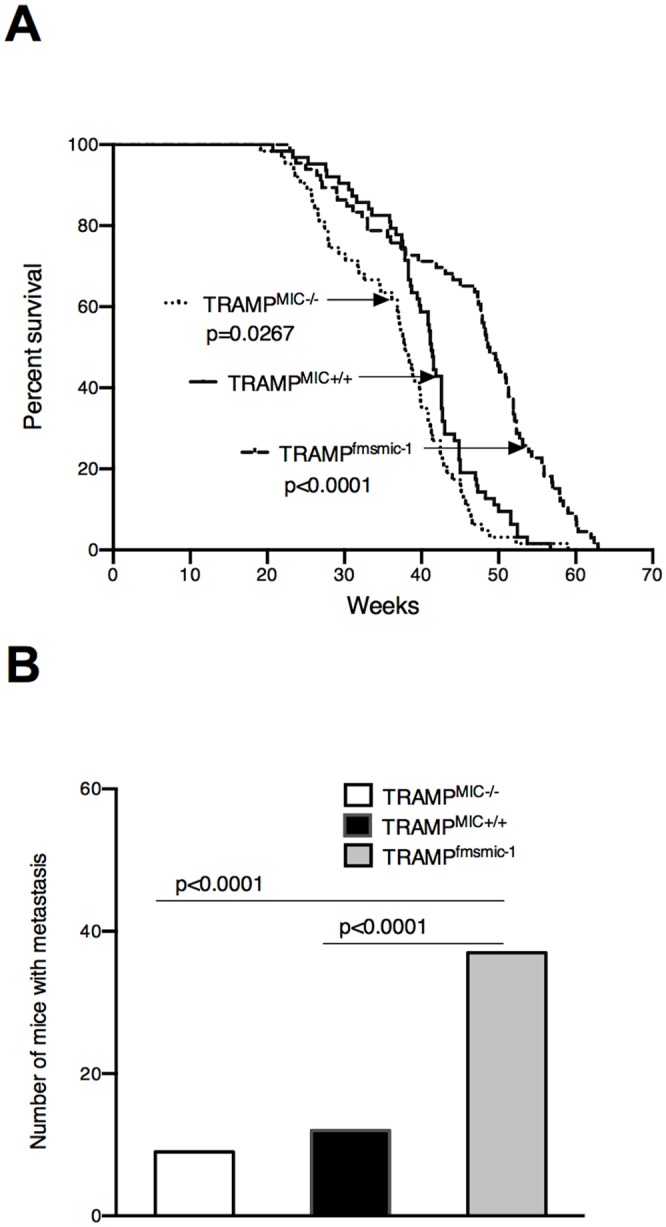
(A) Survival data for TRAMPMIC+/+ (n = 63), TRAMPMIC-/- (n = 63) and TRAMPfmsmic-1 (n = 63) mice is presented as a Kaplan-Meier plot and the log-rank statistic for median survival time is shown. (B) Comparison between number of TRAMPMIC+/+ (n = 63), TRAMPMIC-/- (n = 63) and TRAMPfmsmic-1 (n = 63) mice having distant organ metastases at the time of death has been analyzed using the Chi-squared test.
Discussion
This study clearly indicates that germline deletion of the MIC-1/GDF15 leads to increased local primary tumor growth resulting in earlier death of TRAMP PCa prone mice. These data are consistent with our previous publication indicating that transgenic overexpression of MIC-1/GDF15 decreases local PCa tumor growth and substantially increases survival of TRAMP mice. This result is consistent with results in another transgenic model of early cancer in Apcmin colonic polyp prone mice also overexpressing MIC-1/GDF15 [38]. This reinforces the argument that MIC-1/GDF15 plays a protective role in early local tumor development and growth. Several epidemiological studies suggest that this finding may be translated to at least some human cancers. For example, increased local concentrations of extracellular matrix associated MIC-1/GDF15 in prostate cancer biopsies were associated with reduced risk of disease progression, especially in the subgroup with early cancer and with Gleason grade of 6 or less. In this group MIC-1/GDF15 localised to the tumor matrix was the single best predictor of tumor recurrence [28]. In patients from the polyp prevention trial, a higher polyp free serum MIC-1/GDF15 level afforded protection from polyp recurrence and NSAID mediated protection was lost if MIC-1/GDF15 serum levels were not elevated with NSAID treatment [39].
The apparently discordant effect of MIC-1/GDF15 on local tumor growth and the metastatic process, that we described previously, is again demonstrated here [43]. Transgenic overexpression leads to smaller local tumors and longer survival, as TRAMPcfmsmic-1 mice no longer die early of local disease. Independently of increased survival time, these same TRAMPcfmsmic-1 mice have more metastases. However, we could demonstrate no difference in the proportion of mice with metastases between TRAMPMIC+/+ and TRAMPMIC-/- mice. As few mice of either line developed metastases by the experimental endpoint we may not have studied enough mice to be certain that no differences exist. Additionally, as prostate tumors in TRAMPMIC+/+ mice express little MIC-1/GDF15 [43], it may be that this model is more sensitive to the effects of increased expression compared to gene deletion.
Whilst there was no significant difference in metastases between TRAMPMIC+/+ and TRAMPMIC-/- mice, there may be differences in local invasion. TRAMPMIC-/- mice with larger prostate tumors had smaller SVs (data not shown), which suggests that there may be less SV invasion.
How MIC-1/GDF15 might exert effects on tumor growth and spread is uncertain. Elucidating its mechanisms of action is greatly hampered because the identity of its receptor is unknown. It is presumed that the receptor is a member of the highly conserved hetrotetrameric TGF-b receptor (TBR) superfamily, but there is limited direct evidence for this. There is some indirect evidence, based on antibody blockade, that it may utilise TBRII [26,45] the class 2 receptor used by TGF-b itself, but there is no direct biochemical or genetic evidence supporting this data. The signalling cascade of the receptor complex or class I receptor utilised by MIC-1/GDF15 is to date, completely unknown.
There is now a plethora of published data on the role of MIC-1/GDF15 on tumor growth and spread with a confusing range of results. Data from experiments using human cell lines, in which MIC-1/GDF15 expression has either been induced or knocked down, then xenografted into immunodeficient mice, have provided sometimes, contradictory results [18,29–34]. Studies using transgenic models of spontaneously developing cancer that also bear genetically modified MIC-1/GDF15 expression and an intact immune system all point to a protective role for MIC-1/GDF15 on local tumor development. A similar effect can also be observed in two models of carcinogen induced cancer in transgenic mice overexpressing MIC-1/GDF15, which also have an intact immune system [35,36]. The most parsimonious explanation that may explain these contradictions is that MIC-1/GDF15 regulates anti-cancer immunity, which in turn regulates cancer growth.
Overall, our results support an important protective role for MIC-1/GDF15 in the development and early growth of PCa and probably cancer in general. Unravelling the biological effect of MIC-1/GDF15 on tumor evolution and biology is of practical importance for several reasons. A high proportion of cancers express it, to the extent that serum level can rise up to 10–100 fold and cause cancer anorexia/cachexia. Further, its expression is increased by all cancer treatment modalities including surgery, radiotherapy and chemotherapy. Thus any effect that MIC-1/GDF15 has on local tumor biology, particularly tumor spread is likely to impact most cancer patients, raising the prospect that modulation of MIC-1/GDF15 actions during therapy might reduce the risk of metastatic disease and other complications of cancer.
Data Availability
All relevant data are within the paper.
Funding Statement
This work was supported in part by grants from Cancer Council New South Wales (http://www.cancercouncil.com.au/) and The National Health and Medical Research Council of Australia (NHMRC) (https://www.nhmrc.gov.au/). DAB is a National Health and Medical Research Council Biomedical Career Development Fellow. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. American CS (2014) Cancer Facts & Figures. Atlanta: American Cancer Society; Available: http://www.cancer.org/research/cancerfactsstatistics/cancerfactsfigures2014/. [Google Scholar]
- 2. Bootcov MR, Bauskin AR, Valenzuela SM, Moore AG, Bansal M, et al. (1997) MIC-1, a novel macrophage inhibitory cytokine, is a divergent member of the TGF-beta superfamily. Proc Natl Acad Sci U S A 94: 11514–11519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bauskin AR, Brown DA, Kuffner T, Johnen H, Luo XW, et al. (2006) Role of macrophage inhibitory cytokine-1 in tumorigenesis and diagnosis of cancer. Cancer Res 66: 4983–4986. [DOI] [PubMed] [Google Scholar]
- 4. Breit SN, Johnen H, Cook AD, Tsai VW, Mohammad MG, et al. (2011) The TGF-beta superfamily cytokine, MIC-1/GDF15: a pleotrophic cytokine with roles in inflammation, cancer and metabolism. Growth Factors 29: 187–195. 10.3109/08977194.2011.607137 [DOI] [PubMed] [Google Scholar]
- 5. Welsh JB, Sapinoso LM, Kern SG, Brown DA, Liu T, et al. (2003) Large-scale delineation of secreted protein biomarkers overexpressed in cancer tissue and serum. Proc Natl Acad Sci U S A 100: 3410–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Joiner MC, Thomas RA, Grever WE, Smolinski JM, Divine GW, et al. (2011) Developing point of care and high-throughput biological assays for determining absorbed radiation dose. Radiother Oncol 101: 233–236. 10.1016/j.radonc.2011.05.068 [DOI] [PubMed] [Google Scholar]
- 7. Boyer J, Allen WL, McLean EG, Wilson PM, McCulla A, et al. (2006) Pharmacogenomic identification of novel determinants of response to chemotherapy in colon cancer. Cancer Res 66: 2765–2777. [DOI] [PubMed] [Google Scholar]
- 8. Modlich O, Prisack HB, Munnes M, Audretsch W, Bojar H (2004) Immediate gene expression changes after the first course of neoadjuvant chemotherapy in patients with primary breast cancer disease. Clin Cancer Res 10: 6418–6431. [DOI] [PubMed] [Google Scholar]
- 9. Huang CY, Beer TM, Higano CS, True LD, Vessella R, et al. (2007) Molecular alterations in prostate carcinomas that associate with in vivo exposure to chemotherapy: identification of a cytoprotective mechanism involving growth differentiation factor 15. Clin Cancer Res 13: 5825–5833. [DOI] [PubMed] [Google Scholar]
- 10. Brown DA, Bauskin AR, Fairlie WD, Smith MD, Liu T, et al. (2002) Antibody-based approach to high-volume genotyping for MIC-1 polymorphism. Biotechniques 33: 118–120, 122, 124 passim. [DOI] [PubMed] [Google Scholar]
- 11. Baek KE, Yoon SR, Kim JT, Kim KS, Kang SH, et al. (2009) Upregulation and secretion of macrophage inhibitory cytokine-1 (MIC-1) in gastric cancers. Clin Chim Acta 401: 128–133. 10.1016/j.cca.2008.12.008 [DOI] [PubMed] [Google Scholar]
- 12. Brown DA, Lindmark F, Stattin P, Balter K, Adami HO, et al. (2009) Macrophage inhibitory cytokine 1: a new prognostic marker in prostate cancer. Clin Cancer Res 15: 6658–6664. 10.1158/1078-0432.CCR-08-3126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Brown DA, Ward RL, Buckhaults P, Liu T, Romans KE, et al. (2003) MIC-1 serum level and genotype: associations with progress and prognosis of colorectal carcinoma. Clin Cancer Res 9: 2642–2650. [PubMed] [Google Scholar]
- 14. Brown DA, Stephan C, Ward RL, Law M, Hunter M, et al. (2006) Measurement of serum levels of macrophage inhibitory cytokine 1 combined with prostate-specific antigen improves prostate cancer diagnosis. Clin Cancer Res 12: 89–96. [DOI] [PubMed] [Google Scholar]
- 15. Koopmann J, Buckhaults P, Brown DA, Zahurak ML, Sato N, et al. (2004) Serum macrophage inhibitory cytokine 1 as a marker of pancreatic and other periampullary cancers. Clin Cancer Res 10: 2386–2392. [DOI] [PubMed] [Google Scholar]
- 16. Selander KS, Brown DA, Sequeiros GB, Hunter M, Desmond R, et al. (2007) Serum macrophage inhibitory cytokine-1 concentrations correlate with the presence of prostate cancer bone metastases. Cancer Epidemiol Biomarkers Prev 16: 532–537. [DOI] [PubMed] [Google Scholar]
- 17. Wallin U, Glimelius B, Jirstrom K, Darmanis S, Nong RY, et al. (2011) Growth differentiation factor 15: a prognostic marker for recurrence in colorectal cancer. Br J Cancer 104: 1619–1627. 10.1038/bjc.2011.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Boyle GM, Pedley J, Martyn AC, Banducci KJ, Strutton GM, et al. (2009) Macrophage inhibitory cytokine-1 is overexpressed in malignant melanoma and is associated with tumorigenicity. J Invest Dermatol 129: 383–391. 10.1038/jid.2008.270 [DOI] [PubMed] [Google Scholar]
- 19. Suesskind D, Schatz A, Schnichels S, Coupland SE, Lake SL, et al. (2011) GDF-15: a novel serum marker for metastases in uveal melanoma patients. Graefes Arch Clin Exp Ophthalmol. [DOI] [PubMed] [Google Scholar]
- 20. Koopmann J, Rosenzweig CN, Zhang Z, Canto MI, Brown DA, et al. (2006) Serum markers in patients with resectable pancreatic adenocarcinoma: macrophage inhibitory cytokine 1 versus CA19–9. Clin Cancer Res 12: 442–446. [DOI] [PubMed] [Google Scholar]
- 21. Bauskin AR, Jiang L, Luo XW, Wu L, Brown DA, et al. (2010) The TGF-beta superfamily cytokine MIC-1/GDF15: secretory mechanisms facilitate creation of latent stromal stores. J Interferon Cytokine Res 30: 389–397. 10.1089/jir.2009.0052 [DOI] [PubMed] [Google Scholar]
- 22. Weber F, Shen L, Aldred MA, Morrison CD, Frilling A, et al. (2005) Genetic classification of benign and malignant thyroid follicular neoplasia based on a three-gene combination. J Clin Endocrinol Metab 90: 2512–2521. [DOI] [PubMed] [Google Scholar]
- 23. Fluge O, Bruland O, Akslen LA, Lillehaug JR, Varhaug JE (2006) Gene expression in poorly differentiated papillary thyroid carcinomas. Thyroid 16: 161–175. [DOI] [PubMed] [Google Scholar]
- 24. Staff AC, Bock AJ, Becker C, Kempf T, Wollert KC, et al. (2010) Growth differentiation factor-15 as a prognostic biomarker in ovarian cancer. Gynecol Oncol 118: 237–243. 10.1016/j.ygyno.2010.05.032 [DOI] [PubMed] [Google Scholar]
- 25. Staff AC, Trovik J, Eriksson AG, Wik E, Wollert KC, et al. (2011) Elevated plasma growth differentiation factor-15 correlates with lymph node metastases and poor survival in endometrial cancer. Clin Cancer Res 17: 4825–4833. 10.1158/1078-0432.CCR-11-0715 [DOI] [PubMed] [Google Scholar]
- 26. Johnen H, Lin S, Kuffner T, Brown DA, Tsai VW, et al. (2007) Tumor-induced anorexia and weight loss are mediated by the TGF-beta superfamily cytokine MIC-1. Nat Med 13: 1333–1340. [DOI] [PubMed] [Google Scholar]
- 27. Tsai VW, Husaini Y, Manandhar R, Lee-Ng KK, Zhang HP, et al. (2012) Anorexia/cachexia of chronic diseases: a role for the TGF-beta family cytokine MIC-1/GDF15. J Cachexia Sarcopenia Muscle 3: 239–243. 10.1007/s13539-012-0082-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bauskin AR, Brown DA, Junankar S, Rasiah KK, Eggleton S, et al. (2005) The propeptide mediates formation of stromal stores of PROMIC-1: role in determining prostate cancer outcome. Cancer Res 65: 2330–2336. [DOI] [PubMed] [Google Scholar]
- 29. Baek SJ, Kim KS, Nixon JB, Wilson LC, Eling TE (2001) Cyclooxygenase inhibitors regulate the expression of a TGF-beta superfamily member that has proapoptotic and antitumorigenic activities. Mol Pharmacol 59: 901–908. [PubMed] [Google Scholar]
- 30. Wang X, Chrysovergis K, Bienstock RJ, Shim M, Eling TE (2012) The H6D variant of NAG-1/GDF15 inhibits prostate xenograft growth in vivo. Prostate 72: 677–689. 10.1002/pros.21471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Albertoni M, Shaw PH, Nozaki M, Godard S, Tenan M, et al. (2002) Anoxia induces macrophage inhibitory cytokine-1 (MIC-1) in glioblastoma cells independently of p53 and HIF-1. Oncogene 21: 4212–4219. [DOI] [PubMed] [Google Scholar]
- 32. Roth P, Junker M, Tritschler I, Mittelbronn M, Dombrowski Y, et al. (2010) GDF-15 contributes to proliferation and immune escape of malignant gliomas. Clin Cancer Res 16: 3851–3859. 10.1158/1078-0432.CCR-10-0705 [DOI] [PubMed] [Google Scholar]
- 33. Tsui KH, Chang YL, Feng TH, Chung LC, Lee TY, et al. (2012) Growth differentiation factor-15 upregulates interleukin-6 to promote tumorigenesis of prostate carcinoma PC-3 cells. J Mol Endocrinol 49: 153–163. 10.1530/JME-11-0149 [DOI] [PubMed] [Google Scholar]
- 34. Senapati S, Rachagani S, Chaudhary K, Johansson SL, Singh RK, et al. (2010) Overexpression of macrophage inhibitory cytokine-1 induces metastasis of human prostate cancer cells through the FAK-RhoA signaling pathway. Oncogene 29: 1293–1302. 10.1038/onc.2009.420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cekanova M, Lee SH, Donnell RL, Sukhthankar M, Eling TE, et al. (2009) Nonsteroidal anti-inflammatory drug-activated gene-1 expression inhibits urethane-induced pulmonary tumorigenesis in transgenic mice. Cancer Prev Res (Phila) 2: 450–458. 10.1158/1940-6207.CAPR-09-0057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Baek SJ, Okazaki R, Lee SH, Martinez J, Kim JS, et al. (2006) Nonsteroidal anti-inflammatory drug-activated gene-1 over expression in transgenic mice suppresses intestinal neoplasia. Gastroenterology 131: 1553–1560. [DOI] [PubMed] [Google Scholar]
- 37. Zimmers TA, Jin X, Gutierrez JC, Acosta C, McKillop IH, et al. (2008) Effect of in vivo loss of GDF-15 on hepatocellular carcinogenesis. J Cancer Res Clin Oncol 134: 753–759. 10.1007/s00432-007-0336-4 [DOI] [PubMed] [Google Scholar]
- 38. Zimmers TA, Gutierrez JC, Koniaris LG (2010) Loss of GDF-15 abolishes sulindac chemoprevention in the ApcMin/+ mouse model of intestinal cancer. J Cancer Res Clin Oncol 136: 571–576. 10.1007/s00432-009-0691-4 [DOI] [PubMed] [Google Scholar]
- 39. Brown DA, Hance KW, Rogers CJ, Sansbury LB, Albert PS, et al. (2012) Serum macrophage inhibitory cytokine-1 (MIC-1/GDF15): a potential screening tool for the prevention of colon cancer? Cancer Epidemiol Biomarkers Prev 21: 337–346. 10.1158/1055-9965.EPI-11-0786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mehta RS, Song M, Bezawada N, Wu K, Garcia-Albeniz X, et al. (2014) A prospective study of macrophage inhibitory cytokine-1 (MIC-1/GDF15) and risk of colorectal cancer. J Natl Cancer Inst 106: dju016 10.1093/jnci/dju016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Greenberg NM, DeMayo F, Finegold MJ, Medina D, Tilley WD, et al. (1995) Prostate cancer in a transgenic mouse. Proc Natl Acad Sci U S A 92: 3439–3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gingrich JR, Barrios RJ, Morton RA, Boyce BF, DeMayo FJ, et al. (1996) Metastatic prostate cancer in a transgenic mouse. Cancer Res 56: 4096–4102. [PubMed] [Google Scholar]
- 43. Husaini Y, Qiu MR, Lockwood GP, Luo XW, Shang P, et al. (2012) Macrophage inhibitory cytokine-1 (MIC-1/GDF15) slows cancer development but increases metastases in TRAMP prostate cancer prone mice. PLoS ONE 7: e43833 10.1371/journal.pone.0043833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tsai VW, Macia L, Johnen H, Kuffner T, Manadhar R, et al. (2013) TGF-b superfamily cytokine MIC-1/GDF15 is a physiological appetite and body weight regulator. PLoS One 8: e55174 10.1371/journal.pone.0055174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. de Jager SC, Bermudez B, Bot I, Koenen RR, Bot M, et al. (2011) Growth differentiation factor 15 deficiency protects against atherosclerosis by attenuating CCR2-mediated macrophage chemotaxis. J Exp Med 208: 217–225. 10.1084/jem.20100370 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All relevant data are within the paper.