

Background: d-Glucuronyl C5-epimerase is a crucial modifying enzyme in the heparan sulfate biosynthesis pathway.
Results: We determined the Glce apo-structure and the structure of Glce complexed with a heparin hexasaccharide.
Conclusion: Glce forms a dimer with the active sites located at both C-terminal α-helical domains.
Significance: This work advances understanding of the key epimerization step in heparan sulfate biosynthesis.
Keywords: Crystal Structure, Enzyme Mechanism, Glycosaminoglycan, Heparan Sulfate, Heparin, d-Glucuronyl C5 Epimerase, Glce, Epimerization
Abstract
Heparan sulfate (HS) is a glycosaminoglycan present on the cell surface and in the extracellular matrix, which interacts with diverse signal molecules and is essential for many physiological processes including embryonic development, cell growth, inflammation, and blood coagulation. d-Glucuronyl C5-epimerase (Glce) is a crucial enzyme in HS synthesis, converting d-glucuronic acid to l-iduronic acid to increase HS flexibility. This modification of HS is important for protein ligand recognition. We have determined the crystal structures of Glce in apo-form (unliganded) and in complex with heparin hexasaccharide (product of Glce following O-sulfation), both in a stable dimer conformation. A Glce dimer contains two catalytic sites, each at a positively charged cleft in C-terminal α-helical domains binding one negatively charged hexasaccharide. Based on the structural and mutagenesis studies, three tyrosine residues, Tyr468, Tyr528, and Tyr546, in the active site were found to be crucial for the enzymatic activity. The complex structure also reveals the mechanism of product inhibition (i.e. 2-O- and 6-O-sulfation of HS keeps the C5 carbon of l-iduronic acid away from the active-site tyrosine residues). Our structural and functional data advance understanding of the key modification in HS biosynthesis.
Introduction
Heparan sulfate proteoglycans are macromolecules widely expressed on the cell surface and in the extracellular matrix of all animal tissues. They consist of a core protein with covalently attached heparan sulfate (HS)4 chains (1). HS, a negatively charged polysaccharide, binds to a variety of proteins, including growth factors, chemokines, and interleukins (2). As such, it can regulate a wide variety of biological activities, including embryonic development, cell growth, inflammatory response, blood coagulation, tumor metastasis, and virus infection (2, 3). Heparin, a special form of highly sulfated HS derived from porcine intestine, has been widely used as an injectable anticoagulant since the 1930s.
A heparan sulfate chain is synthesized in vivo by several steps: tetrasaccharide linkage formation, chain elongation, N-deacetylation/N-sulfation, epimerization, and O-sulfation (Fig. 1A). d-Glucuronyl C5-epimerase (Glce) is a key enzyme in HS/heparin synthesis, converting d-glucuronic acid (GlcA) to l-iduronic acid (IdoA) by C5 epimerization at the polymer level (4) (Fig. 1B). The epimerization reaction is reversible in vitro but irreversible in vivo (5). The epimerization step increases the flexibility of the HS chain and is essential for the function of HS in ligand recognition and cell signaling (6). Targeted disruption of the Glce gene (Hsepi) in mice resulted in neonatal death and defects of kidney, lung, and skeletal development (7), which strongly indicates the crucial role of Glce in animal development. It has also been reported that Glce suppresses the proliferation of human breast cancer cells (8) and small-cell lung cancer cells (9), which suggests that Glce may be a tumor suppressor. Quite recently, we showed that Glce depletion promoted PC12 cell neuritogenesis induced by nerve growth factor (10). In addition, the modification of HS by Glce is critical for controlling the binding or activity of molecules that guide early lymphoid tissue morphogenesis and B lymphocyte maturation and differentiation (11, 12).
FIGURE 1.

Glce-catalyzed epimerization is a key step in the heparan sulfate and heparin biosynthesis pathway. A, the heparan sulfate and heparin biosynthesis pathway in vivo. Biosynthesis of HS begins with the transfer of a xylose to specific serine residues within the protein core, followed by the formation of a Xyl-Gal-Gal-GlcA tetrasaccharide. After the addition of the first N-acetylglucosamine (GlcNAc) to the tetrasaccharide by the EXTL3 enzyme, the chain is elongated by the stepwise addition of GlcA and N-acetylglucosamine residues catalyzed by EXT1 and EXT2 enzymes. Next, the polysaccharide undergoes a series of modification reactions, including N-deacetylation/N-sulfation of N-acetylglucosamine residues, epimerization of GlcA units to IdoA, and finally O-sulfation at various positions. B, chemical reaction of C5 carboxyl group epimerization catalyzed by Glce. GlcNS, α-d-N-sulfoglucosamine. Carbon atoms for α-d-N-sulfoglucosamine, GlcA, and IdoA are labeled.
Today, diverse low molecular weight heparins show great potential as anticoagulants and in cancer, antiviral, and anti-inflammatory therapies (13). However, the separation and purification of heparin oligosaccharides from the animal tissues are difficult and inefficient. Thus, a promising approach is to synthesize specific heparin oligosaccharides using rational chemoenzymatic design (14, 15). To unveil the catalytic mechanism and substrate recognition pattern of HS-modifying enzymes is of critical importance to achieve such a goal. Here, we report the first crystal structures of Glce, in apo-form (unliganded) and in complex with heparin hexasaccharide, which is the product of Glce following 2-O-sulfation on iduronic acid and 6-O-sulfation on N-sulfoglucosamine. Based on the structural and functional data, we identify the active site of Glce and propose the mechanism of product inhibition of Glce in vivo. This work advances our understanding of the HS synthesis reaction and will aid in the development of therapeutic heparin mimics as well as the inhibitors of Glce.
EXPERIMENTAL PROCEDURES
Cloning, Expression, and Purification of Glce
Zebrafish Glce (Arg50–Asn585) was expressed as a His6-SUMO fusion protein from the pSUMO expression vector (LifeSensors, Inc.). The fusion protein contains a His6 tag (MKKGHHHHHHG) at the N terminus and a ULP1 protease cleavage site between SUMO and Glce. BL21 (DE3) cells transfected with the expression plasmid were grown in LB broth at 25 °C to an A600 of about 1.0 and were induced with 0.1 mm isopropyl 1-thio-β-d-galactopyranoside for 16 h. Cells were harvested, resuspended in 50 ml of buffer A (20 mm Tris, pH 8.0, 200 mm NaCl, and 10% glycerol) per 2 liters of cells, and lysed using an APV2000 cell homogenizer (SPX Corp.). The lysate was centrifuged, and the supernatant was loaded on a 30-ml nickel High Performance column (GE Healthcare). The column was washed with 250 ml of 90% buffer A plus 10% buffer B (20 mm Tris, pH 8.0, 200 mm NaCl, 500 mm imidazole, and 10% glycerol) and was eluted by 100 ml of 50% buffer A + 50% buffer B. The eluted His6-SUMO-Glce protein was dialyzed against buffer A and cleaved overnight with ULP1 at a protease/protein ratio of 1:1000 at 4 °C. The cleaved His6-SUMO tag was removed by a pass through a 5-ml nickel High Performance column, and the flow-through protein was further purified through a HiLoad 26/60 Superdex 200 gel filtration column in 20 mm Tris, pH 8.0, 200 mm ammonium acetate, 1 mm dithiothreitol, and 1 mm EDTA.
To prepare SeMet-substituted Glce protein, the pSUMO-Glce expression plasmid was transfected into B834 methionine auxotroph cells. A single colony was inoculated into 2 liters of LB medium plus 50 μg/ml ampicillin and 1% glucose and shaken at 30 °C overnight. The 2 liters of cells were spun down and resuspended in 600 ml of filtered H2O. Separately, the following solutions (defined below) were combined step by step and mixed well to make 2 liters of SeMet-substituted expression medium: 40 ml of solution E, 40 ml of solution D, 20 ml of solution C, 100 ml of solution B, 300 ml of solution A, and 1500 ml of autoclaved H2O. Then 100 ml of cells were transferred into each 2 liters of media. The cells were grown at 22–25 °C to an A600 of 1.0–1.2. Protein expression was induced with 0.1 mm isopropyl 1-thio-β-d-galactopyranoside at 16 °C overnight. The cells were harvested the next morning and resuspended in buffer A. The protein was purified using the same protocol as for the native protein.
Solution A contained 12 g of hydrolyzed herring sperm DNA (pH 7.0), 120 g of glucose, 250 mg of vitamin B1, 12 g of NH4Cl·6H2O, and 50 μg/ml ampicillin dissolved in 1.8 liters of H2O and filtered. Solution B contained 0.5 g of each of 19 amino acids (no methionine) plus 0.5 g of selenomethionine, 0.6 g MgCl2, and 0.6 g CaCl2 in 600 ml of H2O. Solution C contained 5 g of EDTA, 0.5 g of FeCl3, 0.1 g of ZnSO4, 0.05 g of CuCl2·2H2O, 0.05 g of CoCl2, and 0.05 g of (NH4)6Mo7O24·4H2O in 1 liter, adjusted to pH 7.0, and was autoclaved and then stored at 4 °C. Solution D contained 50× M9 minimal medium part A plus 300 g of Na2HPO4 in 1 liter of H2O. Solution E contained 50× M9 minimal medium part B plus 150 g of KH2PO4 in 1 liter of H2O.
Heparin Oligosaccharide Purification
Enoxaparin sodium (Hebei Changshan Biochemical Pharmaceutical Co. Ltd.), a low molecular weight heparin, was separated on a Bio-Gel P10 (Bio-Rad) column (2.6 × 90 cm). Samples were eluted with 0.2 m NaCl at a flow rate of 0.33 ml/min. We collected several components with different degrees of polymerization. Following lyophilization, the fractions were desalted using a Bio Gel P2 (Bio-Rad) column (2.6 × 90 cm). The sized fractions were chromatographed using strong anion exchange HPLC fitted with a semipreparative strong anion exchange column (Waters Corp., Spherisorb strong anion exchange column, 10 × 250 mm). A gradient elution (0 min, 40% B; 50 min, 65% B) was performed at a flow rate of 2 ml/min for 65 min using solvent A (H2O, pH 3.5) and solvent B (2 m NaCl, pH 3.5). The major subfractions were desalted on a Bio Gel P2 column and lyophilized.
Crystallization
Purified zebrafish Glce protein (Arg50–Asn585) in the same buffer as was used to elute the HiLoad 26/60 Superdex 200 gel filtration column was concentrated to 15–20 mg/ml prior to crystallization trials. Rod-shaped crystals about 400 μm in length were obtained using 1 μl of the purified protein and 1 μl of well solution. Native apo-Glce crystals were grown using a well solution of 16% (w/v) PEG 3350, 0.1 m sodium citrate tribasic dihydrate, pH 5.6, and 2% (v/v) Tacsimate, pH 5.0. SeMet-substituted Glce crystals were grown using a well solution of 16% (w/v) PEG 3350, 0.06 m citric acid, and 0.04 m Bistris propane, pH 4.1. To prepare crystals of the complex, Glce at a concentration of 5.0 mg/ml was incubated with heparin oligosaccharide at a molar ratio of 1:5 at 4 °C overnight, and crystals were grown at 20 °C in hanging drops by mixing 1 μl of Glce-oligosaccharide complex and 1 μl of well solution consisting of 16% (w/v) PEG 3350, 0.1 m sodium citrate tribasic dehydrate, pH 5.6, and 2% (v/v) Tacsimate, pH 5.0. Crystals of 150–200 μm in length appeared within 3 days.
Data Collection and Structure Determination
All crystals were transferred into well solution plus 22% (v/v) ethylene glycol as a cryoprotectant before flash-freezing in liquid nitrogen. Data collections for native and SeMet crystals were performed at the Life Sciences Collaborative Access Team beamlines of the Advanced Photon Source synchrotron. A native data set was collected to 1.9 Å at sector 21-ID-G. To solve the phase problem, one data set from a SeMet-substituted Glce crystal was collected at sector 21-ID-D at a wavelength of 0.9762 Å (peak wavelength) using an inverse beam strategy to accurately measure the selenium anomalous signal. The data were processed using XDS (16) and scaled using Scala of the Collaborative Computational Project 4 (CCP4) suite (17).
The native and SeMet Glce crystals belong to the P41212 space group with one molecule per asymmetric unit. Initial phases were established using the SHELX program (18) by the SAD phasing method (Table 1). A total of 12 selenium sites were identified by SHELXD with a CCall/CCweak score of 47.2/30.6, and subsequent phasing was performed using SHELXE with a Contrast score of 0.82 for the correct hand solution. CCall is the correlation coefficient between Ecalc and Eobs for all data, whereas CCweak is the correlation coefficient for 30% of reflections that were not used during the dual-space refinement. An initial model including 464 residues was built automatically using the Phenix autobuild program with R/Rfree of 0.30/0.33. The initial model obtained from the SeMet data was used to refine against the native data set at 1.9 Å. The model was further improved by several cycles of manual building using Coot (19) and refinements with the refmac program of CCP4 (20) to an R factor of 0.21 and an Rfree factor of 0.23. The final model has excellent density for most residues except for the N-terminal flexible loop (Pro50–Val71) and a small internal loop (Asp212–Ser215).
TABLE 1.
X-ray data collection and refinement statistics for Glce structures
| Nativea | SeMeta | Heparin complexa | |
|---|---|---|---|
| Data collection | |||
| Space group | P41212 | P41212 | P21212 |
| Cell dimensions | |||
| a, b, c (Å) | 66.4, 66.4, 337.6 | 67.2, 67.2, 337.8 | 152.6, 201.0, 46.6 |
| α, β, γ (degrees) | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 |
| Wavelength | 0.9786 | 0.9762 (peak) | 0.9792 |
| Resolution (Å) | 50–1.9 | 50–2.1 | 50–2.2 |
| Rsym or Rmerge | 0.101 (1.04)b | 0.114 (1.02) | 0.292 (1.37) |
| I/σI | 14.8 (3.2) | 19.1 (3.5) | 9.2 (2.0) |
| Completeness (%) | 100 (99.9) | 100 (99.9) | 100.0 (100.0) |
| Redundancy | 14.0 (14.7) | 28.0 (29.3) | 7.3 (7.4) |
| Refinement | |||
| Resolution (Å) | 50–1.9 | 50–2.2 | |
| No. of reflections | 57,920 | 70,265 | |
| Rwork/Rfree | 0.210/0.244 | 0.194/0.222 | |
| No. of atoms | |||
| Protein | 4086 | 8252 | |
| Ligand/ion | 13 | 210 | |
| Water | 684 | 813 | |
| B-factors | |||
| Protein | 34.3 | 28.6 | |
| Ligand/ion | 56.3 | 41.4 | |
| Water | 46.9 | 34.5 | |
| RMSD | |||
| Bond lengths (Å) | 0.0063 | 0.008 | |
| Bond angles (degrees) | 1.12 | 1.26 | |
a The x-ray diffraction data were obtained from a single crystal.
b Values in parentheses are for the highest resolution shell.
The data set for the complex was collected at Shanghai Synchrotron Radiation Facility beamline BL17U. The data were indexed and integrated using XDS (16) and scaled using Scala of the CCP4 suite (17). The crystal belongs to the P21212 space group with two molecules per asymmetric unit. The structure was solved by molecular replacement using the CCP4 program Phaser with the native Glce structure as a search model. The electron density for the sugar chain became clear after the initial refinement, and a model of heparin hexasaccharide was built based on the electron density map. The complex structure was further improved by several cycles of manual building using Coot (19) and refinements using PHENIX (21) and the CCP4 program Refmac (20). The final structure model was refined to an R factor of 0.20 and an Rfree factor of 0.22 (Table 1). All structure figures were prepared using PyMOL (Schroedinger, LLC, New York).
Mutant Construction and Enzymatic Activity Assay
Mutants of Glce were generated by site-directed mutagenesis in the pSUMO expression vector. The resulting vectors were transformed into the BL21 (DE3) cell line, and all mutant proteins were expressed and purified similarly as the wild type Glce. Glce mutant proteins (10 ng) were mixed with tritium-labeled N-deacetylated/sulfated K5 capsular polysaccharide in a total volume of 100 μl. After incubation at 37 °C for 1 h, the tritium release was analyzed using a biphasic liquid scintillation procedure as described previously (22, 23).
Product Inhibition of Glce by Heparin
Heparin with an average molecular mass of 15 kDa was purchased from Shenzhen Hepalink Biological Technology Co., Ltd. N-Sulfated heparin, desulfated heparin, and heparin oligosaccharides were prepared in the laboratory of Prof. Jin-ping Li (University of Uppsala) as reported previously (5, 23). 10 and 100 μg of heparin, 10 and 100 μg of N-sulfated heparin, 100 μg of desulfated heparin, or 100 μg of heparin oligosaccharides were added into a 100-μl enzymatic reaction system with 10 ng of purified wild type Glce to determine the product inhibition of Glce activity. Various amounts of heparin (0, 10 pg, 100 pg, 10 ng, 100 ng, 1 μg, 10 μg, 100 μg, 1 mg, and 10 mg) and N-sulfated heparin (0, 10 ng, 100 ng, 1 μg, 10 μg, 100 μg, and 1 mg) were added into a 100-μl enzymatic reaction system with 10 ng of purified wild type Glce to obtain the inhibition curve. Enzymatic activity was determined as described above.
AlphaScreen in Vitro Binding Assay
Zebrafish Glce protein (Arg50–Asn585) was cloned into the pSUMO expression vector in fusion with a biotinylation peptide (AviTag) at the N terminus. In addition, the biotin ligase (BirA) gene with a T7 promoter was cloned downstream of zebrafish Glce cDNA. Coexpression of Glce and BirA in BL21 (DE3) in the presence of 60 μm biotin and 100 μm isopropyl 1-thio-β-d-galactopyranoside allowed in vivo biotinylation of zebrafish Glce (24), which was purified similarly as wild type Glce protein. His6-tagged MBP was cloned into pET-22b vector using NdeI and NotI restriction sites, and the protein was purified using an MBP column. His6-tagged human N-deacetylase/N-sulfotransferase 1, 2-O-sulfotransferase 1, and 6-O-sulfotransferase 1 were purchased from R&D Systems.
100 nm biotin-Glce was attached to streptavidin-coated donor beads, and 100 nm His6-tagged MBP and sulfotransferases were attached to nickel-chelated acceptor beads. The interactions were determined by a luminescence-based AlphaScreen assay (PerkinElmer Life Sciences) using a hexahistidine detection kit that our group has used extensively (24). Each data point was an average of triplicate measurements with S.E. values indicated.
RESULTS
The Dimeric Structure of Glce
Protein sequence analysis using the PSIPRED Web server predicted that Glce would contain an α-helical transmembrane region and a highly flexible loop at the N terminus. We chose to express the soluble Glce fragment (i.e. excluding the N-terminal transmembrane α-helix region) from six species (Homo sapiens, Bos taurus, Rattus norvegicus, Gallus gallus, Danio rerio, and Drosophila melanogaster) in Escherichia coli BL21 cells. The Glce protein from D. rerio (zebrafish) formed high quality crystals, which diffracted x-rays to about 1.9 Å (Table 1). The truncated zebrafish Glce (residues 50–585) shares a high sequence identity (80%) with human Glce, which suggests that the structure and function of Glce are highly conserved across species. The zebrafish Glce crystallized in space group P41212 with one molecule per asymmetric unit. Examination of the crystal packing revealed a tight dimer association through a crystallographic 2-fold symmetry (PDB code 4PW2). The overall structure of the dimer is shaped like an upside-down “W” (Fig. 2, A and B). The full-length sequence of Glce also contains a transmembrane α-helix at the N terminus, which presumably anchors the Glce dimer onto the Golgi membrane, where it performs its HS modification (Fig. 2C).
FIGURE 2.

The overall dimeric structure of Glce. A, ribbon structure of the Glce dimer, with the two monomers shown in green and cyan. Left, side view; right, view from the bottom. B, surface charge distribution on the Glce dimer. The color-coded bar indicates an electrostatic scale from −5 eV (red) to +5 eV (blue). The highly charged clefts are indicated by arrows. C, Glce dimer showing the domain organization. The N-terminal β-hairpin domain is anchored to the Golgi membrane through an N-terminal transmembrane helix (blue cylinder), which is not present in the current Glce structure. D, the domain structure of the Glce monomer. E, the dimer interface at the C-terminal α-helical domain. Carbon atoms for the two monomers are shown in green and cyan. F, the dimer interface at the N-terminal β-hairpin domain. Carbon atoms for the two monomers are shown in green and cyan. G, size column profile of zebrafish Glce protein. Glce protein was run on an 120-ml Superdex column and eluted at 72.3 ml, corresponding to the size of a dimer.
A soluble Glce monomer can be divided into three domains: an N-terminal β-hairpin domain, a β-barrel domain, and a C-terminal α-helical domain (Fig. 2D). The N-terminal β-hairpin domain, which crosses into its counterpart in the dimer, consists of two β-hairpins that are connected through a short α-helix. The C-terminal α-helical domain (Ser129–Pro200 and Thr367–Asn585) consists of two β-strands forming a β-hairpin and eight α-helixes in four pairs of anti-parallel helices. One helix from each pair faces the concave surface, forming a cleft; the other four helices face the convex surface. The α-helical domain is the most conserved region according to the sequence alignment. The β-barrel domain (His201–Thr366) consists of one α-helix and 13 β-strands arising as an insertion within the sequence of the α-helical domain (Fig. 2, C and D).
Two Glce molecules interact through the N-terminal β-hairpin and C-terminal α-helical domains to form a tight dimer, and the dimer formation buries a total surface area of 6020 Å2. At the interface, the C-terminal domains interact through hydrophobic packing interactions involving residues Met459, Val515, Phe458, Leu519, Phe533, and Thr522 (Fig. 2E). For the N-terminal domains, the interactions include both hydrophobic packing and ionic interactions, with hydrophobic interactions being dominant (Fig. 2F). The hydrophobic packing interactions involve residues Phe106, Trp101, Phe123, Met102, Phe121, Met97, Tyr96, and Tyr73, and the ionic interactions include two ionic pairs: Asp119 with Arg90 and Glu91 with Arg89. These extensive interactions observed in the crystal structure provide strong evidence that Glce is a stable dimer. In agreement with the structure, Glce protein eluted with the expected molecular mass of about 125 kDa in size exclusion column chromatography, suggesting that it is a dimer in solution (the predicted monomer mass is 60.6 kDa) (Fig. 2G).
To locate the substrate binding site, we analyzed the surface charge potential of the apo-Glce dimeric structure. The charge distribution calculation of the dimer revealed that the cleft enclosed by four α-helices in the α-helical domain has a strong positive charge potential (Fig. 2B), which is probably the binding site for the highly negatively charged HS chain.
Structure of the Glce Dimer in Complex with Heparin Hexasaccharide
To actually locate the substrate binding site and reveal the substrate recognition mechanism, we pursued a Glce complex structure. We purified heparin hexasaccharide from enoxaparin sodium and used it for complex formation and crystallization (Fig. 3A). Compared with the actual substrate of Glce, the hexasaccharide has excess 2,6-O-sulfations. The Glce-heparin complex crystallized in space group P21212 with two molecules per asymmetric unit, and the crystals diffracted x-rays to 2.2 Å resolution (PDB code 4PXQ; Table 1). The complex contains two Glce protein molecules and two heparin hexasaccharides with a stoichiometry of 2:2 (Fig. 3B); each negatively charged heparin hexasaccharide binds to the positively charged cleft within the C-terminal α-helical domain of one monomer (Fig. 3, C and E). The heparin binding site is mostly contained within the cleft but also includes residues from the other monomer, suggesting that the dimer conformation is crucial for forming the heparin binding site (Fig. 3D). This heparin binding mode is well supported by the excellent electron density of the hexasaccharide in the substrate binding site (Fig. 3F).
FIGURE 3.

The Glce dimer complexed with heparin hexasaccharide. A, the chemical structure of heparin hexasaccharide. B, Glce dimer with two heparin hexasaccharides bound. Monomers are shown in green (subunit A) and cyan (subunit B); heparin hexasaccharides (arrows) are shown as stick models. C, surface charge distribution of the Glce complex. The color-coded bar indicates an electrostatic scale from −5 eV (red) to +5 eV (blue). The negatively charged heparin hexasaccharide lies in the positively charged cleft within the α-helical domain. D, close-up view of the heparin-binding cleft with the monomers shown as green and cyan. E, close-up view of the heparin-binding cleft with charge distribution. F, close-up view of the heparin-binding cleft with heparin hexasaccharide shown as a stick model and the 2Fo − Fc map contoured at 1σ (blue mesh). G, stereo view of the detailed interactions between Glce dimer and heparin hexasaccharide. Overall structures are shown in schematic representations. Residues closely interacting with heparin are shown as stick models; charge and hydrogen bond interactions are indicated by black dotted lines. The carbon atoms are green and cyan for the two Glce monomers; heparin carbon atoms are white.
The heparin hexasaccharide residues, from the non-reducing end to the reducing end, are ΔUAP1-SGN2-IDS3-SGN4-IDS5-SGN6 (Fig. 3A). The molecule has an extended conformation and interacts with the substrate-binding cleft in Glce through extensive charge interactions and hydrogen bonds (Fig. 3G). The sugar rings of residues 1–4 are roughly perpendicular to the binding surface and so have more interactions, whereas the sugar rings of residues 5 and 6 are roughly parallel to the binding surface and have fewer interactions. The 2-O-sulfate of ΔUAP1 interacts through hydrogen bonds with Tyr149, Gln185, Tyr482, and Thr549 (Fig. 3G). The carboxyl group and the ring oxygen of ΔUAP1 form ionic interactions with Arg154. The 3-OH group and the O1 atom interact with Arg543 through hydrogen bonds. N,O6-disulfoglucosamine (SGN2) exhibits a 4C1 chair conformation. The 6-O-sulfate is inserted between two positively charged residues (Arg154 and Arg156) and has ionic interactions with the Arg154 and Arg156 side chains; it also forms a hydrogen bond with the Gln182 side chain. The N-sulfate of SGN2 is on the opposite site of the 6-O-sulfate and has fewer interactions, with only two hydrogen bonds with the backbone amides of the Asp155 and Arg156 residues.
Iduronic acid-2-O-sulfate (IDS3) exhibits a 1C4 chair conformation and lies in the center of the substrate binding site with its carboxyl group facing the residues in the substrate binding site (Fig. 3G). The ring oxygen atom of IDS3 has a charge interaction with the Arg543 side chain. The carboxyl group has an ionic interaction with Arg543 and forms a hydrogen bond with Tyr546. In addition, it also forms a water-mediated ionic interaction with Arg396 and a water-mediated hydrogen bond with Tyr468. The 2-O-sulfate of IDS3 faces the solvent and does not interact with any protein residues. Residue SGN4 has a 4C1 chair conformation. The N-sulfate has ionic interactions with Arg531 and hydrogen bonds with Asn540 (Fig. 3G). Interestingly, the N-sulfate also has one hydrogen bond with the Asn585 residue from the adjacent Glce molecule in the dimer. The 6-O-sulfate has no interactions with protein residues. IDS5 exhibits a 2S0 skew-boat conformation and has only one ionic interaction between its carboxyl group and the Arg396 side chain (Fig. 3G). Residue SGN6 adopts a 4C1 chair conformation and makes few interactions with active site residues (Fig. 3G). The N-sulfate has one hydrogen bond with the backbone amide of the Lys397 residue, and the 6-O-sulfate has one ionic interaction with the side chain of Lys397 and one hydrogen bond with the backbone amide of Glu400.
Heparin Hexasaccharide-induced Conformational Changes
Compared with the apo-structure, there was a small shift of the N-terminal β-hairpin domain and the β-barrel domain in the complex, both of which moved closer to the α-helical domain upon heparin binding (Fig. 4A). Furthermore, a flexible loop (Asp212–Ser215) at the outside of the β-barrel domain, which was disordered in the apo-structure, became ordered and had clear electron density in the complex. The most significant change involved the Gln171–Gln175 loop within the α-helical domain, which was flipped out of the substrate binding site to accommodate the heparin hexasaccharide (Fig. 4A).
FIGURE 4.

Heparin hexasaccharide binding induces conformational changes in Glce. A, a superposition of the Glce apo-structure (green) and heparin-bound structure (magenta). Only the subunit A of the complex structure is shown; heparin hexasaccharide is shown as a stick model. Small shifts in the N-terminal β-hairpin and β-barrel domains are indicated by black arrows. Two regions with major conformational changes are shown in the expanded views. B, close-up stereo view of the active site showing the conformational changes in Glce upon heparin binding. The residues with major conformational changes and the heparin hexasaccharide are shown as stick models. The conformational changes are indicated by black arrows.
Detailed structural analysis provided an explanation for the dramatic conformational change of the Gln171–Gln175 loop. Heparin binding resulted in major movements of residues Arg396, Arg154, Asp155, and Arg156 (Fig. 4B). Arg396 moved closer to the bound heparin to interact with the C5 carboxyl group of IDS5. The side chains of Arg156 and Arg154 residues moved up and down, respectively, to accommodate the bound heparin, whereas the backbone amides of Asp155 and Arg156 moved closer to interact with the N-sulfate of SGN2. The side chain movement of Arg156 displaced the side chains of Gln171 and Trp172, hence the large movement of the entire Gln171–Gln175 loop outward.
Active Site of Glce and Mutational Analysis
Because the heparin hexasaccharide binds to the cleft in α-helical domain, we hypothesize that this is the actual catalytic site of Glce. We used site-directed mutagenesis to examine the roles of specific residues in the binding cleft in substrate recognition and catalytic function. The activities of wild type Glce and mutants were determined by an 3H/1H exchange approach, which measures the release of a 3H proton from 3H-labeled substrate during the epimerization reaction, as reported previously (25). We identified eight mutants (Y149F, R156A, Y468A, Y528A, Y528F, R543A, Y546A, and Y546F) that had 10% or less activity compared with wild type (Fig. 5A), and all residues are located close to the substrate binding site (Fig. 3G). Six other mutants (R154A, R396A, Y468F, R531A, H584A, and N585A) retained partial enzymatic activity (less than 40%).
FIGURE 5.
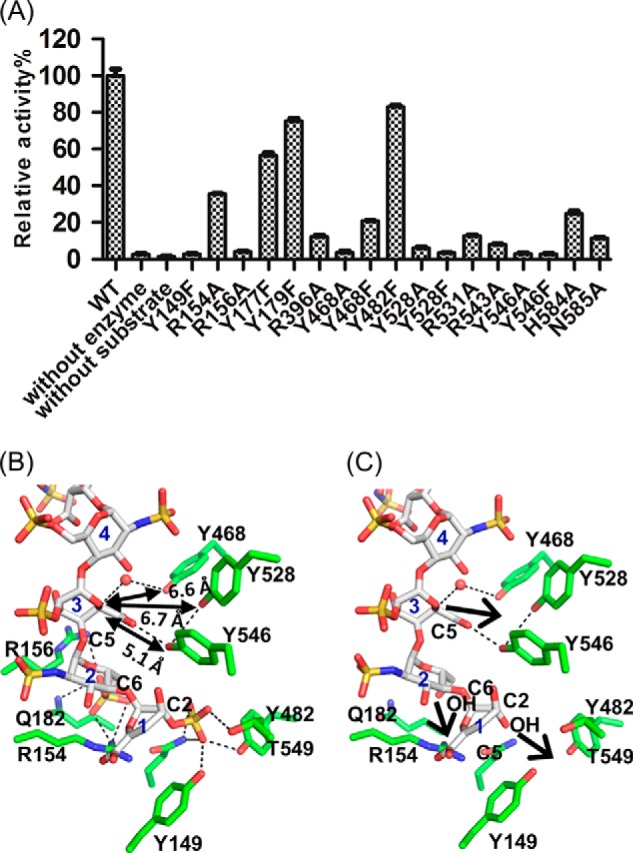
Structural and functional analyses of Glce active-site residues. A, enzymatic activity measurements of Glce wild type (WT) and mutants toward polysaccharide substrates. The activity was determined by a 3H/1H exchange method (n = 3; error bars indicate S.D.). Equal amounts of proteins were used for activity assays based on quantification using a Qubit fluorometer. B, Glce active site. The structured water molecule close to Tyr468 is shown as a sphere. Carbon atoms are white for heparin hexasaccharide and green for Glce residues. Charge and hydrogen bond interactions are shown as black dotted lines. The distances from three tyrosine residues to the C5 carbon atom are indicated. C, heparin hexasaccharide without O-sulfation may bind closer to the active-site tyrosine residues to allow catalysis of C5 epimerization reaction. In this panel, O-sulfations at C2 of residue 1 and C6 of residue 2 have been removed. The arrows indicate the likely movements of the heparin chain toward the active-site tyrosine residues because of fewer O-sulfations.
Previous studies suggested that tyrosine residues may be involved in the catalytic function of Glce and heparin lyases, which share a similar carbon anion intermediate (26, 27). According to our crystal structures and mutant analysis, Tyr468, Tyr528, and Tyr546, near the carboxyl group of IDS3 (Fig. 5B), probably participate in the catalytic reaction as bases for proton abstraction and donation. However, they are still relatively far from the C5 carbon (6.6, 6.7, and 5.1 Å, respectively) in the complex structure, which prevents catalysis. This is due to the fact that heparin hexasaccharide is not the actual substrate of Glce because heparin has excess O-sulfation. Thus, it is predicted that an actual substrate without 2-O- and 6-O-sulfation will bind closer to these tyrosine residues to allow catalysis (Fig. 5C).
The charged residues at the substrate binding site are also crucial for the enzyme's activity. Arg543 interacts with the carboxyl group and the ring oxygen atom of IDS3 and with the 3-OH group and the O1 atom of ΔUAP1 (Fig. 3G). Arg154 and Arg156 interact with the 6-O-sulfate of SGN2; Arg154 also interacts with the carboxyl group of ΔUAP1. Arg396 interacts with the carboxyl of IDS5 and forms a water-mediated interaction with the carboxyl of IDS3, and Arg531 interacts with the N-sulfate of SGN4. These charged residues all play an important role in substrate recognition. Asn585 from the adjacent monomer is also important for substrate recognition by forming a hydrogen bond with the N-sulfate of SGN4 (Fig. 3G). The extensive interactions with the SGN4 N-sulfate group support the concept of an absolute requirement for the N-sulfate group on the glucosamine residue linked to the GlcA for substrate recognition (5, 25).
Mechanism of Product Inhibition of Glce
Glce recognizes both GlcA and IdoA as substrates and catalyzes a reversible reaction in vitro. Interestingly, the Glce-catalyzed reaction is irreversible in vivo (5). After epimerization by Glce, the product undergoes further 2-O-sulfation of IdoA by 2-O-sulfotransferase and 6-O-sulfation of α-d-N-sulfoglucosamine by 6-O-sulfotransferase (Fig. 1A), resulting in products that are no longer recognized as substrates for Glce (29). Because heparin hexasaccharide can still bind to the active site of Glce (Fig. 3G), we examined whether the product after consecutive modification by Glce and O-sulfotransferases might influence the activity of Glce using a soluble in vitro reaction system. Upon incubation of the wild type Glce with 3H-labeled substrate in the presence of heparin, N-sulfated heparin, desulfated heparin, or heparin oligosaccharides in different degrees of polymerization, we found that heparin, N-sulfated heparin, and heparin oligosaccharides efficiently inhibited Glce activity, and desulfated heparin could not inhibited Glce activity (Fig. 6A). The data indicate that O-sulfation on heparin inhibits Glce activity and prevents the back-conversion of IdoA to GlcA through an end product inhibition mechanism. Heparin inhibited Glce activity with an IC50 of 225 μg/ml, whereas N-sulfated heparin had an IC50 of 10 μg/ml (Fig. 6, B and C). This indicated that a higher ratio of N-sulfate group on glucosamine had a better inhibitory effect on Glce activity, which further demonstrated that the N-sulfate group played an important role in substrate recognition by Glce.
FIGURE 6.

Mechanism of product inhibition of Glce. A, heparin, N-sulfated heparin, and heparin oligosaccharides in different degrees of polymerization (dp) could inhibit Glce activity, whereas desulfated heparin could not inhibit Glce activity. B, heparin inhibits the activity of Glce with an IC50 of 225 μg/ml (n = 4; error bars indicate S.D.). C, N-sulfated heparin inhibits the activity of Glce with an IC50 of 10 μg/ml (n = 4; error bars indicate S.D.). D, a schematic diagram for the principle of the AlphaScreen binding assay. E, interactions of Glce with N-deacetylase/N-sulfotransferase (NDST) and downstream O-sulfotransferase enzymes 2-O-sulfotransferase (2OST) and 6-O-sulfotransferase (6OST) were measured by an AlphaScreen binding assay using 100 nm biotin-tagged zebrafish Glce and 100 nm His6-tagged MBP (negative control) and human N-deacetylase/N-sulfotransferase, 2-O-sulfotransferase, or 6-O-sulfotransferase enzymes (n = 3; error bars show S.D.).
What is the structural basis of product inhibition of Glce by O-sulfated HS or heparin? In our structure, the O-sulfates on IDS3, SGN4, and IDS5 face solvent and do not interact with active-site residues (Fig. 3G). The O-sulfate of SGN6 makes few interactions: one ionic interaction with the Lys397 side chain and one hydrogen bond with the Glu400 backbone amide. However, the O-sulfates on ΔUAP1 and SGN2 face the active site and have extensive, important interactions with active-site residues. The 2-O-sulfate of ΔUAP1 interacts with the Gln185, Tyr149, Tyr482, and Thr549 side chains, and the 6-O-sulfate of SGN2 interacts with the Arg156, Gln182, and Arg154 side chains (Fig. 5B). Importantly, these interactions keep the C5 atom of IDS3 away from the critical tyrosine residues (Tyr468, Tyr528, and Tyr546), with the closest distance being 5.1 Å for Tyr546. These distances are too far for proton abstraction/donation, a structural basis for the lack of activity of Glce for the O-sulfated HS or heparin, which leads to the accumulation of IdoA residues in vivo. On the other hand, the hydroxyl groups without O-sulfation would move closer to the active site and form optimal hydrogen bonds with the active-site residues (Fig. 5C), which in turn would cause the C5 atom of IDS3 to move closer to the crucial catalytic tyrosine residues. This allows proton abstraction and readdition and a reversible conversion between GlcA and IdoA in vitro toward the substrate without O-sulfation.
In addition, we found that Glce could physically interact with 2-O-sulfotransferase and 6-O-sulfotransferase, but not N-deacetylase/N-sulfotransferase, by an AlphaScreen in vitro binding assay (Fig. 6, D and E), which further indicated that these proteins form a complex to allow coupling of 2-O-sulfation and 6-O-sulfation with C5 epimerization in HS/heparin biosynthesis.
DISCUSSION
Here, we report that zebrafish Glce has a dimeric structure. Recently, it has been reported that Glce from the marine bacterium Bermanella marisrubri is also a dimer (30). Together, these findings strongly support the concept that Glce functions as a dimer. Each Glce dimer contains two active sites at the C-terminal α-helical domains (Fig. 3B). A long HS chain may thread through the two active sites, or two such polysaccharide chains may simultaneously bind to two active sites of the Glce dimer to allow HS modifications. There are three enzymes involved in epimerization of hexuronic acid at the polymer level: Glce, C5-mannuronan epimerase, and DS-epimerase 1. Our Glce structure revealed that three tyrosine residues (Tyr468, Tyr528, and Tyr546) are crucial for enzymatic activity and are close to the C5-carboxyl group of IDS3, which probably participates in the catalytic reaction as bases for proton abstraction and donation. A Dali structural alignment search (31) revealed many polysaccharide hydrolases and lyases whose structures are distantly related to the Glce C-terminal α-helical domain (the catalytic domain in Glce), including endo-β-1,4-glucanase (PDB code 1WZZ, a superposition RMSD of 3.3 Å of 215 residues), family 8 xylanase (PDB code 1XWQ, a superposition RMSD of 3.8 Å of 229 residues), chitosanase (PDB code 1V5D, a superposition RMSD of 3.6 Å of 217 residues), and pectate lyase (PDB code 1GXM, a superposition RMSD of 4.0 Å of 211 residues) with Z-scores from 14.4 to 11.7. The C5-mannuronan epimerase has a right-handed parallel β-helix structure (32) (PDB code 2PYG), which is completely different from the Glce structure. Dali structural analysis of C5-mannuronan epimerase showed high structural similarity to β-helix polysaccharide hydrolases and lyases, and it catalyzes its reaction using a tyrosine and a histidine acting as a proton acceptor and donor, respectively (32). In a computational model of DS-epimerase 1, a tyrosine and two histidine residues were identified as potential proton acceptor/donor residues (33). These three enzymes do not share any sequence and structural homology that would suggest a common evolutionary origin, but all three share a similar catalytic mechanism of proton abstraction and readdition using different amino acids (34, 35). Also, both the lyases and the polymer-level epimerases have essentially a common mechanism of action, sharing a similar proton abstraction mechanism and an enolate anion intermediate.
It was previously reported that the three sugars at the non-reducing end in front of the epimerization site could influence the reversibility of epimerization (36). This suggests that Glce could recognize at least one more sugar residue at the non-reducing end in front of ΔUAP1, which would be adjacent to the Tyr149 residue. Tyr149 is far from the Tyr468, Tyr528, and Tyr546 residues; it forms a hydrogen bond with the 2-O-sulfate of ΔUAP1. It should be noted that the actual substrate for Glce does not have 2-O-sulfation. Thus, it is likely that Tyr149 forms a crucial hydrogen bond with the 2-hydroxyl group of the residue in the actual substrate that is at the ΔUAP1 position and is probably involved in recognition of the sugar residue (GlcA or IdoA) at that position.
The epimerization reaction is irreversible in vivo (5, 25), thereby increasing the number of IdoA units in the HS chain. O-Sulfation by the downstream O-sulfotransferases is probably a key means of maintaining the epimerization reaction in one direction, through inhibition of Glce activity. The structure of the complex suggests that the mechanism of inhibition is through a binding conformation that is not conducive to catalysis (i.e. the crucial catalytic residues are kept away from the C5 atom of IDS3 due to 2-O-sulfation and 6-O-sulfation). To allow efficient, consecutive modifications of HS by Glce and O-sulfotransferases, these enzymes need to be in close proximity and may interact. The interaction of Glce with 2-O-sulfotransferase in mammalian cultured cells has been reported, and lack of the 2-O-sulfotransferase caused a decrease of epimerase activity and protein stability (37). Further, it was reported that Drosophila Glce, 2-O-sulfotransferase, and 6-O-sulfotransferase could physically interact (28). Here, we used the AlphaScreen in vitro binding assay to examine whether Glce and O-sulfotransferases could physically interact (Fig. 6D). We found that the 2-O-sulfotransferase strongly interacted with Glce, and the 6-O-sulfotransferase proteins also clearly interacted with Glce (Fig. 6E). The data support a model in which Glce and O-sulfotransferases associate to form a protein complex that allows efficient coupling of the epimerization and O-sulfation reactions. The enzymes' association promotes the accumulation of IdoA in the final HS polysaccharide product.
In summary, we report the first dimeric structures of Glce in apo-form and in complex with heparin hexasaccharide. Detailed structural analysis revealed that Tyr468, Tyr528, and Tyr546 at the active site are crucial for catalysis. We provide structural evidence that heparin with O-sulfation binds to the Glce active site in a conformation that is not conducive for catalysis. Finally, we demonstrated the direct interaction of Glce with O-sulfotransferases, which couples the epimerization and O-sulfation reactions to ensure efficient HS chain modification in one direction. The structural and functional data advance understanding of the key epimerization step in the HS synthesis and will facilitate the use of Glce in rational biosynthesis of bioactive heparin oligosaccharides for therapeutic applications.
Acknowledgments
We thank staff members of the Life Science Collaborative Access Team of the Advanced Photon Source for assistance in data collection at the beam lines of sector 21. In addition, we thank Dr. David Nadziejka for editorial assistance in preparing the manuscript.
This work was supported by National Natural Science Foundation of China (NSFC) Grant 31230022, National Science Fund for Distinguished Young Scholars Grant 81125025, New Drug Creation and Manufacturing Program Grant 2012ZX09301001-003, the Swedish Research Council and Swedish Cancer Foundation (to J. P. L.), the Michigan Economic Development Corporation, and Michigan Technology Tri-Corridor Grant 085P1000817.
- HS
- heparan sulfate
- Glce
- d-glucuronyl C5-epimerase
- GlcA
- d-glucuronic acid
- IdoA
- l-iduronic acid
- ΔUAP
- 4-deoxy-2-O-sulfo-α-l-threo-hex-4-enopyranuronic acid
- IDS
- 2-O-sulfo-α-l-idopyranuronic acid
- SGN
- N,O6-disulfoglucosamine
- SUMO
- small ubiquitin-like modifier
- SeMet
- selenomethionine
- Bistris propane
- 1,3-bis[tris(hydroxymethyl)methylamino]propane
- MBP
- maltose-binding protein
- RMSD
- root mean square deviation
- PDB
- Protein Data Bank.
REFERENCES
- 1. Li J. P. (2010) Glucuronyl C5-epimerase an enzyme converting glucuronic acid to iduronic acid in heparan sulfate/heparin biosynthesis. Prog. Mol. Biol. Transl. Sci. 93, 59–78 [DOI] [PubMed] [Google Scholar]
- 2. Bishop J. R., Schuksz M., Esko J. D. (2007) Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature 446, 1030–1037 [DOI] [PubMed] [Google Scholar]
- 3. Hallak L. K., Collins P. L., Knudson W., Peeples M. E. (2000) Iduronic acid-containing glycosaminoglycans on target cells are required for efficient respiratory syncytial virus infection. Virology 271, 264–275 [DOI] [PubMed] [Google Scholar]
- 4. Feyerabend T. B., Li J. P., Lindahl U., Rodewald H. R. (2006) Heparan sulfate C5-epimerase is essential for heparin biosynthesis in mast cells. Nat. Chem. Biol. 2, 195–196 [DOI] [PubMed] [Google Scholar]
- 5. Hagner-McWhirter A., Li J. P., Oscarson S., Lindahl U. (2004) Irreversible glucuronyl C5-epimerization in the biosynthesis of heparan sulfate. J. Biol. Chem. 279, 14631–14638 [DOI] [PubMed] [Google Scholar]
- 6. Jia J., Maccarana M., Zhang X., Bespalov M., Lindahl U., Li J. P. (2009) Lack of l-iduronic acid in heparan sulfate affects interaction with growth factors and cell signaling. J. Biol. Chem. 284, 15942–15950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li J. P., Gong F., Hagner-McWhirter A., Forsberg E., Abrink M., Kisilevsky R., Zhang X., Lindahl U. (2003) Targeted disruption of a murine glucuronyl C5-epimerase gene results in heparan sulfate lacking l-iduronic acid and in neonatal lethality. J. Biol. Chem. 278, 28363–28366 [DOI] [PubMed] [Google Scholar]
- 8. Prudnikova T. Y., Mostovich L. A., Domanitskaya N. V., Pavlova T. V., Kashuba V. I., Zabarovsky E. R., Grigorieva E. V. (2010) Antiproliferative effect of d-glucuronyl C5-epimerase in human breast cancer cells. Cancer Cell Int. 10, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Grigorieva E. V., Prudnikova T. Y., Domanitskaya N. V., Mostovich L. A., Pavlova T. V., Kashuba V. I.c, Zabarovsky E. R. (2011) d-Glucuronyl C5-epimerase suppresses small-cell lung cancer cell proliferation in vitro and tumour growth in vivo. Br. J. Cancer 105, 74–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li J., Fang J., Qin Y., Liao W., Liu H., Zhou Y., Ding K. (2014) Glce regulates PC12 cell neuritogenesis induced by nerve growth factor through activating Smad/Id3 signaling. Biochem. J. 459, 405–415 [DOI] [PubMed] [Google Scholar]
- 11. Reijmers R. M., Vondenhoff M. F., Roozendaal R., Kuil A., Li J. P., Spaargaren M., Pals S. T., Mebius R. E. (2010) Impaired lymphoid organ development in mice lacking the heparan sulfate modifying enzyme glucuronyl C5-epimerase. J. Immunol. 184, 3656–3664 [DOI] [PubMed] [Google Scholar]
- 12. Reijmers R. M., Groen R. W., Kuil A., Weijer K., Kimberley F. C., Medema J. P., van Kuppevelt T. H., Li J. P., Spaargaren M., Pals S. T. (2011) Disruption of heparan sulfate proteoglycan conformation perturbs B-cell maturation and APRIL-mediated plasma cell survival. Blood 117, 6162–6171 [DOI] [PubMed] [Google Scholar]
- 13. Chen J., Jones C. L., Liu J. (2007) Using an enzymatic combinatorial approach to identify anticoagulant heparan sulfate structures. Chem. Biol. 14, 986–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kuberan B., Lech M. Z., Beeler D. L., Wu Z. L. L., Rosenberg R. D. (2003) Enzymatic synthesis of antithrombin III-binding heparan sulfate pentasaccharide. Nat. Biotechnol. 21, 1343–1346 [DOI] [PubMed] [Google Scholar]
- 15. Xu Y., Masuko S., Takieddin M., Xu H., Liu R., Jing J., Mousa S. A., Linhardt R. J., Liu J. (2011) Chemoenzymatic synthesis of homogeneous ultralow molecular weight heparins. Science 334, 498–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Collaborative Computational Project, Number 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 18. Sheldrick G. M. (2010) Experimental phasing with SHELXC/D/E: combining chain tracing with density modification. Acta Crystallogr. D Biol. Crystallogr. 66, 479–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 20. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 21. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D. Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Campbell P., Feingold D. S., Jensen J. W., Malmström A., Rodén L. (1983) New assay for uronosyl 5-epimerases. Anal. Biochem. 131, 146–152 [DOI] [PubMed] [Google Scholar]
- 23. Hagner-McWhirter A., Hannesson H. H., Campbell P., Westley J., Rodén L., Lindahl U., Li J. P. (2000) Biosynthesis of heparin/heparan sulfate: kinetic studies of the glucuronyl C5-epimerase with N-sulfated derivatives of the Escherichia coli K5 capsular polysaccharide as substrates. Glycobiology 10, 159–171 [DOI] [PubMed] [Google Scholar]
- 24. Ke J., Harikumar K. G., Erice C., Chen C., Gu X., Wang L., Parker N., Cheng Z., Xu W., Williams B. O., Melcher K., Miller L. J., Xu H. E. (2013) Structure and function of Norrin in assembly and activation of a Frizzled 4-Lrp5/6 complex. Genes Dev. 27, 2305–2319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hagner-Mcwhirter A., Lindahl U., Li J. (2000) Biosynthesis of heparin/heparan sulphate: mechanism of epimerization of glucuronyl C-5. Biochem. J. 347, 69–75 [PMC free article] [PubMed] [Google Scholar]
- 26. Shaya D., Tocilj A., Li Y., Myette J., Venkataraman G., Sasisekharan R., Cygler M. (2006) Crystal structure of heparinase II from Pedobacter heparinus and its complex with a disaccharide product. J. Biol. Chem. 281, 15525–15535 [DOI] [PubMed] [Google Scholar]
- 27. Li K., Bethea H. N., Liu J. (2010) Using engineered 2-O-sulfotransferase to determine the activity of heparan sulfate C-5-epimerase and its mutants. J. Biol. Chem. 285, 11106–11113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dejima K., Takemura M., Nakato E., Peterson J., Hayashi Y., Kinoshita-Toyoda A., Toyoda H., Nakato H. (2013) Analysis of Drosophila glucuronyl C5-epimerase: implications for developmental roles of heparan sulfate sulfation compensation and 2-O-sulfated glucuronic acid. J. Biol. Chem. 288, 34384–34393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jacobsson I., Lindahl U., Jensen J. W., Rodén L., Prihar H., Feingold D. S. (1984) Biosynthesis of heparin. Substrate specificity of heparosan N-sulfate d-glucuronosyl 5-epimerase. J. Biol. Chem. 259, 1056–1063 [PubMed] [Google Scholar]
- 30. Raedts J., Lundgren M., Kengen S. W., Li J. P., van der Oost J. (2013) A novel bacterial enzyme with d-glucuronyl C5-epimerase activity. J. Biol. Chem. 288, 24332–24339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Holm L., Sander C. (1993) Protein structure comparison by alignment of distance matrices. J. Mol. Biol. 233, 123–138 [DOI] [PubMed] [Google Scholar]
- 32. Rozeboom H. J., Bjerkan T. M., Kalk K. H., Ertesvåg H., Holtan S., Aachmann F. L., Valla S., Dijkstra B. W. (2008) Structural and mutational characterization of the catalytic A-module of the mannuronan C-5-epimerase AlgE4 from Azotobacter vinelandii. J. Biol. Chem. 283, 23819–23828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pacheco B., Maccarana M., Goodlett D. R., Malmström A., Malmström L. (2009) Identification of the active site of DS-epimerase 1 and requirement of N-glycosylation for enzyme function. J. Biol. Chem. 284, 1741–1747 [DOI] [PubMed] [Google Scholar]
- 34. Hannesson H. H., Hagner-McWhirter A., Tiedemann K., Lindahl U., Malmström A. (1996) Biosynthesis of dermatan sulphate: defructosylated Escherichia coli K4 capsular polysaccharide as a substrate for the d-glucuronyl C-5 epimerase, and an indication of a two-base reaction mechanism. Biochem. J. 313, 589–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jerga A., Stanley M. D., Tipton P. A. (2006) Chemical mechanism and specificity of the C5-mannuronan epimerase reaction. Biochemistry 45, 9138–9144 [DOI] [PubMed] [Google Scholar]
- 36. Sheng J., Xu Y., Dulaney S. B., Huang X., Liu J. (2012) Uncovering biphasic catalytic mode of C5-epimerase in heparan sulfate biosynthesis. J. Biol. Chem. 287, 20996–21002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pinhal M. A., Smith B., Olson S., Aikawa J., Kimata K., Esko J. D. (2001) Enzyme interactions in heparan sulfate biosynthesis: uronosyl 5-epimerase and 2-O-sulfotransferase interact in vivo. Proc. Natl. Acad. Sci. U.S.A. 98, 12984–12989 [DOI] [PMC free article] [PubMed] [Google Scholar]