

Background: Enhancement of L-type calcium channel trafficking by β-subunits (CaVβ) remains unclear.
Results: CaVβ associates directly with actin filaments. In cardiomyocytes, overexpression of CaVβ2 stimulates L-type currents without altering their voltage dependence of activation. Actin cytoskeleton disruption inhibited channel up-regulation.
Conclusion: CaVβ anchors the channel to actin filaments for plasma membrane delivery.
Significance: Novel insights into L-type channel anterograde trafficking are presented.
Keywords: Actin, Calcium Channel, Cardiac Muscle, Fluorescence Resonance Energy Transfer (FRET), Trafficking
Abstract
Expression of the β-subunit (CaVβ) is required for normal function of cardiac L-type calcium channels, and its up-regulation is associated with heart failure. CaVβ binds to the α1 pore-forming subunit of L-type channels and augments calcium current density by facilitating channel opening and increasing the number of channels in the plasma membrane, by a poorly understood mechanism. Actin, a key component of the intracellular trafficking machinery, interacts with Src homology 3 domains in different proteins. Although CaVβ encompasses a highly conserved Src homology 3 domain, association with actin has not yet been explored. Here, using co-sedimentation assays and FRET experiments, we uncover a direct interaction between CaVβ and actin filaments. Consistently, single-molecule localization analysis reveals streaklike structures composed by CaVβ2 that distribute over several micrometers along actin filaments in HL-1 cardiomyocytes. Overexpression of CaVβ2-N3 in HL-1 cells induces an increase in L-type current without altering voltage-dependent activation, thus reflecting an increased number of channels in the plasma membrane. CaVβ mediated L-type up-regulation, and CaVβ-actin association is prevented by disruption of the actin cytoskeleton with cytochalasin D. Our study reveals for the first time an interacting partner of CaVβ that is directly involved in vesicular trafficking. We propose a model in which CaVβ promotes anterograde trafficking of the L-type channels by anchoring them to actin filaments in their itinerary to the plasma membrane.
Introduction
L-type calcium channels (LCC)4 initiate myofilament contraction in cardiac cells (1). Normal channel function requires the association of the α1 pore-forming subunit (CaVα1) with the regulatory β-subunit (CaVβ) (2, 3). Cardiomyocytes also express T-type calcium channels, which appear more prominent at the embryonic stages (4, 5). They differ from LCCs in their voltage sensitivity and quaternary structure. Whereas CaVα1 of LCC associates with high affinity to CaVβ through a highly conserved motif named α1-interaction domain (6), T-type calcium channels lack this site. CaVβ is a membrane-associated guanylate kinase-like protein encompassing an Src homology 3 (SH3) domain and a guanylate kinase (GK) domain that are highly conserved among the four isoforms (CaVβ1 to CaVβ4) (7–9). All subtypes are expressed in cardiac tissue, but CaVβ2 is the most prominent in the mouse and human embryonic heart, as well as adult heart (10, 11). The relevance of this subunit for normal cardiac function and pathological conditions is highlighted by the fact that mice lacking CaVβ2 die during embryogenesis from heart malformations (12) and that its expression is altered in some myocardial dysfunctions (13–15). Nevertheless, inactivation of its gene at adult ages has a moderate effect, suggesting the existence of compensatory mechanisms (3, 16).
The association of CaVβ2 with CaV1.2α1, the main LCC in heart (4) increases calcium current density by two different mechanisms: it facilitates the voltage-dependent opening of the channel (15, 17, 18), and it augments the number of channels in the plasma membrane (19). The molecular mechanism by which CaVβ controls the density of channels is the most controversial and least understood (20). Several often conflicting hypotheses have been set forth to explain the molecular mechanism by which CaVβ regulates CaVα1 surface expression. It has been proposed that binding of CaVβ occludes an endoplasmic reticulum retention signal located close to the α1-interaction domain site within the intracellular loop joining repeats I and II of CaVα1 (21). Other studies suggested that the I-II loop contains an endoplasmic reticulum export signal that is suppressed by retention signals within the other cytoplasmic loops (22). Additionally, binding of CaVβ may also increase the density of the channels at the cell surface by reducing CaVα1 degradation via the endoplasmic-reticulum associated protein complex (23). Despite the mechanism postulated, it appears that association of CaVβ at early stages of the channel biogenesis is a requirement for CaVα1 targeting to the plasma membrane. It still remains elusive whether CaVβ dissociates from the channel complex during their way to the plasma membrane. To arrive to the cell surface from their place of synthesis, ion channels are incorporated into vesicles that move along microtubules and actin filaments toward the plasma membrane (24).
The fact that several SH3-containing proteins interact with actin prompted us to study the functional relationship between the CaVβ and actin. Our results reveal a direct molecular interaction that leads to an increase of L-type calcium currents in the mouse cardiomyocyte HL-1 cell line (25) that depends on an intact actin cytoskeleton. We propose that association between CaVβ and actin filaments facilitates the forward trafficking of the channel complex along actin filaments. Our results provide a novel framework for understanding the mechanism by which the CaVβ regulates the anterograde trafficking of L-type calcium channels and offer new insights into the molecular basis underlying CaVβ altered expression in the diseased heart.
EXPERIMENTAL PROCEDURES
cDNA Constructs
The coding region of the rat CaVβ2-N3 (3) (accession number Q8VGC3-2) was either fused to mRFP (CaVβ2-N3-mRFP) and subcloned into the lentiviral expression vector p156RRL or fused to CFP and subcloned in pcDNA3.1 (CaVβ2-N3-CFP (26)). pEYFP-actin vector (Clontech) was used to express YFP-actin. For protein expression, the coding regions of SH3-GK (residues 24–422 from CaVβ2-N3) and fusion proteins SH3-GST and GST-GK were subcloned in pRSET (Invitrogen) and pGEX (GE Healthcare) vectors, respectively.
HL-1 Cell Culture
The murine atrial cardiomyocyte cell line, HL-1, was kindly provided by Dr. W. C. Claycomb (Louisiana State University Health Sciences Center). Cell culture was carried on according to the specifications for the cell line (25). The medium contained 10% fetal bovine serum, 0.1 mm norepinephrine, 2 mm l-glutamine, and 100 units/ml penicillin/streptomycin in Claycomb medium. Exchange of media was done every 24–48 h. For downstream applications such as virus infection, immunolabeling, imaging, and patch clamp recordings cells were split onto fibronectin precoated dishes or coverslips after showing the macroscopic beating phenotype (25).
Viral Production and Transduction
Lentiviral production and transduction were performed as described (27). Helper plasmids were kindly provided by Dr. Thomas Südhof (Howard Hughes Medical Institute, Stanford University). Briefly, the lentiviral expression vectors and the three helper plasmids (pRSV-REV, pMDLg/pRRE, and vesicular stomatitis virus G protein-expressing plasmid) were co-transfected into HEK293FT cells using calcium phosphate transfection method. The HEK293FT cell culture medium containing viruses was collected 48 h after transfection, concentrated by ultracentrifugation, and stored at −80 °C. Lentiviral particles were added directly to the medium of cultured HL-1 cells maintained in 3.5-cm dishes (150–200 μl of viral suspension per dish).
Immunofluorescence
HL-1 cells were seeded onto 8-well plates (ibidi) and maintained at 37 °C for 24 h. All the steps during the labeling procedure were done at room temperature. The cells were washed with PBS and fixed with 4% paraformaldehyde in PBS for 10 min. After several washes, the cells were permeabilized by incubation in PBS supplemented with 0.5% Triton X-100 for 10 min and blocked for 45 min with 5% normal goat serum (Sigma) dissolved in PBS. For immunostaining of endogenous CaVβ2, the cells were incubated for 1 h with anti-CaVβ2 antibody (Abcam, S8b-1, 1:250) diluted in blocking solution. Following several washes with PBS supplemented with 0.1% Tween 20, the cells were incubated for 1 h with anti-mouse antibody conjugated to Alexa Fluor 647 (Invitrogen, A21237, 1:5000) diluted in blocking buffer. After additional washes, actin filaments were stained using 100 nm phalloidin-ATTO 488 (Sigma) for 45 min. Following several washes with 0.1% Tween 20, PBS, the cells were fixed again with paraformaldehyde 4% for 5 min and stored at 4 °C until the next day for single molecule localization super-resolution microscopy analysis.
Fluorescence Lifetime Imaging Microscopy (FLIM)
HEK293 cells expressing either CaVβ2-N3-CFP alone or together with YFP-actin were visualized using an upright fluorescence microscope (A1 MP; Nikon Instruments Europe, Amsterdam, The Netherlands) and observed through a 25× water immersion objective (NA = 1.1; Nikon) at room temperature. Fluorescence was excited with 100-fs light pulses (λexc = 880 nm) by two-photon excitation. Excitation light pulses were generated at a frequency of 80 MHz by a mode-locked Titan-Sapphire laser (MaiTai DeepSee; output power 2.3 W at 880 nm; Newport (Spectra Physics, Irvine, CA). The laser light was directed through the lens onto the HEK293 cells with reduced power (∼2–10 mW) and scanned over the sample. Fluorescence was recorded by a GaAsP hybrid photodetector (HPM-100-40; Becker & Hickl, Berlin, Germany) using appropriate filters for detecting the CFP fluorescence well separated from the YFP fluorescence (short pass filter, 500 nm; λobs < 500 nm; Omega Optical, Brattleboro, VT). For the recording of YFP-actin alone before and after treatment with cytochalasin D (see Fig. 6A), the short pass filter was removed. The use of the short pass filter allowed recording virtually only the donor emission while rejecting the emission from the YFP.
FIGURE 6.

Disruption of the actin cytoskeleton by cytochalasin D inhibits CaVβ2-actin interaction and the increase of ICa,L induced by this subunit in HL-1 cells. A, representative images of HEK293 cells expressing only YFP-actin acquired using two-photon excitation microscopy before and after incubation in 100 μm cytochalasin D for 1 h. Scale bars, 10 μm. B, representative images of HEK293 cells co-expressing CaVβ2-N3-CFP and YFP-actin acquired using two-photon excitation microscopy after incubation in 100 μm cytochalasin D for 1 h. Scale bars, 10 μm. C, representative plots of the decay of CaVβ2-N3-CFP fluorescence intensity following incubation with cytochalasin D from cells expressing this construct alone or together with YFP-actin. D, histogram of the donor fluorescence lifetime (τCFP) for cells expressing CaVβ2-N3-CFP alone or together with YFP-actin after cytochalasin D treatment. E, representative current traces from HL-1 cells showing L-type calcium currents (ICa,L) recorded with the indicated protocol pulse from a holding potential of −40 mV (HP −40) and average current density-to-voltage (I-V) curves. Native HL-1 cells (−β2) or cells transduced with CaVβ2-N3-mRFP (+β2) are shown after incubation with 100 μm cytochalasin D. F, fraction of activated channels versus voltage following incubation with cytochalasin D as calculated from B is shown for cells overexpressing CaVβ2-N3 (black circles). For comparison, the fit to a Boltzmann function obtained from nontransduced, noncytochalasin treated cells (dotted line) is shown. G, graph of the ICa,L peak amplitudes calculated from the I-V plots shown in E. Black bars show values from nontreated cells (from Fig. 5C), whereas white bars show values from cells incubated with cytochalasin D. The difference after treatment with cytochalasin is significant (p ≤ 0.01) only for cells overexpressing CaVβ2. CytD, cytochalasin D.
Fluorescence lifetime imaging was performed using electronics for time-correlated single photon counting (Simple-Tau 152; Becker & Hickl) and acquisition software (SPCM 9.55; Becker & Hickl) as described before (28, 29). Lifetime images were analyzed using SPCImage 4.8 (Becker & Hickl) by fitting a bi-exponential model equation to the fluorescence decay in every pixel of the image. The bi-exponential function described best the fluorescence intensity decay for all experiments including the different combinations of the donor/acceptor pair shown in Figs. 2 and 6. The fit quality parameter χ2 improved by 15% when using a bi-exponential fit function instead of a mono-exponential one (data not shown). The addition of a third component resulted in no significant improvement of χ2 (less than 2%). The program uses an iterative reconvolution of the exponential function with an instrument response function and a least square algorithm for finding parameters for a satisfactory fit. To include all possible populations and conformations of the CFP donor (those that do and those that eventually do not do FRET), the fluorescence lifetime displayed in FLIM images or lifetime histograms corresponds to the average fluorescence lifetime calculated as the amplitude weighted mean value using the data from the bi-exponential fit. For cytochalasin treatment, HEK293 cells were incubated for 1 h with 10 μm cytochalasin D (Sigma) dissolved in DMSO. The final DMSO concentration did not exceed 0.1% (v/v).
FIGURE 2.

Fluorescence lifetime imaging of CaVβ2-N3 and actin. A, representative images of cells expressing CaVβ2-N3-CFP (donor) alone or together with YFP-actin (acceptor) acquired using two-photon excitation microscopy. The intensity and lifetime values for both combinations are shown. Scale bars, 10 μm. B, representative plots of the decay of CaVβ2-N3-CFP fluorescence intensity in the presence (red) or absence (black) of YFP-actin. Representative plots for control conditions are shown in the right panel as indicated. For comparison we included the fit of the decay of CaVβ2-N3-CFP fluorescence in the presence of YFP-actin (red dashed line). C, histogram of the donor fluorescence lifetime (τCFP) in the presence (red) or absence (black) of YFP-actin. The histograms for control conditions are shown on the right. The shift in τCFP toward shorter values demonstrates the association between CaVβ2-N3 and actin.
Single-molecule Localization Microscopy (SMLM)
All measurements were done on our custom-built wide field and TIRF fluorescence microscope based on an Olympus IX-71 inverted microscope body. It uses an AOTF (AOTF nC-VIS-TN 1001; AA Opto-Electronic) to control the throughput of the two continuous wave laser sources that we used, i.e. an argon ion laser (488 nm; Coherent Innova 70C) and a 642-nm diode laser (Oxxius LBX-642-130 CIR-PP). All measurements were done with an Olympus ApoN 60× oil TIRF objective (NA 1.49). TIRF was achieved by repositioning the laser beam on the back of the objective using a motorized mirror. Excitation and emission light were separated via a multiband dichroic mirror (F73-866, BS R405/488/561/633; AHF Analysentechnik) in combination with a multiple band pass filter (F72-866, 466/523/600/677; AHF Analysentechnik). Images were recorded with an EMCCD camera (Andor iXon DU897E-C00-#BV) cooled to −75 °C using a resolution of 512 × 512 pixels. The image from the microscope was additionally magnified via an achromatic lens (focal point, 50 mm; AC254-050-A-ML; Thorlabs). By adjusting the lens and camera position (motorized), the pixel size can be adjusted between 65 and 130 nm/pixel.
For the measurement of the HL-1 cells, the sample chamber (8-well plate; ibidi) was completely filled with oxygen scavenger containing imaging buffer at room temperature. The buffer was composed of 20 μl of oxygen scavenger solution (1 mg/ml glucose oxidase, 22 μg/ml catalase, 4 mm Tris(2-carboxyethyl)phosphine hydrochloride, 25 mm KCl, 20 mm Tris-HCl, pH 7.5, 50% (v/v) glycerol) added to the imaging buffer (50 mg/ml glucose, 5% (v/v) glycerol, 90 mm β-mercaptoethylamine in PBS). Two color single-molecule localization microscopy measurements were performed by first measuring anti-body stained CaVβ2 (excitation, 642 nm), followed by measuring phalloidin ATTO 488-stained F-actin (excitation, 488 nm). For both, 8000 images were recorded using an exposure time of 20 ms and a pixel size of 70 nm/pixel.
For the measurement of the HeLa cells, they were transfected with Cavβ-N3-YFP and seeded onto 8-well plates (ibidi) and maintained at 37 °C for 24 h. All the steps during the labeling procedure were done at room temperature unless otherwise noted. Cells were washed with cytoskeleton buffer containing: 60 mm PIPES free base, 27 mm HEPES, 10 mm EGTA, 4 mm MgSO4, pH 7.0, preheated to 37 °C, and then fixed with 4% paraformaldehyde in cytoskeleton buffer for 15 min. After several washes with PBS supplemented with 50 mm glycine, the cells were permeabilized with 0.2% Triton-X and stained for 15 min with 50 nm Alexa Fluor® 647 phalloidin (Invitrogen, A22287) in PBS. After several washing steps with 0.1% Tween 20, PBS followed by a wash with PBS alone, the samples were measured. The sample chamber was completely filled with an alternate oxygen scavenger containing imaging buffer at room temperature. This buffer was composed of 100 mm NaPO4, 50 mm NaCl, 5 mm KCl, 2 mm Tris(2-carboxyethyl)phosphine hydrochloride, 50 mm β-mercaptoethylamine, 10% (w/v) glucose, 0.5 mg/ml glucose oxidase, 40 μg/ml catalase and had a pH of 7.0. Two color single-molecule localization microscopy measurements were performed by first measuring F-actin stained with Alexa Fluor® 647 phalloidin (excitation, 642 nm), followed by measuring Cavβ2a-YFP (excitation, 488 nm). For both, 4000 images were recorded using an exposure time of 50 ms and a pixel size of 80 nm/pixel. Localization information for SMLM was performed using the RapidSTORM (30) and SNSMIL software. SNSMIL (Shot Noise-based Single Molecule Identification and Localization) was developed by Y. Tang and L. Dai in the labs of Prof. J. Li (National Center of Nanoscience and Technology, Beijing, China) and Dr. T. Gensch.
Protein Purification
GST alone and fusion proteins SH3-GST and GST-GK and the N-terminal hexahistidine SH3-GK were expressed in bacteria and purified as previously described (31, 32). Briefly, the proteins were expressed in Escherichia coli (BL-21) by 2 h of induction with 1 mm isopropyl β-d-thiogalactopyranoside. After lysis, the soluble fraction was loaded onto a metal (Ni2+) affinity column (SH3-GK) or a gluthathione-based column (GST, SH3-GST, and GST-GK) attached to an AKTA-FPLC system (GE Healthcare). The eluates were collected, pooled, and loaded onto a 26/60 Superdex 200 column (GE Healthcare). After size exclusion chromatography, the eluates were collected, concentrated by ultracentrifugation, and stored in aliquots at −80 °C until use except for the GST-GK protein that was used fresh.
F-actin Co-sedimentation Assay
The assay was performed as described in the actin-binding protein biochem kit (Cytoskeleton, Inc.). Briefly, rabbit muscle actin was incubated with either SH3-GK, SH3-GST, GST-GK or GST alone in actin polymerization buffer for 30 min. After centrifugation (150,000 × g for 1.5 h), the pellet fraction was diluted in the same volume as the supernatant. The fractions were resolved by reducing SDS-PAGE, and the proteins were stained with Coomassie Blue.
Electrophysiology
Calcium currents were recorded using whole cell patch clamp 24–72 h after splitting with a HEKA EPC 10 amplifier using patch master software as described (33). The extracellular solution contained 157 mm tetraethylammonium Cl, 5 mm CaCl2, 0.5 mm MgCl2, and 10 mm HEPES, pH 7.4. The pipette solution contained 125 mm CsCl, 20 mm tetraethylammonium Cl, 3.6 mm PCr-Na2, 10 mm EGTA, 5 mm Mg-ATP, 0.2 mm Na-GTP, and 10 mm HEPES, pH 7.35 (34). Borosilicate pipettes with resistances of 1–3 MΩ were pulled on a Sutter P-97 puller (Harvard Apparatus) and fire-polished using a Narishige MF-900 microforge. Ca2+ currents were measured during 200-ms test pulses ranging from −70 to +70 mV in 10-mV increments using two different protocol pulses: (i) from holding potential of −90 mV (HP −90) and (ii) from a holding potential of −40 mV (HP −40). The total calcium current (ICa,L+T) was recorded with HP −90, whereas the L-type currents (ICa,L) were recorded using HP −40 where most of the T-type current component (ICa,T) is inactivated. ICa,T is obtained then by subtracting from the current trace recorded at HP −90 the one recorded with HP −40 at a given voltage step (35, 36). Because the overexpression of CaVβ2-N3 might cause a shift in the voltage dependence of ICa,L (18, 31), we compared the electrophysiological dissection of ICa,L and ICa,T to a pharmacological separation of the calcium currents using the L-type calcium channel blocker amlodipine. The relative contribution of L-type calcium channels in cells overexpressing CaVβ2 estimated by subtracting the remaining current after amlodipine application from the total calcium current was 0.32. ± 0.02 (n = 3), which was similar to 0.44 ± 0.05 (n = 15; p = 0.276) as estimated by a comparison of the peak current amplitudes using HP −40.
Currents were leak-subtracted using a P/4 protocol. For cytochalasin treatment, HL-1 cells were incubated for 1 h with 10 μm cytochalasin D (Sigma) dissolved in DMSO. The final DMSO did not exceed 0.1% (v/v). Measurements were done at room temperature (22 °C).
Data Analysis
L-type calcium peak current densities were compared using Student's t test in SigmaPlot 11.0 (Systat Software Inc.). The voltages of half-maximal activation were obtained by fitting a plot of the current-voltage relation for each cell according to Equation 1,
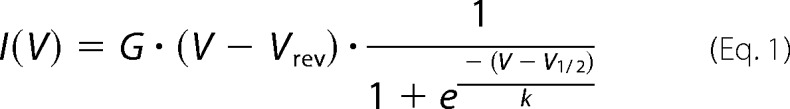 |
where I is the macroscopic current, G is the maximum conductance, V is the voltage, Vrev is the reversal potential, V½ is the half-maximal activation, and k the slope. Plots of the fraction of activated channels were calculated by dividing peak current amplitudes by the normalized maximum conductance (Ipeak/(V − Vrev)) and were fitted with the Boltzmann function according to Equation 2,
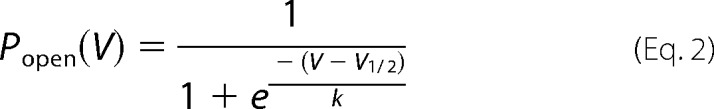 |
with Popen being the open probability at the desired voltage. A two-tailed Student's t test was used to compare the samples. The values are expressed as means ± S.E.
RESULTS
CaVβ Associates with Actin Filaments in Vitro via the SH3 and GK Domains
We first studied the ability of CaVβ2 to interact with actin filaments in vitro using a co-sedimentation assay (Fig. 1). The assay consists of dissolving actin in a polymerization buffer to promote the formation of the filamentous form (F-actin). After ultracentrifugation, F-actin is found in the pellet, whereas only a minor amount of monomeric actin could be seen in the supernatant (Fig. 1A). Proteins that associate with F-actin like α-actinin are then recovered together in the pellet fraction, whereas noninteracting proteins are found in the supernatant (Fig. 1A). The gene for CaVβ2 encodes different splice variants that are identical except for a short segment at the N terminus (Fig. 1B) (3, 11). Because no full-length CaVβ2 was found to be stable in the actin-polymerization buffer, we constructed a derivative encompassing the highly conserved SH3 and GK domains that are identical among all CaVβ2 splice variants (Fig. 1B, SH3-GK). SH3-GK alone was found exclusively in the supernatant, but it pelleted in the presence of F-actin without perturbing the apparent equilibrium between monomeric and F-actin (Fig. 1C). This result demonstrates that CaVβ2 associates preferentially with F-actin and suggests that actin association is a common property of the members of the CaVβ family that share a common SH3/GK domain structure (7–9).
FIGURE 1.

CaVβ associates with actin filaments in vitro. A, F-actin co-sedimentation assay. Actin is dissolved in a polymerization buffer that promotes its filament formation and then is either incubated or not with the test proteins. BSA and α-actinin were used as negative and positive controls, respectively. After centrifugation, the supernatant (S) and pellet (P) fractions are separated and resolved by SDS-PAGE. Although BSA is always found in the supernatant, the actin-binding protein α-actinin is found in the supernatant in the absence of actin but pelleted after incubation with F-actin. B, schematic of CaVβ2 domain structure and protein constructs used in the co-sedimentation assays. The variable N-terminal segment among the CaVβ2 splice variants is shown in gray. The SH3-GK construct used here is identical for all CaVβ2 variants. C–E, F-actin co-sedimentation assay for the CaVβ2-derived constructs indicated on the right margin. F, GST is not found in the pellet fraction. The numbers denote the molecular mass of standards in kDa. Each assay was repeated three times.
Next we assessed the contribution to actin binding of the SH3 and GK domains in isolation. To circumvent the problem of reduced protein stability observed for recombinant GK domains (37), we replaced each domain separately by GST to generate GST-GK and SH3-GST (Fig. 1B). Although the SH3-GST construct pelleted only with F-actin, a small fraction of GST-GK was found in the pellet in the absence of actin, indicating that replacement of the SH3 domain by GST still reduced GK stability (Fig. 1, D and E). Nevertheless, the amount of GST-GK found in the pellet increased after being incubated with F-actin. GST alone was excluded from the pellet in all conditions (Fig. 1F). Together, these results indicate that both SH3 and GK contribute to actin binding and highlight the idea that binding to F-actin is a property shared by all CaVβs.
CaVβ Associates with Actin in Vivo
Next, we investigated the existence of CaVβ2·actin complexes in vivo using FLIM (38). The fluorescence lifetime of a given fluorophore (donor) is affected by FRET if it is in close physical proximity—below a few nanometers—to another fluorophore (acceptor) with an excitation spectrum overlapping the donor emission spectrum. When FRET occurs, the lifetime of the donor decreases because of its quenching.
To perform FLIM-FRET experiments, CaVβ2-N3 and actin were fused to CFP and YFP acting as donor and acceptor, respectively. Fig. 2A shows fluorescence intensity (left panel) and color-coded lifetime (right panel) images obtained by two-photon excitation microscopy of HEK cells expressing CaVβ2-N3-CFP alone or together with YFP-actin. The apparent membrane staining given by CaVβ2-N3 is due to its ability of being palmitoylated and to associate with the plasma membrane even in the absence of the α1-subunit (39). Co-expression of CaVβ2-N3-CFP with YFP-actin resulted in a decrease of the mean lifetime constant of the donor fluorescence by ∼24% (Fig. 2, B and C). Mean fluorescence decay time constants of CaVβ2-N3-CFP alone were similar to those obtained for CFP alone. Control experiments using either CaVβ2-N3-CFP/YFP or CFP/YFP-actin also resulted in time constants that were indistinguishable from the values for cells expressing CFP alone (Fig. 2, B and C, and Table 1). These results demonstrate a specific co-localization between CaVβ2-N3 and actin in the cell interior on a nanometer scale.
TABLE 1.
Parameters describing the distribution of the CFP fluorescence lifetimes in cells expressing various combinations of donor and acceptor constructs
The values were obtained from FLIM-FRET recordings of cells expressing the donor/acceptor combinations indicated. Mean, maximum, and minimum values were calculated over all values for each condition.
| Mean | S.E. | Maximum | Minimum | n | |
|---|---|---|---|---|---|
| CaVβ2-N3-CFP | 2.66 | 0.01 | 2.77 | 2.51 | 79 |
| CaVβ2-N3-CFP + cytochalasin D | 2.68 | 0.01 | 2.85 | 2.46 | 132 |
| CaVβ2-N3-CFP + YFP-actin | 2.05 | 0.03 | 2.71 | 1.57 | 86 |
| CaVβ2-N3-CFP + YFP-actin + cytochalasin D | 2.60 | 0.02 | 2.80 | 1.84 | 117 |
| CaVβ2-N3-CFP + YFP | 2.69 | 0.01 | 2.46 | 2.84 | 89 |
| CFP | 2.69 | 0.01 | 2.93 | 2.49 | 118 |
| CFP + YFP-actin | 2.68 | 0.01 | 2.92 | 2.43 | 102 |
SMLM Demonstrates That the CaVβ2-N3-YFP Spatially Overlaps with Actin Filaments
We used SMLM to resolve the spatial organization of CaVβ and actin inside of HeLa cells (30, 40). SMLM is based on the detection of single photoswitchable molecules from a vast population by activating a small subset and determining their locations. A final image is reconstituted by summing all the encountered molecular positions (localizations). To detect CaVβ, we fused it to YFP and expressed the fusion CaVβ2-N3-YFP protein in HeLa cells. The actin filaments were stained with the high affinity probe phalloidin coupled to Alexa Fluor® 647. SMLM images from these cells reveal an extensive degree of spatial overlap between CaVβ and F-actin (Fig. 3A). The CaVβ·F-actin structures extend to different lengths and are broadly distributed throughout the cell from nearby the plasma membrane to the nuclear periphery (Fig. 3, B and C, left panels). A projection of the localizations within a cross-section along these structures in two different intracellular regions shows similar widths for the distribution of CaVβ2 and F-actin that fully overlap (Fig. 3, B and C, right panels).
FIGURE 3.

SMLM image of CaVβ2-N3-YFP expressed in HeLa cells. A, wide field SMLM image of HeLa cells with the focus around the midsection of the cell. F-actin was labeled with Alexa Fluor® 647 phalloidin (green). CaVβ2-N3 was visualized via a fused YFP (magenta). The overlay of F-actin and CaVβ2-N3-YFP is shown in white. B and C, 4 × 4-μm subsection indicated by the yellow squares in A. The right panels show the projections of the single molecule localizations delineated by the yellow lines indicated by arrows in B and C. Green lines represent localizations from F-actin, and magenta lines represent localizations from CaVβ2-N3-YFP. The scale bar for A represents 4 μm. The scale bars for B and C represent 1 μm. Norm., normalized.
SMLM Demonstrates That the CaVβ2 Subunit Is Distributed along Actin Filaments in HL-1 Cardiomyocytes
To reconstitute the native arrangement of CaVβ2 and actin, we used the HL-1 cardiac cell line. Endogenous CaVβ2 was immunodetected using an antibody against a sequence conserved among all CaVβ2 splice variants. In control experiments, no signal was detected when the primary anti-CaVβ2 antibody was omitted (Fig. 4A). The specificity of the antibody was tested in HEK293 cells that do not express endogenous CaVβ2. Although a diffuse background signal was observed in an image of HL-1 cells, the SMLM analysis resulted in only few localizations that did not show any spatial correlation relative to phalloidin labeled F-actin (Fig. 4B).
FIGURE 4.

CaVβ2 is distributed along actin filaments in HL-1 cells. HL-1 cells were labeled for F-actin using phalloidin coupled to ATTO 488 and for CaVβ2 subunits using anti-CaVβ2 detected with anti-mouse IgG coupled to Alexa Fluor® 647. A, control images of HL-1 cells labeled for F-actin and the secondary antibody coupled to Alexa Fluor® 647 in the absence of the anti-CaVβ2 antibody. The left panel shows detection of F-actin, and the right panel shows detection of Alexa Fluor® 647. No signal was obtained using the secondary antibody alone. Scale bar, 5 μm. B, control images of HEK293 cells lacking endogenous CaVβ2 subunits detected for F-actin (left panel) and CaVβ2 (middle panel). Although unspecific background signal can be seen in conventional TIRF images (scale bar, 5 μm), SMLM analysis of the region delimited with a white box (right panel) shows few localizations for CaVβ2 that do not correlate with F-actin (scale bar, 0.5 μm). C, TIRF and SMLM images of a HL-1 cell labeled for F-actin and CaVβ2 showing detection of F-actin alone (left top panel) or CaVβ2 (left bottom panel). Scale bar, 5 μm. The SMLM analysis of the section enclosed by the white rectangle is shown in the right panel. Each localized molecule is represented by a 30-nm diameter disk, and co-localization is shown in white. Scale bar, 0.5 μm. A projection of the single molecule localizations delineated by the green line indicated by an arrow demonstrates the co-localization (lower right panel). Each projected point in the graph was obtained by summing the intensities of all localizations 10 nm to the left and right of the projection point. Norm., normalized.
In contrast, TIRF images from HL-1 cells subjected to the same labeling procedure for CaVβ2 and F-actin showed a large number of streaklike structures formed by CaVβ2 that closely follow the actin filaments (Fig. 4C). SMLM images demonstrate that CaVβ2 is distributed along actin filaments over distances covering several micrometers including furcations and branches. The full width at half-maximum obtained from a fit of a Gaussian function to the cross-sectional distribution of the localizations yielded widths for the CaVβ2-based structures ranging from 80 to 250 nm correlating nicely with the width of actin bundles (41) (Fig. 4C) as observed in HeLa cells. The CaVβ2-actin streaklike structures recall cargo transport along actin tracks (24), giving rise to the idea that the CaVβ may serve to anchor the channel complex to F-actin during its intracellular trafficking itinerary.
Overexpression of CaVβ2-N3 in HL-1 Cardiomyocytes Up-regulates L-type Calcium Currents
Whole cell patch clamp recordings from native HL-1 cells held at −90 mV (HP −90) show a sum of two types of calcium currents, a large fast inactivating current, attributed to the T-type channel mediated current (ICa,T), and a relatively small slowly inactivating current, corresponding to L-type currents (ICa,L) (34) (Fig. 5A). T-type calcium channels differ from LCC in their voltage sensitivity and do not associate with CaVβ (2). Dissection of ICa,L from ICa,T was done by using a holding potential of −40 mV (HP −40), where T-type calcium channels are already inactivated, and thus their contribution to the recorded calcium current is negligible (35, 36) (Fig. 5A, right panel).
FIGURE 5.

Overexpression of CaVβ2-N3 increases L-type calcium currents in HL-1 cells. A, pulse protocol and representative current traces from HL-1 cells showing the superposition of T- and L-type calcium currents (ICa,L+T) recorded from a holding potential of −90 mV (HP −90) are shown in the left panel. LCC current traces (ICa,L) recorded from a holding potential of −40 mV (HP −40) are shown in the right panel. Native HL-1 cells (−β2) or cells transduced with CaVβ2-N3-mRFP (+β2) were recorded using both protocol pulses. B, average current density to voltage (I-V) curves for ICa,L from cells as shown in A. C, comparison of the peak amplitudes of ICa,L obtained from the I-V plots shown in B. D, fraction of activated channels versus voltage as calculated from B for cells overexpressing CaVβ2-N3 (black circles). For comparison the fit to a Boltzmann function obtained from nontransduced cells (dotted line) is shown. The number of cells is given in parentheses. **, p ≤ 0.01.
Lentiviral transduction of CaVβ2-N3 fused to mRFP significantly increased ICa,L at all voltages as shown in the current density versus voltage (I-V) plot (Fig. 5B). Transduction of mRFP alone did not alter ICa,L (data not shown). Overexpression of CaVβ2-N3 induced a 2-fold increase in the peak ICa,L current obtained from the I-V curves (Fig. 5, B and C).
ICa,L is given by the product of the probability that the channel is open, the single channel current amplitude, and the number of channels in the plasma membrane. Therefore, the increased current density may result from alterations in the electrical activity and/or the surface expression of the channel.
CaVβ does not alter the single-channel current amplitude of L-type calcium channels (42, 43), and changes in the open probability of CaV1.2 induced by CaVβ2 are caused by a shift in the voltage dependence of the activation toward more hyperpolarized voltages (18, 44). We found that the increase in the L-type current density upon overexpression of CaVβ2-N3 occurs without alterations in the voltage-dependent activation of the channel. The relative fractions of activated channels at each potential are virtually identical between native HL-1 cells and transduced with CaVβ2-N3 (Fig. 5D). The voltage at which the half-maximal activation (V½) takes place in native cells is statistically equal for cells expressing CaVβ2-N3 (Table 2). We conclude that CaVβ2-N3 up-regulates ICa,L by increasing the number of channels. This is consistent with previous studies showing that overexpression of different CaVβ, including the CaVβ2-N3 variant, in cultured rat cardiomyocytes augments the number of functional channels in the cell surface (18).
TABLE 2.
Parameters describing the best fit of a modified Boltzmann equation to the I-V plot for the indicated conditions
The values were obtained from a statistical analysis of values obtained from a fit of a modified Boltzmann function as stated under “Experimental Procedures.” There is no statistical difference in either V½ or the slope (k). Cyt D, cytochalasin D.
| Cyt D | n | Mean V½ | Mean k | p value | |
|---|---|---|---|---|---|
| mV | mV | ||||
| Native | − | 12 | −0.2 ± 2.1 | 10.6 ± 0.9 | |
| + | 6 | 1.9 ± 3.1 | 15.0 ± 2.5 | 0.57 | |
| CaVβ2-N3 | − | 14 | −2.1 ± 2.0 | 12.3 ± 0.9 | 0.52 |
| + | 11 | −2.1 ± 1.9 | 9.6 ± 0.4 | 0.52 |
The Actin Filament Disrupter Cytochalasin D Inhibits the Association of CaVβ2-N3 with Actin and the Up-regulation of L-type Calcium Currents
To test our hypothesis, we next studied L-type-mediated currents in native HL-1 cells and cells overexpressing β2a after pharmacological disruption of the actin cytoskeleton using cytochalasin D. We repeated the FLIM-FRET experiments shown in Fig. 2 but following incubation of the cells with 100 μm cytochalasin D for 1 h. The actin cytoskeleton was severely disturbed upon this treatment as seen by the spotted actin pattern in cells expressing YFP-actin alone (Fig. 6A). The decay of donor fluorescence intensity in the presence of actin is no longer accelerated after cytochalasin D treatment (Fig. 6, B and C). Mean donor fluorescence time constants obtained for the CaVβ2-N3-CFP/YFP-actin pair were also similar to those obtained from nontreated cells expressing CaVβ2-N3-CFP only (Fig. 6D and Table 1). This absence of FRET indicates a loss of the CaVβ·F-actin interaction following the disruption of the actin cytoskeleton.
Whole cell recordings from native HL-1 cells or cells transduced with CaVβ2-N3-mRFP following the same treatment with cytochalasin D showed only small currents through L-type calcium channels (Fig. 6E). There was a slight tendency toward a reduction of the already low ICa,L amplitude when compared with native HL-1 cells that were not treated with cytochalasin D. More pronounced, the increase in ICa,L induced by overexpression of CaVβ2-N3 was significantly reduced by disruption of the actin cytoskeleton without alterations in the voltage dependence of activation (Fig. 6, F and G, and Table 2). These results support our initial hypothesis of a role of the CaVβ·F-actin complex in sustaining the forward trafficking of LCC in HL-1 cells and emphasize the notion that the availability of CaVβ is the rate-limiting step for the channel surface expression in native cells (45).
DISCUSSION
Here we demonstrate a direct association between CaVβ and actin filaments that increases L-type currents in HL-1 cells. Given that the single channel conductance is not altered by CaVβ (42, 43), an increase in the current density might arise from an increase in the channel's open probability and/or in the number of channels expressed at the plasma membrane (17, 19, 46). Increases in gating currents that reflect a rise in the number of channels have been observed upon overexpression of CaVβ2-N3 and other CaVβ isoforms in cardiomyocytes (18).
All experimental data published so far show that CaVβ-induced increase of the open probability of CaV channels is associated with a shift in the voltage dependence of the activation threshold toward more hyperpolarized voltages (44). The absence of such changes in the voltage dependence of activation of the channels (Figs. 5 and 6) indicates that open probabilities are unchanged upon CaVβ overexpression and/or cytochalasin treatment. Although we cannot formally exclude the occurrence of changes in absolute open probabilities that are not reflected in the voltage-dependent activation of the channel, it appears very unlikely that CaVβ affects voltage-independent gating transitions.
Forward trafficking of vesicular cargo, particularly in the final transport step to the plasma membrane, is sustained by actin filaments (24, 47). Within this framework, the findings presented here suggest a model of actin-dependent anterograde LCC trafficking mediated by CaVβ. Newly synthesized channels are transported from the trans-Golgi network to the vicinity of the plasma membrane along microtubules (48). In the vicinity of the cell surface where the microtubule network is mostly replaced by actin cytoskeleton (24), CaVβ would promote the recruitment of channel complexes via its direct association with F-actin that serve as tracks for transporting the channels to their final destination in the plasma membrane. It has been proposed that in cardiomyocytes vesicles containing CaV1.2 L-type channels are transported along microtubules to BIN1 scaffolding protein for their specific delivery (48). It will be of interest to investigate whether the interaction between CaVβ and actin contributes to this directed targeting of L-type channel complexes.
The TIRF-based super-resolution microscopy reveals streaklike structures formed by CaVβ along actin filaments over several micrometers. Because fluorescent excitation and, subsequently, emission is restricted to the plasma membrane and the regions immediately nearby, we cannot precisely determine the spatial arrangement of protein complexes deeper into the cell. However, our data from FRET-FLIM predict a formation of CaVβ·F-actin complexes also in the interior of the cell. Recently it has been suggested that actin filaments can also sustain long range vesicle transport (49). Whether or not actin filaments support long range transport of vesicles containing CaVα1 is not inferable from our experiments.
The direction of the changes in calcium channel activity caused by overexpression of the CaVβ2-N3 in HL-1 cells resembles those alterations during the transition of the myocyte toward the adult phenotype. This suggests a role of the CaVβ in cardiomyocyte maturation that is consistent with the observation that CaVβ2 expression levels increase from the embryonic to early postnatal stages (11).
Despite the fact that the minute L-type component recorded from nontransduced cells limited the detection of changes in the current amplitude, treatment with cytochalasin D by itself did not significantly alter ICa,L in nontransduced cells. This confirms previous suggestions that the expression of the β-subunit is the rate-limiting step for L-type current expression in heart (18, 46). Although unlikely, we cannot discard the existence of a set of functionally active LCC not associated with actin that gives rise to ICa,L in nontransduced cells. Increases in LCC and CaVβ expression underlie several myocardial dysfunctions (13, 15). Thus, the up-regulation of LCC by CaVβ·actin complexes in cells overexpressing CaVβ2-N3 mimics a state observed in certain pathological conditions. Our study provides novel insights into the puzzling mechanism by which CaVβ regulates the number of channels in the plasma membrane and new theoretical considerations for understanding CaVβ altered expression in diseased heart.
Acknowledgments
We thank Christoph Fahlke and Alan Neely for insightful discussions and Petra Kilian and Joachim Schmitz for valuable assistance with HL-1 cells.
This work was supported by Deutsche Forschungsgemeinschaft Grant Hi 800/3-1 (to P. H.).
- LCC
- voltage-gated L-type calcium channel
- SH3
- Src homology 3
- mRFP
- monomeric red fluorescent protein
- CFP
- cyan fluorescent protein
- FLIM
- fluorescence lifetime imaging microscopy
- SMLM
- single-molecule localization microscopy
- GK
- guanylate kinase.
REFERENCES
- 1. Bodi I., Mikala G., Koch S. E., Akhter S. A., Schwartz A. (2005) The L-type calcium channel in the heart: the beat goes on. J. Clin. Invest. 115, 3306–3317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Catterall W. A. (2011) Voltage-gated calcium channels. Cold Spring Harb. Perspect. Biol. 3, a003947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hofmann F., Flockerzi V., Kahl S., Wegener J. W. (2014) L-type CaV1.2 calcium channels: from in vitro findings to in vivo function. Physiol. Rev. 94, 303–326 [DOI] [PubMed] [Google Scholar]
- 4. Larsen J. K., Mitchell J. W., Best P. M. (2002) Quantitative analysis of the expression and distribution of calcium channel alpha 1 subunit mRNA in the atria and ventricles of the rat heart. J. Mol. Cell Cardiol. 34, 519–532 [DOI] [PubMed] [Google Scholar]
- 5. Cribbs L. L., Martin B. L., Schroder E. A., Keller B. B., Delisle B. P., Satin J. (2001) Identification of the t-type calcium channel (CaV3.1d) in developing mouse heart. Circ. Res. 88, 403–407 [DOI] [PubMed] [Google Scholar]
- 6. Pragnell M., De Waard M., Mori Y., Tanabe T., Snutch T. P., Campbell K. P. (1994) Calcium channel β-subunit binds to a conserved motif in the I-II cytoplasmic linker of the α1-subunit. Nature 368, 67–70 [DOI] [PubMed] [Google Scholar]
- 7. Opatowsky Y., Chen C. C., Campbell K. P., Hirsch J. A. (2004) Structural analysis of the voltage-dependent calcium channel beta subunit functional core and its complex with the alpha 1 interaction domain. Neuron 42, 387–399 [DOI] [PubMed] [Google Scholar]
- 8. Van Petegem F., Clark K. A., Chatelain F. C., Minor D. L., Jr. (2004) Structure of a complex between a voltage-gated calcium channel β-subunit and an α-subunit domain. Nature 429, 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen Y. H., Li M. H., Zhang Y., He L. L., Yamada Y., Fitzmaurice A., Shen Y., Zhang H., Tong L., Yang J. (2004) Structural basis of the α1-β subunit interaction of voltage-gated Ca2+ channels. Nature 429, 675–680 [DOI] [PubMed] [Google Scholar]
- 10. Foell J. D., Balijepalli R. C., Delisle B. P., Yunker A. M., Robia S. L., Walker J. W., McEnery M. W., January C. T., Kamp T. J. (2004) Molecular heterogeneity of calcium channel β-subunits in canine and human heart: evidence for differential subcellular localization. Physiol. Genomics 17, 183–200 [DOI] [PubMed] [Google Scholar]
- 11. Link S., Meissner M., Held B., Beck A., Weissgerber P., Freichel M., Flockerzi V. (2009) Diversity and developmental expression of L-type calcium channel β2 proteins and their influence on calcium current in murine heart. J. Biol. Chem. 284, 30129–30137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Weissgerber P., Held B., Bloch W., Kaestner L., Chien K. R., Fleischmann B. K., Lipp P., Flockerzi V., Freichel M. (2006) Reduced cardiac L-type Ca2+ current in CaVβ2−/− embryos impairs cardiac development and contraction with secondary defects in vascular maturation. Circ. Res. 99, 749–757 [DOI] [PubMed] [Google Scholar]
- 13. Haase H., Kresse A., Hohaus A., Schulte H. D., Maier M., Osterziel K. J., Lange P. E., Morano I. (1996) Expression of calcium channel subunits in the normal and diseased human myocardium. J. Mol. Med. 74, 99–104 [DOI] [PubMed] [Google Scholar]
- 14. Hullin R., Asmus F., Ludwig A., Hersel J., Boekstegers P. (1999) Subunit expression of the cardiac L-type calcium channel is differentially regulated in diastolic heart failure of the cardiac allograft. Circulation 100, 155–163 [DOI] [PubMed] [Google Scholar]
- 15. Hullin R., Matthes J., von Vietinghoff S., Bodi I., Rubio M., D'Souza K., Friedrich Khan I., Rottländer D., Hoppe U. C., Mohacsi P., Schmitteckert E., Gilsbach R., Bünemann M., Hein L., Schwartz A., Herzig S. (2007) Increased expression of the auxiliary β2-subunit of ventricular L-type Ca2+ channels leads to single-channel activity characteristic of heart failure. PLoS One 2, e292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Meissner M., Weissgerber P., Londoño J. E., Prenen J., Link S., Ruppenthal S., Molkentin J. D., Lipp P., Nilius B., Freichel M., Flockerzi V. (2011) Moderate calcium channel dysfunction in adult mice with inducible cardiomyocyte-specific excision of the cacnb2 gene. J. Biol. Chem. 286, 15875–15882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hullin R., Khan I. F., Wirtz S., Mohacsi P., Varadi G., Schwartz A., Herzig S. (2003) Cardiac L-type calcium channel β-subunits expressed in human heart have differential effects on single channel characteristics. J. Biol. Chem. 278, 21623–21630 [DOI] [PubMed] [Google Scholar]
- 18. Colecraft H. M., Alseikhan B., Takahashi S. X., Chaudhuri D., Mittman S., Yegnasubramanian V., Alvania R. S., Johns D. C., Marbán E., Yue D. T. (2002) Novel functional properties of Ca2+ channel β subunits revealed by their expression in adult rat heart cells. J. Physiol 541, 435–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bourdin B., Marger F., Wall-Lacelle S., Schneider T., Klein H., Sauvé R., Parent L. (2010) Molecular determinants of the CaVβ-induced plasma membrane targeting of the CaV1.2 channel. J. Biol. Chem. 285, 22853–22863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Simms B. A., Zamponi G. W. (2012) Trafficking and stability of voltage-gated calcium channels. Cell. Mol. Life Sci. 69, 843–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bichet D., Cornet V., Geib S., Carlier E., Volsen S., Hoshi T., Mori Y., De Waard M. (2000) The I-II loop of the Ca2+ channel α1 subunit contains an endoplasmic reticulum retention signal antagonized by the β subunit. Neuron 25, 177–190 [DOI] [PubMed] [Google Scholar]
- 22. Fang K., Colecraft H. M. (2011) Mechanism of auxiliary β-subunit-mediated membrane targeting of L-type (CaV1.2) channels. J. Physiol. 589, 4437–4455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Altier C., Garcia-Caballero A., Simms B., You H., Chen L., Walcher J., Tedford H. W., Hermosilla T., Zamponi G. W. (2011) The Cavβ subunit prevents RFP2-mediated ubiquitination and proteasomal degradation of L-type channels. Nat. Neurosci. 14, 173–180 [DOI] [PubMed] [Google Scholar]
- 24. Ross J. L., Ali M. Y., Warshaw D. M. (2008) Cargo transport: molecular motors navigate a complex cytoskeleton. Curr. Opin. Cell Biol. 20, 41–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Claycomb W. C., Lanson N. A., Jr., Stallworth B. S., Egeland D. B., Delcarpio J. B., Bahinski A., Izzo N. J., Jr. (1998) HL-1 cells: a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc. Natl. Acad. Sci. U.S.A. 95, 2979–2984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Miranda-Laferte E., Gonzalez-Gutierrez G., Schmidt S., Zeug A., Ponimaskin E. G., Neely A., Hidalgo P. (2011) Homodimerization of the Src homology 3 domain of the calcium channel β-subunit drives dynamin-dependent endocytosis. J. Biol. Chem. 286, 22203–22210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Barde I., Salmon P., Trono D. (2010) Production and titration of lentiviral vectors. Curr. Protoc. Neurosci. 53, 4.21:4.21.1–4.21.23 [DOI] [PubMed] [Google Scholar]
- 28. Gilbert D., Franjic-Würtz C., Funk K., Gensch T., Frings S., Möhrlen F. (2007) Differential maturation of chloride homeostasis in primary afferent neurons of the somatosensory system. Int. J. Dev. Neurosci. 25, 479–489 [DOI] [PubMed] [Google Scholar]
- 29. Kaneko H., Putzier I., Frings S., Kaupp U. B., Gensch T. (2004) Chloride accumulation in mammalian olfactory sensory neurons. J. Neurosci. 24, 7931–7938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wolter S., Schüttpelz M., Tscherepanow M., van de Linde M., Heilemann M., Sauer M. (2010) Real-time computation of subdiffraction-resolution fluorescence images. J. Microsc. 237, 12–22 [DOI] [PubMed] [Google Scholar]
- 31. Hidalgo P., Gonzalez-Gutierrez G., Garcia-Olivares J., Neely A. (2006) The α1-β subunit interaction that modulates calcium channel activity is reversible and requires a competent α-interaction domain. J. Biol. Chem. 281, 24104–24110 [DOI] [PubMed] [Google Scholar]
- 32. Gonzalez-Gutierrez G., Miranda-Laferte E., Neely A., Hidalgo P. (2007) The Src homology 3 domain of the β-subunit of voltage-gated calcium channels promotes endocytosis via dynamin interaction. J. Biol. Chem. 282, 2156–2162 [DOI] [PubMed] [Google Scholar]
- 33. Scholl U. I., Goh G., Stölting G., de Oliveira R. C., Choi M., Overton J. D., Fonseca A. L., Korah R., Starker L. F., Kunstman J. W., Prasad M. L., Hartung E. A., Mauras N., Benson M. R., Brady T., Shapiro J. R., Loring E., Nelson-Williams C., Libutti S. K., Mane S., Hellman P., Westin G., Åkerström G., Björklund P., Carling T., Fahlke C., Hidalgo P., Lifton R. P. (2013) Somatic and germline CACNA1D calcium channel mutations in aldosterone-producing adenomas and primary aldosteronism. Nat. Genet. 45, 1050–1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xia M., Salata J. J., Figueroa D. J., Lawlor A. M., Liang H. A., Liu Y., Connolly T. M. (2004) Functional expression of L- and T-type Ca2+ channels in murine HL-1 cells. J. Mol. Cell Cardiol. 36, 111–119 [DOI] [PubMed] [Google Scholar]
- 35. Hagiwara N., Irisawa H., Kameyama M. (1988) Contribution of two types of calcium currents to the pacemaker potentials of rabbit sino-atrial node cells. J. Physiol. 395, 233–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bean B. P. (1985) Two kinds of calcium channels in canine atrial cells: differences in kinetics, selectivity, and pharmacology. J. Gen. Physiol. 86, 1–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gonzalez-Gutierrez G., Miranda-Laferte E., Nothmann D., Schmidt S., Neely A., Hidalgo P. (2008) The guanylate kinase domain of the β-subunit of voltage-gated calcium channels suffices to modulate gating. Proc. Natl. Acad. Sci. U.S.A. 105, 14198–14203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sun Y., Day R. N., Periasamy A. (2011) Investigating protein-protein interactions in living cells using fluorescence lifetime imaging microscopy. Nat. Protoc. 6, 1324–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chien A. J., Gao T., Perez-Reyes E., Hosey M. M. (1998) Membrane targeting of L-type calcium channels. Role of palmitoylation in the subcellular localization of the β2a subunit. J. Biol. Chem. 273, 23590–23597 [DOI] [PubMed] [Google Scholar]
- 40. Patterson G., Davidson M., Manley S., Lippincott-Schwartz J. (2010) Superresolution imaging using single-molecule localization. Annu. Rev. Phys. Chem. 61, 345–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Heilemann M., van de Linde S., Schüttpelz M., Kasper R., Seefeldt B., Mukherjee A., Tinnefeld P., Sauer M. (2008) Subdiffraction-resolution fluorescence imaging with conventional fluorescent probes. Angew. Chem. Int. Ed. Engl. 47, 6172–6176 [DOI] [PubMed] [Google Scholar]
- 42. Dzhura I., Neely A. (2003) Differential modulation of cardiac Ca2+ channel gating by β-subunits. Biophys. J. 85, 274–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Luvisetto S., Fellin T., Spagnolo M., Hivert B., Brust P. F., Harpold M. M., Stauderman K. A., Williams M. E., Pietrobon D. (2004) Modal gating of human CaV2.1 (P/Q-type) calcium channels: I. the slow and the fast gating modes and their modulation by β subunits. J. Gen. Physiol 124, 445–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Neely A., Wei X., Olcese R., Birnbaumer L., Stefani E. (1993) Potentiation by the beta subunit of the ratio of the ionic current to the charge movement in the cardiac calcium channel. Science 262, 575–578 [DOI] [PubMed] [Google Scholar]
- 45. Obermair G. J., Tuluc P., Flucher B. E. (2008) Auxiliary Ca2+ channel subunits: lessons learned from muscle. Curr. Opin. Pharmacol. 8, 311–318 [DOI] [PubMed] [Google Scholar]
- 46. Wei S. K., Colecraft H. M., DeMaria C. D., Peterson B. Z., Zhang R., Kohout T. A., Rogers T. B., Yue D. T. (2000) Ca2+ channel modulation by recombinant auxiliary β subunits expressed in young adult heart cells. Circ. Res. 86, 175–184 [DOI] [PubMed] [Google Scholar]
- 47. Lanzetti L. (2007) Actin in membrane trafficking. Curr. Opin. Cell Biol. 19, 453–458 [DOI] [PubMed] [Google Scholar]
- 48. Hong T. T., Smyth J. W., Gao D., Chu K. Y., Vogan J. M., Fong T. S., Jensen B. C., Colecraft H. M., Shaw R. M. (2010) BIN1 localizes the L-type calcium channel to cardiac T-tubules. PLoS Biol. 8, e1000312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Schuh M. (2011) An actin-dependent mechanism for long-range vesicle transport. Nat. Cell Biol. 13, 1431–1436 [DOI] [PMC free article] [PubMed] [Google Scholar]