

Background: Animal and plant matrix metalloproteinases (MMPs) are kept zymogenic through large prodomains and a cysteine-switch mechanism.
Results: Bacterial MMP karilysin has only a short N-terminal peptide upstream of the catalytic domain, which lacks cysteines.
Conclusion: This peptide inhibits through an aspartate-switch mechanism and also exerts other functions of authentic prodomains.
Significance: Karilysin is kept latent by a novel mechanism for MMPs.
Keywords: Matrix Metalloproteinase (MMP), Peptidase, Periodontal Disease, Protease, Protein Evolution, X-ray Crystallography
Abstract
The matrix metalloproteinases (MMPs) are a family of secreted soluble or membrane-anchored multimodular peptidases regularly found in several paralogous copies in animals and plants, where they have multiple functions. The minimal consensus domain architecture comprises a signal peptide, a 60–90-residue globular prodomain with a conserved sequence motif including a cysteine engaged in “cysteine-switch” or “Velcro” mediated latency, and a catalytic domain. Karilysin, from the human periodontopathogen Tannerella forsythia, is the only bacterial MMP to have been characterized biochemically to date. It shares with eukaryotic forms the catalytic domain but none of the flanking domains. Instead of the consensus MMP prodomain, it features a 14-residue propeptide, the shortest reported for a metallopeptidase, which lacks cysteines. Here we determined the structure of a prokarilysin fragment encompassing the propeptide and the catalytic domain, and found that the former runs across the cleft in the opposite direction to a bound substrate and inhibits the latter through an “aspartate-switch” mechanism. This finding is reminiscent of latency maintenance in the otherwise unrelated astacin and fragilysin metallopeptidase families. In addition, in vivo and biochemical assays showed that the propeptide contributes to protein folding and stability. Our analysis of prokarilysin reveals a novel mechanism of latency and activation in MMPs. Finally, our findings support the view that the karilysin catalytic domain was co-opted by competent bacteria through horizontal gene transfer from a eukaryotic source, and later evolved in a specific bacterial environment.
Introduction
The matrix metalloproteinases (MMPs)5 are a family of zinc- and calcium-dependent peptidases, which are grouped into the metzincin clan of metallopeptidases (MPs) together with other separate families such as the ADAMs/adamalysins, astacins, fragilysins, and serralysins (1–8). MMPs are found throughout animals and plants (9–12), where their distribution is consistent with a Darwinian tree-based pathway. In addition, polyplication has led to several paralogous MMP genes being present in the same organism: 24 in humans, 26 in sea urchin, 26 in zebrafish, seven in sea squirt, and two in fruit fly (11). In contrast, only a patchy phylogenetic distribution of genes encoding hypothetical orthologs has been found in viruses, Bacteria, Archaea, and fungi. Earlier studies of the relationship between mammalian MMPs and supposed prokaryotic orthologs included, as we now know, bacterial members of other metzincin families such as serralysins, fragilysins, and astacins (13–16). Accordingly, it was suggested that a primordial MMP may have arisen from an ancestor that is common to vertebrates, invertebrates, and plants but is not shared by earlier stages in evolution (11, 16–18). This entails that the hypothetical prokaryotic, viral, and fungal MMPs are incongruent with the tree of life or, more accurately, xenologs, i.e. the result of direct or indirect horizontal gene transfer from eukaryotic donors (9, 19, 20). This is reminiscent of the evolutionary origin postulated for fragilysin, which is the only molecular virulence factor described for enterotoxigenic Bacteroides fragilis and for which no similar proteins have been reported, not even from other B. fragilis strains (21). Structural studies supported the view that the catalytic domain of this MP is the result of horizontal gene transfer of a member of the ADAM/adamalysin family, which has 38 orthologs in humans (8, 22–25), from a mammalian host to this bacterium, which thrives in the intestinal tract (26, 27).
Returning to MMPs, karilysin from the human periodontopathogen Tannerella forsythia is the only bacterial family member to have been analyzed biochemically to date (9, 28–33). In addition to karilysin, only MmpZ from Bacillus anthracis has been functionally assessed at the genetic level through knock-out studies in B. anthracis cells, but it has not been isolated or characterized (34). Similarly to vertebrate MMPs, karilysin showed preference for medium-sized to bulky hydrophobic residues (leucine, tyrosine and methionine) in the specificity pocket, S1′ (Ref. 30; for active-site cleft subsite nomenclature, see Ref. 35). It inactivates antimicrobial peptide LL-37 and integrants of the complement system, including ficolin-2, ficolin-3, C4, and C5, by proteolysis and may thus contribute to evasion of the innate host immune response (29, 31). Karilysin is sequentially and evolutionarily closer to MMPs from winged insects that are transmission vectors of human diseases (47% sequence identity with Dm1 from Aedes aegypti and Anopheles gambiae; (9)) and mammals (44% identity with human MMP-11, -13, and -20 (9)) than to the few other bacterial sequences found in genomic sequences. Accordingly it was likewise suggested that it may be the result of horizontal gene transfer of an MMP gene from an animal to an intimate bacterial pathogen, which inhabits a biofilm on the tooth surface in humans (9).
The metzincins are characterized by a consensus sequence responsible for binding of the catalytic zinc ion (CSBZ), H-E-X-X-H-X-X-(G/N)-X-X-(H/D) (amino acid one-letter code; X stands for any residue), and a conserved methionine-containing turn, the “Met-turn” (1–5, 36). In MMPs, the CSBZ encompasses three histidine zinc ligands, the general base/acid glutamate for catalysis, and a structurally relevant glycine (3). In addition, the distinct MMP paralogs are multidomain proteins that display a disparate domain organization that is the result of successive polyplication, gene fusion, and exon shuffling (11). The only domains common to all animal and plant MMPs are a signal peptide, which is removed after secretion, a prodomain and a catalytic domain, as found, e.g. in human MMP-7 and MMP-26, and in plant MMPs (12, 16, 18).
Most peptidases are biosynthesized as zymogens containing prosegments, which are required for latency maintenance to prevent unbridled activity but also sometimes to assist in proper folding of the usually downstream catalytic moieties (37–40). Metzincin exceptions lacking prosegments include the archaemetzincins, for which no hydrolytic activity has so far been reported, i.e. they might not need to be kept latent (41, 42); the toxilysin EcxA from Escherichia coli, whose soluble expression requires co-expression with its cognate EcxB subunit, thus pointing to a chaperone-like function for this ancillary subunit (43–45); the cholerilysin StcE from E. coli, for which an N-terminal immunoglobulin-like domain may assist the downstream catalytic moiety in proper folding (46); and igalysins, where an all-β-domain of similar topology to immunoglobulin-like domains is likewise found at the N terminus of the catalytic moiety (see Protein Data Bank (PDB) access codes 4DF9 and 3P1V and Ref. 5).
MMP prodomains (see Table 1 in Ref. 47) span 60–90 residues and include a conserved sequence motif, P-R-C-G-(V/N)-P-D, engaged in a “cysteine-switch” or “Velcro” mechanism of latency (10, 16, 48–51). It has been suggested that this mechanism may be shared by variants within other metzincin families, for which conserved cysteines were described upstream of the catalytic domain, such as the ADAMs/adamalysins (motif P-K-M-C-G-V (8, 52–54)), leishmanolysins (motif H-R-C-I-H-D (2)), and pappalysins (motif C-G (55)). In contrast, the 472 residues encoded by the karilysin gene (see UniProt sequence database access code D0EM77) only comprise a short 14-residue potential propeptide, which lacks cysteines, between the 20-residue signal peptide and the 161-residue mature catalytic moiety (Fig. 1A). A C-terminal domain of 277 residues of unknown function and sequence unrelated to any domain found in eukaryotic MMPs completes the protein. This strongly suggests a potentially different mechanism of latency maintenance, hitherto unseen not only in MMPs but also in metzincins in general, as the shortest prosegments described to date are those of members of the astacin family, which span >34 residues (7, 56–58).
TABLE 1.
Vectors and constructs
| Name | Original vector | Antibiotic resistancea | Restriction sites | Insert | Fusion construct (N-terminal) | Additional N-terminal residues |
|---|---|---|---|---|---|---|
| pKAR1 | pGEX-GP-1 (GE Healthcare) | amp | BamHI/XhoI | wt Gln21-Lys472 (pKly) | GST + HR3CPr | G-P-l-G-Sb |
| pKAR2 | pGEX-GP-1 (GE Healthcare) | amp | BamHI/XhoI | Y35A mutant Gln21-Lys472 (pKly-Y35A) | GST + HR3CPr | G-P-l-G-Sb |
| pKAR3 | pGEX-GP-1 (GE Healthcare) | amp | BamHI/XhoI | E156A mutant Gln21-Lys472 (pKly-E156A) | GST + HR3CPr | G-P-l-G-Sb |
| pKAR4 | pGEX-GP-1 (GE Healthcare) | amp | BamHI/XhoI | D25A/Y35A mutant Gln21-Lys472 (pKly-D25A/Y35A) | GST + HR3CPr | G-P-l-G-Sb |
| pKAR5 | pGEX-GP-1 (GE Healthcare) | amp | BamHI/XhoI | E156A mutant Gln21-Ser201 (pKly18-E156A) | GST + HR3CPr | G-P-b |
| pKAR6 | pGEX-GP-1 (GE Healthcare) | amp | BamHI/XhoI | E156A mutant Tyr35-Ser201 (Kly18-E156A) | GST + HR3CPr | G-P-b |
| pKAR7 | pGEX-GP-1 (GE Healthcare) | amp | BamHI/XhoI | wt Gln21-Ser201 (pKly18) | GST + HR3CPr | G-P-b |
| pKAR8 | pGEX-GP-1 (GE Healthcare) | amp | BamHI/XhoI | Y35A mutant Gln21-Ser201 (pKly18-Y35A) | GST + HR3CPr | G-P-b |
| pKAR9 | pGEX-GP-1 (GE Healthcare) | amp | BamHI/XhoI | D25A/Y35A mutant Gln21-Ser201 (pKly18-D25A/Y35A) | GST + HR3CPr | G-P-b |
| pKAR10 | pCRI-7a (59) | kan | NcoI/XhoI | wt Gln21-Ser201 (pKly18) | None | G-P-b |
| pKAR11 | pCRI-7a (59) | kan | NcoI/XhoI | E156A mutant Gln21-Ser201 (pKly18-E156A) | None | M-G- |
| pKAR12 | pCRI-7a (59) | kan | NcoI/XhoI | wt Tyr35-Ser201 (Kly18) | None | M-G- |
| pKAR13 | pCRI-7a (59) | kan | NcoI/XhoI | E156A mutant Tyr35-Ser201 (Kly18-E156A) | None | M-G- |
a amp, ampicillin; GST, glutathione S-transferase; HR3CPr, recognition sequence for human rhinovirus 3C peptidase; kan, kanamycin; mut., mutant; wt, wild-type.
b After cleavage with HR3CP.
FIGURE 1.

Effect of the Kly18 propeptide in vitro and in vivo. A, scheme depicting the domain structure of T. forsythia karilysin. Numbering according to UniProt D0EM77. B, SDS-PAGE of cultures of wild-type and E156A variants of pKly18 and Kly18. Lanes 1 and 2, insoluble and soluble fractions of wild-type pKly18 (from pKAR10), respectively. Lanes 3 and 4, insoluble and soluble fractions of wild-type Kly18 (pKAR12), respectively. Lanes 5 and 6, insoluble and soluble fractions of pKly18-E156A (pKAR11), respectively. Lanes 7 and 8, insoluble and soluble fractions of Kly18-E156A (pKAR13), respectively. Overexpressed proteins are labeled with an asterisk. C, unfolding transition curves showing temperature-dependent change in fluorescence of pKly18-E156A (pKAR5; solid line) and Kly18-E156A (pKAR6; dashed line) in the absence (black curve) and presence of CaCl2 (1 mm, red curve; 5 mm, green curve). The blank curve is indicated with a dotted line. D, proteolytic activity of Kly18 (pKAR1) at 37 °C using substrate Mca-R-P-K-P-V-E-Nva-W-R-K(dnp)-NH2 at 10 μm in the absence (0) and presence of 0.1, 0.5, 1, 2, and 5 mm propeptide mimic. E and F, stability of mutant pKly-Y35A (E) and mutant pKly-Y35A/D25A (F) over time. Kly48, Kly38, and Kly18 are indicated by arrows.
We had previously determined the structure of the catalytic domain of karilysin (termed Kly18 (9)). To shed light on the molecular determinants of the first mechanism of latency maintenance of a bacterial MMP, in this work we assayed the possible function of the propeptide in folding, stability, and activity inhibition of Kly18. We further solved the x-ray crystal structure of an active-site mutant of a construct spanning the propeptide and Kly18 affecting the catalytic glutamate, pKly18-E156A, to circumvent autolysis. The mechanism derived was supported by site-directed mutagenesis and it is discussed in the context of general MMP latency maintenance.
EXPERIMENTAL PROCEDURES
Protein Production and Purification
The gene coding for full-length wild-type T. forsythia prokarilysin without the 20-residue signal peptide (hereafter pKly; 52 kDa; residues Gln21-Lys472 according to UP D0EM77, see also Fig. 1A) was cloned at BamHI and XhoI restriction sites into vector pGEX-6P-1 (GE Healthcare) as described elsewhere (30). The resulting vector, pKAR1 (see Table 1 for an overview of vectors and constructs used), confers resistance toward ampicillin and attaches an N-terminal glutathione S-transferase (GST) moiety followed by a human rhinovirus 3C proteinase (HR3CP) recognition site (L-E-V-L-F-Q-↓-G-P; HR3CP cleavage leaves two extra residues, underlined, at the N terminus of the recombinant protein after digestion; three extra residues, L-G-S, are further present due to the cloning strategy). Single-residue point mutants pKly-Y35A and pKly-E156A (pKAR2 and pKAR3, respectively) were generated using the QuikChange Site-directed Mutagenesis Kit (Stratagene) according to the manufacturer's instructions as described (30). Double mutant pKly-D25A/Y35A (pKAR4) was similarly generated using pKAR2 as a template. Genes coding for the E156A-mutated catalytic domain of karilysin, with and without the propeptide (hereafter pKly18-E156A and Kly18-E156A; 20 and 18 kDa; residues Gln21-Ser201 and residues Tyr35-Ser201, respectively), were also cloned into vector pGEX-6P-1 (pKAR5 and pKAR6, respectively). Genes coding for pKly18 and its mutant proteins pKly18-Y35A and pKly18-D25A/Y35A were cloned into the same vector (pKAR7, pKAR8, and pKAR9, respectively) following a strategy previously described (59). Genes coding for pKly18, pKly18-E156A, Kly18, and Kly18-E156A were, furthermore, cloned at NcoI and XhoI restriction sites into vector pCRI-7a (59), which confers resistance toward kanamycin and does not attach fusion proteins (pKAR10-pKAR13, respectively). In these cases, the cloning strategy entailed that residues M-G were attached at the N terminus. All constructs were verified by DNA sequencing.
Proteins encoded by vectors pKAR1-pKAR9 were produced by heterologous overexpression in E. coli BL21(DE3) cells, which were grown at 37 °C in Luria-Bertani medium supplemented with 100 μg/ml of ampicillin. Cultures were induced at an A600 of 0.8 with 0.2 mm isopropyl β-d-thiogalactopyranoside and incubated overnight at 18 °C. Purification of wild-type and mutant pKly, and subsequent autolysis of the former to obtain Kly18, was achieved as described elsewhere (30). In turn, pKly18-E156A, Kly18-E156A, pKly18-Y35A, and pKly18-D25A/Y35A were purified as follows. After centrifugation at 7,000 × g for 30 min at 4 °C, the pellet was washed twice in 1× PBS, and resuspended in the same buffer supplemented with EDTA-free protease inhibitor mixture tablets and DNase I (both Roche Diagnostics). Cells were lysed using a cell disrupter (Constant Systems, Ltd.) at 1.35 Kbar, and the cell debris was removed by centrifugation at 40,000 × g for 1 h at 4 °C. The supernatant was filtered (0.22 μm pore size; Millipore), and incubated with glutathione-Sepharose 4B resin (GE Healthcare). The sample was washed first in 1× PBS and then in buffer A (50 mm Tris-HCl, 150 mm NaCl, pH 7.5), and eluted by incubation and cleavage with HR3CP at a 1:20 enzyme:substrate (w/w) ratio for 48 h at 4 °C. The protein was concentrated by ultrafiltration, and finally purified by size-exclusion chromatography on 16/600 or 10/300 Superdex 75 columns (GE Healthcare) previously equilibrated with buffer B (20 mm Tris-HCl, pH 8.0) or buffer C (20 mm Tris-HCl, 150 mm NaCl, pH 7.5).
Proteins encoded by vectors pKAR10–pKAR13 were produced in E. coli BL21(DE3) cells, which were grown at 37 °C in Luria-Bertani medium supplemented with 30 μg/ml of kanamycin. Cultures were induced at an A600 of 0.8 with 0.2–1 mm isopropyl β-d-thiogalactopyranoside and incubated either for 5 h at 37 °C or overnight at 18 °C. Cells were harvested by centrifugation at 7,000 × g for 30 min at 4 °C, washed in buffer A, resuspended in the same buffer, and further lysed in an ice-bath using a digital sonifier (Branson). After centrifugation at 15,000 × g for 30 min at 4 °C, both cell debris and supernatant were analyzed by 15% Tricine-SDS-PAGE stained with Coomassie Blue.
Protein identity and purity were assessed by mass spectrometry using an Autoflex Bruker apparatus and N-terminal sequencing through Edman degradation at the Proteomics Facility of Centro de Investigaciones Biológicas (Madrid, Spain). Ultrafiltration steps were performed with Vivaspin 15 and Vivaspin 4 filter devices of 5-kDa cut-off (Sartorius Stedim Biotech). Approximate protein concentration was determined by measuring A280 in a spectrophotometer (NanoDrop) using the calculated absorption coefficients E0.1% = 2.32 and 2.42 for pKly18-E156A and Kly18-E156A, respectively.
Autolytic Activation and Propeptide Inhibitory Activity Assays
Mutants pKly-Y35A (from pKAR2), pKly-D25A/Y35A (pKAR4), and pKly18-Y35A (pKAR8) were incubated in buffer B at 37 °C and at 0.4 mg/ml final protein concentration for up to 120 h to assay autolysis. Reactions were stopped at specific time points by boiling aliquots in reducing/denaturing buffer, and samples were further analyzed by 10% or 15% Tricine-SDS-PAGE stained with Coomassie Blue. Kly18, obtained by autolysis from pKAR1-encoded protein, was incubated at 0.025 μg/ml of final protein concentration for 30 min with 0.1–10 mm peptide Q-R-L-Y-D-N-G-P-L-T (purchased from GL Biochem Ltd.), which mimics the propeptide sequence. Proteolytic activity was subsequently measured at 37 °C in buffer C on substrate Mca-R-P-K-P-V-E-Nva-W-R-K(dnp)-NH2 (Bachem; at 10 μm) in a microplate fluorimeter (Infinite M200, Tecan).
Thermal Shift Assays
Aliquots were prepared by mixing 7.5 μl of ×300 Sypro Orange dye (Molecular Probes) and 42.5 μl of either pKly18-E156A (from pKAR5) or Kly18-E156A (pKAR6) at 1–2 mg/ml in buffer C in the absence and presence of 1–5 mm CaCl2. Four replicates of each aliquot were analyzed in an iQ5 Multi-Color Real Time PCR Detection System (Bio-Rad) in 96-well PCR plates sealed with optical tape. Samples were heated from 30 to 95 °C at 0.5 °C/min, and the change in absorbance (λex = 490 nm; λem = 575 nm) was monitored over time. The temperature of midtransition (Tm (60)) was determined for both proteins from the inflection point of each curve using iQ5 software.
Crystallization and Data Collection
Crystallization assays of pKAR5-encoded pKly18-E156A protein were carried out at the IBMB/IRB Crystallography Platform by the sitting-drop vapor diffusion method using 96 × 2-well MRC plates (Innovadyne). A TECAN Freedom EVO robot was used to prepare reservoir solutions, and a Phoenix/RE (Art Robbins) robot or a Cartesian Microsys 4000 XL (Genomic Solutions) was used for nanodrop dispensing. Crystallization plates were stored in Bruker steady-temperature crystal farms at 4 and 20 °C. Successful conditions were scaled up to the microliter range in 24-well Cryschem crystallization dishes (Hampton Research). The best crystals were obtained at 20 °C from drops containing protein solution (3.75 mg/ml in buffer B) and 100 mm Bistris propane, 200 mm potassium thiocyanate, 20% (w/v) polyethylene glycol 3350 (pH 7.5) as reservoir solution from 2:1-μl drops. Crystals were cryo-protected with 20% (v/v) glycerol. Diffraction datasets were collected at 100 K from liquid-N2 flash cryo-cooled crystals (Oxford Cryosystems 700 series cryostream) on a Pilatus 6M pixel detector (from Dectris) at beam lines ID23-1 and ID29 of the European Synchrotron Radiation Facility (ESRF, Grenoble, France) within the Block Allocation Group “BAG Barcelona.” Crystals contained two molecules per asymmetric unit. Diffraction data were integrated, scaled, merged, and reduced with programs XDS (61) and XSCALE (62) (see Table 2 for data processing statistics of the best dataset).
TABLE 2.
Crystallographic data
| Data | |
|---|---|
| Space group/cell constants (a, b, c in Å and β in °) | P21/36.18, 121.69, 41.88; 105.26 |
| Wavelength (Å) | 0.97242 |
| No. of measurements/unique reflections | 104,406/22,975 |
| Resolution range (Å) (outermost shell) | 60.8–2.01 (2.06–2.01) |
| Completeness (%) | 99.8 (96.2) |
| Rmergea | 0.105 (0.534) |
| Rr.i.m. (=Rmeas)a /CC(1/2)b | 0.116 (0.616)/99.6 (84.3) |
| Average intensity | 10.3 (2.5) |
| B-factor (Wilson) (Å2)/average multiplicity | 29.0/4.5 (4.0) |
| Resolution range used for refinement (Å) | ∞–2.01 |
| No. of reflections used (test set) | 22,047 (741) |
| Crystallographic Rfactor (free Rfactor)c | 0.165 (0.194) |
| No. of protein atomsd/solvent molecules/ligands/ions | 2,843/226/3 glycerols/4 zinc, 2 calcium |
| Root mean square deviation target values bonds (Å)/angles (°) | 0.010/0.98 |
| Overall average B-factor (Å2) | 30.8 |
| Model validatione | |
| Residues in favored regions/outliers/all residues | 345 (97.5%)/0/355 |
| Residues with bad bonds/bad angles/poor rotamers/Cβ deviation >0.25 Å | 0/0/7(2.4%)/0 |
Values in parentheses refer to the outermost resolution shell.
b According to Karplus and Diederichs (78).
c Crystallographic Rfactor = Σhkl ‖Fobs| − k |Fcalc‖/Σhkl |Fobs|, where k is a scaling factor, and Fobs and Fcalc are the observed and calculated structure factor amplitudes, respectively. This factor is calculated for the working-set reflections; free Rfactor, same for a test-set of reflections (>500) not used during refinement.
d Including atoms with double occupancy.
e According to MOLPROBITY (76).
Structure Solution and Refinement
The structure of pKly18-E156A was solved by likelihood-scoring molecular replacement with the program PHASER (63) using the coordinates of the protein part only of mature wild-type Kly18 (PDB access code 4IN9 (64, 65)) as searching model. Two solutions were found at final Eulerian angles (α, β, γ, in °) of 285.8, 56.7, 97.2, and 76.9, 91.3, 284.2; and fractional cell coordinates (x, y, z) of 0.120, −0.017, 0.100, and 0.997, 0.332, and 0.608, respectively. These solutions gave initial Z-scores of 8.5 and 8.3 for the rotation functions, and 6.6 and 6.2 for the translation functions, respectively, as well as a final log-likelihood gain of 1,120. A subsequent density improvement step with ARP/wARP (66) rendered an electron density map that enabled straightforward chain tracing. Thereafter, manual model building with COOT (67, 68) alternated with crystallographic refinement with programs PHENIX (69) and BUSTER/TNT (70, 71), which included TLS refinement and automatically determined non-crystallographic restraints, until completion of the model. Both final model chains A and B contained residues Gln21-Pro199, as well as two zincs and one calcium each. Segment Val36-Gly39 of chain A was continuous in the final Fourier map but ambiguous as to the position of the side chains. In addition, segments Gln38-Ser40 and Ser54-His57 of chain B were traced based on weak electron density to preserve chain continuity. Pro122 and Pro123 were linked by a cis-peptide bond. Three glycerol molecules and 226 solvent molecules completed the structure (see Table 2).
Miscellaneous
Figures were prepared with CHIMERA (72). Structural superpositions were performed with SSM (73) within COOT, and with LSQKAB (74) and ROTMAT within the CCP4 suite of programs (75). Model validation was performed with MOLPROBITY (76). The interaction surface buried at the interface between the propeptide and the mature enzyme moiety was calculated with CNS version 1.3 (77). The final coordinates of pKly18-E156A are deposited with the PDB with code 4R3V.
RESULTS AND DISCUSSION
Roles of the Propeptide in Vitro and in Cellula
Wild-type karilysin is secreted as a zymogen with a 14-residue N-terminal propeptide (21Q-R-L-Y-D-N-G-P-L-T-G-D-N-N34), which is cleaved off at position Asn34-Tyr35 during maturation (Fig. 1A). This is the primary activation cleavage and it releases an active 48-kDa form (Kly48 (30)). In recombinant protein production, subsequent cleavages within the C-terminal domain give rise to Kly38 and, finally, to a stable form of 18 kDa (Kly18), which corresponds to the isolated mature catalytic domain (CD) (5, 28, 30, 33). These cleavages were shown to be autolytic as activation was repressed by general chelating MP inhibitors and in the inactive active-site variant, E156A, which ablated the catalytic glutamate of the CSBZ (1, 5, 30, 79, 80). In addition, cleavage-site mutant Y35A, which does not match the specificity of the enzyme, was activated only slowly when compared with the wild-type (30, 33).
To assess whether the propeptide had a chaperone-like function on the downstream catalytic moiety, we cloned the genes encoding pKly18-E156A and Kly18-E156A in a vector that does not attach a fusion protein at the N terminus that would assist in proper folding (pKAR11 and pKAR13, respectively; see “Experimental Procedures”). We found that the active-site mutant pKly18-E156A was successfully overexpressed in soluble form (Fig. 1B). In contrast to the zymogen, Kly18-E156A was produced only in insoluble form (Fig. 1B). Moreover, when expressed from the pKAR6 vector, which attaches an N-terminal glutathione S-transferase fusion protein (see Table 1), Kly18-E156A was obtained with ∼10 times lower yield than the proprotein (vector pKAR5). We conclude that the propeptide plays a major role in proper folding of Kly18 as previously described for other MPs such as fragilysin (26, 27), funnelin metallocarboxypeptidases (79, 81, 82), and ADAMs/adamalysins (54) but not for mammalian MMPs (83).
We further examined the effect of the propeptide in response to denaturation by a thermal shift assay following the thermofluor approach (60). Purified pKly18-E156A (pKAR5) showed two unfolding transitions compatible with unfolding of propeptide and CD, with Tm values of 60 ± 0.5 and 74 ± 0.5 °C (Fig. 1C). In contrast, the unfolding of purified Kly18-E156A (pKAR6) showed a single transition, with a Tm of 49 ± 2.2 °C. The addition of a physiological concentration of calcium resulted in a substantial increase in stability of both pKly18-E156A and Kly18-E156A. Accordingly, the former showed Tm values of 67.5 ± 1.7 and 76.5± 1.2 °C, and 69.5 ± 1.7 and 79 ± 2.2 °C, in the presence of 1 and 5 mm CaCl2, respectively, whereas those of Kly18-E156A were 52.5 ± 1.2 and 54.5 ± 1 °C. This result is in agreement with the important role of calcium in Kly18 activity, as addition of 2–5 mm CaCl2 is reported to enhance activity about three times (30). Thus, regardless of calcium, the 14-residue propeptide redounded to a dramatic increase in Tm, underpinning that it plays a major role in the thermal stability of the zymogen. Finally, we assayed the effect of a decapeptide spanning propeptide sequence Gln21-Thr30 on the activity of purified mature Kly18 (from pKAR1) in the presence of a fluorogenic peptide substrate (Fig. 1D). We observed a weak but consistently concentration-dependent inhibitory effect as previously shown for other MPs when their propeptides or prodomains were added in trans, among others funnelins (79, 81), ADAMs/adamalysins (84), and mammalian MMPs (85–87). Summarizing, the propeptide of karilysin is the shortest currently described to date for an MP, and it exerts all roles, which collectively or selectively had been previously described for peptidase propeptides or prodomains: latency maintenance, folding assistance during biosynthesis, stability to thermal denaturation, and inhibition of peptidolytic activity (38, 39, 81).
Overall Structure of pKly18
Due to rapid autolytic processing of recombinant wild-type prokarilysin (30), crystals of pKly18 could only be obtained for an inactive variant affecting the catalytic glutamate (pKly18-E156A), as already reported for other MP zymogens (88–92). This protein crystallized as monoclinic crystals diffracting to 2-Å resolution with two molecules per asymmetric unit. These were essentially identical (Cα-atom root mean square deviation = 0.53 Å) except for segments Asn53-His57 and Asn34-Gly39. The latter flanks the primary activation cleavage point and is flexible. It is stabilized through an interaction with segment Asn87-Asn89 of the second molecule present in the asymmetric unit of the crystal although in different conformations in molecules A and B, so the discussion hereafter is centered on molecule A if not otherwise stated. When two values are indicated, they refer to both molecules.
The protein reveals a compact, almost spherical shape of ∼40 Å in diameter and is subdivided into three moieties (Fig. 2A): the N-terminal propeptide (Gln21-Asn34), and a CD split into a larger N-terminal upper subdomain moiety (Tyr35-Gly162; NTS) and a smaller C-terminal lower subdomain moiety (Ile163-Pro199; CTS, see also Refs. 9 and 28)) if viewed in the standard orientation for MPs (35). NTS and CTS conform to the overall fold of vertebrate MMPs (47, 93) and are separated by a shallow active-site cleft. The NTS is an α/β-sandwich consisting of a twisted five-stranded pleated β-sheet (strands βI-βV; see Fig. 2A), which is parallel for its first four strands and antiparallel for its lowermost one, βIV. The sheet accommodates on its concave side two α-helices (the “backing helix” αA and the “active-site helix” αB; for numbering and extension of repeating secondary structure elements, see Fig. 2C of Ref. 9). The right-handed twist of the helices coincides with the right-handed twist of the sheet and both helices' axes intersect the strands of the sheet at an angle Ω ≈ −35° (94). The two helices pack against each other interacting through Ala66-Ala70 of αA and Leu149-Ala154 of αB at a crossing angle Ω ≈ −50°, which corresponds to a class II helix interaction (94). The loop connecting strands βIII and βIV (LβIIIβIV) contains the “S-loop” (Gly100-Leu115), which encompasses first a binding site for a structural zinc (Zn998) and, downstream, a binding site for a structural calcium (Ca997; see Fig. 2B). The zinc is tetrahedrally coordinated by His102 Nϵ2, Asp104 Oδ2, His117 Nϵ2, and His133 Nδ1, whereas the calcium is octahedrally coordinated by six oxygens: Asp109 Oδ1, Gly110 O, Thr112 O, Ile114 O, Asp135 Oδ2, and Glu138 Oϵ2 (see Fig. 2B and its legend for details). The presence of calcium is consistent with its crucial role in catalysis (30) and in protein stability (see Fig. 1C). Such calcium is found in mammalian MMP structures (47, 93), but it was not found in previous mature Kly18 structures (see below and Refs. 9 and 28). At Gly162 of the CSBZ, the polypeptide chains take a sharp turn and enters the CTS (Fig. 2A), which mainly contains the “C-terminal helix” αC and the Met-turn, centered on Met173, which forms a hydrophobic base for the catalytic metal-binding site and is required for its integrity in MMPs and metzincins in general (47).
FIGURE 2.

Overall structure of pKly18-E156A. A, ribbon-type plot of pKly18-E156A in standard orientation (35). Depicted are the propeptide (ribbon and carbon atoms in turquoise) and the mature enzyme moiety (β-strands in yellow labeled βI-βV; α-helices in salmon labeled αA-αC; and coils and carbon atoms in tan). Further shown are the catalytic zinc ion (Zn999; bottom magenta sphere), the structural zinc ion (Zn998; top magenta sphere), and the structural calcium ion (Ca997; red sphere), as well as the side chains of the three catalytic zinc ligands (His155, His159, and His165), the Met turn methionine (Met173), the alanine replacing the catalytic glutamate (Ala156), and residues flanking the primary activation cleavage point, Asn34-Tyr35. B, close-up of the window of A as stick model highlighting the structural zinc and calcium sites. Protein segments depicted are Asn101-Gly105 from the first part of the S-loop (carbons in gold), Asp109-Phe119 from the second part of the S-loop (carbons in sandy brown), His131-Glu138 from βV-LβVαB (carbons in tan), and the side chain of Arg22 from the propeptide (carbons in turquoise). The zinc is bound by His102 Nϵ2, Asp104 Oδ2, His117 Nϵ2, and His133 Nδ1 at distances 1.99–2.06 Å, and the calcium is bound by Asp109 Oδ1, Gly110 O, Thr112 O, Ile114 O, Asp135 Oδ2, and Glu138 Oϵ2 at distances 2.34–2.39 Å. These distances agree with standard zinc- (1.99–2.09 Å; (109)) and calcium-binding (2.36–2.39 Å; (109)) distance values for oxygens and nitrogens. C, close-up of A in wall-eye stereo centered on the catalytic zinc after a horizontal ∼30° rotation upwards. Selected hydrogen and ionic bonds (see also Table 3) are depicted as green lines. Residues and ions labeled in A are not labeled here for clarity. The propeptide is shown in cyan to distinguish it from the mature catalytic moiety (in tan/yellow/orange) and its chain direction is pinpointed by a cyan arrow and labels of the N- and C-terminal parts depicted. D, superposition in wall-eye stereo of pKly18-E156A (ribbon in tan for the mature enzyme moiety and in brown for the propeptide, zinc ions in magenta, and calcium ion in red; stick model for the side chains of Ser20-Tyr35 with carbons in brown) and Kly18 (ribbon and zinc ions in pink, see PDB 2XS3, molecule A (9)), which was obtained in a product complex with peptide A-F-T-S bound to the primed side of the cleft (stick model with carbons in gold). Tyr35 is shown for both structures. E, detail of D in wall-eye stereo depicting the large rearrangement of the N terminus at Tyr35 after maturation cleavage at Asn34-Tyr35. The α-amino group of Tyr35 makes a salt bridge with the side chain of Asp187 in the mature enzyme. Aside from Tyr120 and Glu164 (significantly) and Pro122-Ala129 (slightly; see black arrows), maturation does not entail major conformational rearrangement of the rest of the structure.
The active-site cleft contains the catalytic zinc ion (Zn999) at half-width coordinated by the three histidines of the CSBZ (His155, His159, and His165) through their Nϵ2 atoms at distances 2.00–2.05Å (Fig. 2, A and C). The cleft is top-framed on its non-primed side (see Refs. 35 and 95)) by the “upper-rim strand” βIV of the NTS β-sheet, which in MMPs binds substrates in extended conformation from left to right through antiparallel β-ribbon-like interactions. On its primed side, the cleft is top-framed by the final stretch of the S-loop, termed the “bulge-edge segment” (Thr112-Ley115), and bottom-framed by the segment bridging the Met-turn and helix αC. This segment includes the “S1′-wall forming segment” (Pro175-Tyr177) at the front and the “specificity loop” (Gly179-Gln183) at the back. Together with the first turn of the active-site helix αB, the latter structural elements contribute to the size and chemical nature of the S1′ pocket, which confers specificity to Kly18 and also MMPs in general (47, 93), here for medium-sized to bulky hydrophobic residues (30). Side chains participating in pocket shaping include Leu115, Ala116, Thr151, Val152, His155, Leu172, Tyr177, and Lys181.
Inhibition by the Propeptide
The 14-residue propeptide starts at the front right and runs in extended conformation across the active-site cleft, thus blocking access to the cleft, though in the opposite direction to a substrate, i.e. right to left (Fig. 2, A and C). This reverse orientation of the propeptide in the cleft may contribute to attenuate autolysis, as previously suggested for zymogens of cysteine peptidases and mammalian MMPs (39). The interaction with the CD buries a surface of 2,100 ± 35 Å2, which is much larger than the average of monomeric protein-protein domain intra-chain interfaces (1,193 Å2 (96)) but is slightly lower than the range of typical MMP-protein inhibitor interaction surfaces (2,400–2,700 Å2; see Ref. 97). The interaction includes 13 hydrogen bonds, a double salt bridge, one metallorganic bond, and hydrophobic carbon-carbon contacts between eight residues from the propeptide and 11 from the CD (see Table 3). Segments involved include almost the entire propeptide (Arg22-Gly31) and, from the CD, mainly Asn111-Tyr120 from the bulge-edge segment and the upper-rim strand, and Pro175-Tyr177 from the S1′-wall forming segment. Further involved are Tyr106, Ala124, and Glu138 and the zinc-liganding histidine side chains. Four inter-main chain hydrogen bonds form on the primed side of the cleft (two with the S1′-wall forming segment and two with the bulge-edge segment and strand βIV) and three more on the upstream non-primed side (with βIV and LβIVβV; Fig. 2C). In particular, Arg22 contacts the base of the S-loop: it doubly salt bridges Glu138, which is also one of the calcium ligands (see above, Table 3 and Fig. 2B), and hydrogen bonds three carbonyl oxygens of the S-loop, Asn111, Gly113, and Thr112, which, again, is also a calcium ligand. In addition, the Arg22 carbonyl oxygen binds the S1′-wall forming segment and its side chain performs a hydrophobic interaction with Leu115. Accordingly, this residue plays a major role in the stabilization of the Ca997 site and, thus, the zymogen in general, which explains its enhanced stability in response to thermal denaturation (see above). In addition, superposition of pKly18-E156A onto mature Kly18 in complex with a tetrapeptidic cleavage product in the primed side (see below) and human MMP-8 with a modeled substrate traversing its cleft based on inhibitor structures (98) indicates that Arg22 occupies the S3′ position of the cleft.
TABLE 3.
Direct interactions between the propeptide (PP) and the catalytic domain (CD)
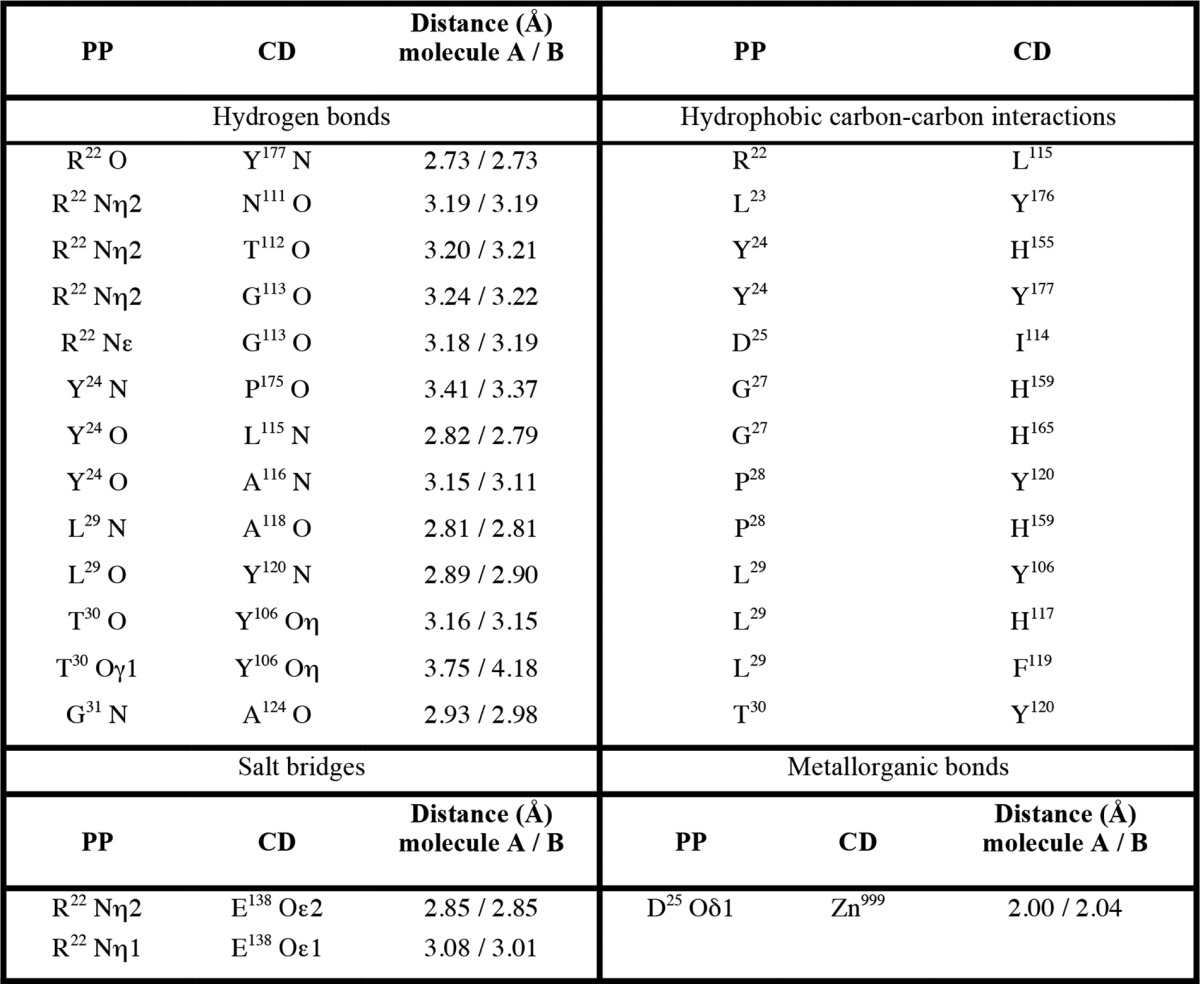
However, the most important interaction of the propeptide with the CD is exerted by Asp25, which approaches the catalytic zinc from the top and monodentately occupies through its Oδ1 atom the fourth position of the tetrahedral coordination sphere of the metal (2.00/2.04 Å apart; Fig. 2C) further to His155, His159, and His165 Nϵ2 atoms. The preceding carbonyl group of Tyr24 binds strand βIV, and its aromatic side chain penetrates the deep hydrophobic S1′ pocket, mainly interacting with the His155 ring face-to-face. The π-rings are ∼3.5 Å apart and parallel but slightly displaced along the ring planes to form a half-overlapping sandwich, which gives rise to an optimal π-stacked structure (99). Downstream in the chain, Pro28 is in a pocket, probably S2, framed by His159, Glu164, and Tyr120, the latter two interact through a tight hydrogen bond (Tyr120 Oη-Glu164 Oϵ2, 2.61 Å). Residue Leu29 is surrounded by the side chains of Tyr106, His117, and Phe119, which may feature S3 (Fig. 2C). After Gly31, the polypeptide abandons the active-site cleft moving outward to reach the primary activation cleavage point, Asn34-Tyr35 (Fig. 2A), after which the chain folds back toward the molecular moiety and enters strand βI of the NTS β-sheet.
A Novel Activation Mechanism in MMPs
Previous work had yielded three structures of mature wild-type Kly18 in complexes with tri- and tetrapeptidic cleavage products, as well as an inhibitory tetrapeptide in the non-primed side of the cleft (PDB 2XS3, 2XS4, and 4IN9 (9, 28)). These were obtained both in the presence and absence of magnesium and showed deviating chain traces for segment Asn53-His57 (LβIαA) in the two molecules found in the asymmetric unit of the magnesium unbound structure (PDB 2XS3 (9)) and in the single molecules found in magnesium-bound (PDB 2XS4 (9)) and inhibitor-bound crystals (PDB 4IN9 (28)). In addition, significant differences were also found in the second half of the S-loop including the bulge-edge segment, which was metal-free in all structures, as the aforementioned magnesium, which coincides with a potassium site in the inhibitor-bound form, was found on the opposite surface of the CD (see Fig. 1, A and C, in Ref. 9, and Fig. 1A in Ref. 28), in a place that suggests little if any functional or structural relevance. In these structures, either an outward- or an inward-folded flap was found for the S-loop (Fig. 1E in Ref. 9 and Fig. 1D in Ref. 28), which suggests intrinsic flexibility of this protein segment to adapt to different substrates. Among the distinct mature Kly18 coordinates, molecule A of the magnesium-unbound structure (PDB 2XS3) was chosen here for comparison with pKly18-E156A as it showed the lowest divergence in the overall chain trace (Fig. 2, D and E).
Superposition revealed that the mature CD is preformed in the zymogen and, with some notable local exceptions (see below), is simply uncovered during maturation by removal of the propeptide, as found in mammalian MMPs (47) and other MPs such as funnelins (79, 82). Removal occurs through cleavage at Asn34-Tyr35, which is solvent exposed on the molecular surface and thus readily accessible for processing (Fig. 2A). This explains why the wild-type zymogen undergoes rapid autolysis, so it cannot be isolated intact (see Ref. 30 and first section of “Results and Discussion”). This was the first cleavage observed in vitro, thus termed primary activation cleavage site, and no further cleavage was detected either within the propeptide or in the CD. The site is consistent with most vertebrate MMPs being activated at X-F/Y bonds, which are found at similar regions in all structures (10). Propeptide removal occurs under loss of a number of protein-protein interactions (see Table 3 and the preceding section), which explains why the mature enzyme is less stable to thermal denaturation (see first section of “Results and Discussion”). In particular, Arg22 plays a key role in stabilizing the Ca997 site (see above), and its removal may contribute to cation-site and S-loop flexibility, leading to metal loss. This site is easily created from the unbound form by two glycine-mediated main chain rotations (peptide flip of bond Thr112-Gly113 and ∼70° rotation of peptide bond Gly110-Asn111), so as to orient the carbonyl oxygens toward the interior, and cation binding should largely compensate for the energetic cost of such minor rearrangement. However, the finding that none of the mature Kly18 structures, which were partially obtained in the presence of calcium (9), contained an intact calcium site supports the requirement of Arg22 as an additional stabilizing factor for site integrity.
Activation further entails that the position occupied by Asp25 Oδ1 in the ligand sphere of the catalytic zinc (see the preceding section) is taken over by a catalytic solvent molecule, which renders a competent active site following an “aspartate-switch” mechanism. Such a competent zinc environment has also been reported for several mature MPs (see e.g. Refs. 64, 80, and 100). To date, aspartate-switch zymogenic mechanisms have been described only for astacins (7, 88) and fragilysins (26), which are only distantly related MPs grouped with MMPs within the metzincins. To verify the function of Asp25 in latency in pKly18, we used mutant pKly18-Y35A (from pKAR8), as the wild-type form (pKAR7) was insoluble. Although this mutant was produced with a yield similar to that of pKly18-E156A and was stable for several days, mutant pKly18-D25A/Y35A (pKAR9) was insoluble. We further assessed the function of Asp25 in full-length karilysin using the slowly autolytic mutant pKly-Y35A (pKAR2), as the reaction in the wild-type is too rapid (30). While pKly-Y35A was essentially intact after 5 days at 37 °C, pKly-D25A/Y35A (pKAR4) had been entirely transformed into the 38- and 18-kDa forms after this time (Fig. 1, E and F). Taken together, these results support the essential role of Asp25 in latency maintenance.
As to further changes upon maturation, segment Pro122-Ala129 from LβIVβV is slightly shifted downwards by ∼2 Å and the side chains of Tyr120 and Glu164 rotate toward the zinc site (Fig. 2E). Activation only entails major rearrangement of the new N-terminal segment Tyr35-Ser40, on the left surface (Fig. 2, D and E), which is rotated downward around bonds C-Cα and Cα-N of Ser40. In this way, this segment nestles in a surface cavity framed by helix αC and the first segment of the CTS between Gly162, and the “family specific residue,” which is a serine in MMPs (1, 101) (here Ser166). This entails that the new α-amino group of Tyr35, which is translated 25 Å, establishes an intra-molecular salt bridge with Asp187 of αC, which is vaguely reminiscent of the activation of trypsin-like serine peptidases (102). Asp187, in turn, is itself further bound to Ser166 and is adjacent to a second aspartate, Asp188, which binds two main chain amides of the Met turn. This electrostatic network is characteristic of physiologically relevant mature MMPs, also referred to as “superactive forms” (47, 103). With the exception of the mature N-terminal fragment, the rest of this electrostatic network is already present in the zymogen (Fig. 2E).
Intensive studies of the activation of mammalian MMPs have produced the structures of pro-MMP-1 (PDB 1SU3 (90)), pro-MMP-3 (PDB 1SLM (104)), pro-MMP-9 (PDB 1L6J(105)), and pro-MMP-2 (PDB 1EAK; (89)). These studies revealed that the mammalian MMP zymogens contain a pre-formed competent protease moiety and true prodomains, which span between 66 and 91 residues, as shown for pro-MMP-2 (Fig. 3A) (47). The prodomains include elongated N-terminal extensions that may interact with ancillary domains, such as the fibronectin type II insertions found in MMP-2 and MMP-9, followed by globular cores of ∼55 residues. These are made up of three α-helices that are arranged around a 3-fold axis with a left-handed twist.
FIGURE 3.

Structural comparison with mammalian pro-MMPs. A, schematic depicting of the structure of pro-MMP-2 (PDB 1EAK (89); MMP-2 residues in italics with superscripted numbering), shown only for its CD (Tyr110-Asp52 in cyan; the fibronectin type-II domains spanning Gln219-Asp92 have been omitted, the black arrows pinpoint the insertion points) and prodomain (Pro43-Asn109 in pink, without the first 11 residues in extended conformation). The orientation displayed corresponds to that of Fig. 2A after applying a horizontal rotation of 15°. Residues of the conserved motif (Pro100-Asp106) key for structural integrity of the inhibitory segment are depicted for their side chains. B, close-up of A after removal of prodomain segment Pro43-Asn66 to provide insight into the interactions of the conserved motif. Key electrostatic interactions are shown as green lines. The catalytic glutamate, Glu404, is replaced by a glutamine, the histidines from the CSBZ are His403, His407, and His413. C and D, scheme depicting the interaction modi of the propeptides of pro-MMP-2 through a cysteine-switch mechanism (C) and pKly18 through an aspartate-switch mechanism (D). The catalytic zinc ions are shown as magenta spheres and relevant interactions are shown as yellow dashed lines.
The prodomain globular core serves as a scaffold to place a downstream peptide, which runs in extended conformation in the opposite direction to a bound substrate and thus blocks the active-site cleft (Fig. 3, A and B). This peptide encompasses the conserved motif involved in cysteine-switch or Velcro latency characteristic of animal and plant MMPs (48–50), 100P-R-C-G-N-P-D106 (MMP-2 residues in italics; see PDB 1EAK and UP P08253), which is equivalent to pKly18 segment 23L-Y-D-N-G-P-L29 (Fig. 3, C and D). Both the cysteine- and aspartate-switch motif show an intricate electrostatic network producing a unique scaffold to interact with the mature catalytic domain moiety. In contrast to pKly18, where the first cleavage occurs in the primary activation cleavage site, however, classical mammalian pro-MMPs are activated by conformational changes in the prodomain induced by cleavage in a so-called “bait region” by several peptidases such as trypsin, plasmin, and other MMPs. Activation follows a “stepwise activation” process to eventually yield the final cleavage site X-F/Y accessible for processing and dissociation of cysteine and zinc to generate a functional active site (48, 49, 51, 106–108). As in Kly18, after cleavage at Asn109-Tyr110, the new N terminus is rearranged and participates in the electrostatic network centered on the conserved aspartate of helix αC, Asp346 in MMP-2.
Conclusions
This examination of the structure and function of the zymogen of the first bacterial MMP to be studied biochemically has uncovered several features of the activation mechanism of pKly18, which are shared with animal and plant MMPs: (i) the relevant cleavage site is X-F/Y; (ii) the scissile bond is located in similar regions of the structure; (iii) activation entails rearrangement of the segment equivalent to Tyr35-Ser40 to yield a salt bridge between the new α-amino group and the first of two conserved aspartates in helix αC; (iv) this aspartate is bound to the family-specific serine; (v) the aspartate immediately downstream binds two main chain amides of the Met turn; (vi) the inhibitory segments run across the cleft in the opposite direction to a genuine substrate and metal blockage occurs through the side chain of an intervening residue, not through a chain terminus; and (vii) the catalytic moiety is largely preformed in the zymogen. All these features are related to the highly conserved CD itself. In contrast, all features of the mechanism related to the segment preceding this conserved CD diverge: (i) in pKly the propeptide spans just 14 residues and does not contain repetitive secondary structure elements, whereas eukaryotic MMPs feature a true protein prodomain that folds into a pseudosymmetric three-helix bundle followed by a segment in extended conformation; (ii) no relevant sequence similarity is found between the proregions; (iii) in eukaryotic MMPs activation occurs through a cysteine-switch mechanism exerted by residues from a conserved sequence motif, whereas in pKly18 this motif is absent and activation follows an aspartate-switch mechanism; (iv) multiple cleavages are apparently required in eukaryotic MMPs to liberate the CD, whereas a single cleavage suffices in pKly; and (v) the prodomain is not required for (re)folding of the catalytic moieties in eukaryotic MMPs, whereas it is in karilysin. In addition, pKly shares parts of its mechanism of latency with otherwise unrelated MPs from the astacin and fragilysin families. Accordingly, this overall novel mechanism unveiled for MMPs supports previous hypotheses, according to which Kly18 originated from an animal MMP CD co-opted through horizontal gene transfer by T. forsythia. This transfer was fostered by the intimate coexistence of the latter with the human blood-irrigated gingival crevice. Subsequently, Kly18 would have evolved in a bacterial environment, where it was furnished with unique flanking domains that contribute to a mechanism of zymogenicity similar to distantly related MPs only (9).
Acknowledgments
We are grateful to the joint IBMB/IRB Automated Crystallography Platform for assistance during crystallization experiments and Robin Rycroft for very helpful contributions to the manuscript. We acknowledge the help provided by local contacts at the ESRF synchrotron. The clone for production of human rhinovirus 3C proteinase was a generous gift from Arie Gerlof (EMBL, Hamburg). Funding for data collection was provided in part by ESRF. The Faculty of Biochemistry, Biophysics and Biotechnology of the Jagiellonian University is a beneficiary of structural funds from the European Union (POIG.02.01.00-12-064/08).
This work was supported in part by European, United States American, Polish, Spanish, and Catalan Grants UMO-2012/04/A/NZ1/00051, UMO-2013/08/T/NZ1/00315, 2137/7.PR-EU/2011/2, 2975/7.PR/13/2014/2, DE09761, DE022597, FP7-HEALTH-2010-261460 “Gums&Joints,” FP7-PEOPLE-2011-ITN-290246 “RAPID,” FP7-HEALTH-2012-306029-2 “TRIGGER,” BFU2012-32862, BIO2013-49320-EXP, CSD2006-00015, and 2014SGR9).
The atomic coordinates and structure factors (code 4R3V) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- MMP
- matrix metalloproteinase
- MP
- metallopeptidase
- ADAM
- a disintegrin and a metallopeptidase
- HR3CP
- human rhinovirus 3C proteinase
- GST
- glutathione S-transferase
- Bistris propane
- 1,3-bis[tris(hydroxymethylmethylamino]propane
- Tm
- temperature of midtransition
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- CSBZ
- consensus sequence for zinc binding
- NTS
- N-terminal upper subdomain moiety
- CD
- catalytic domain
- CTS
- C-terminal lower subdomain
- PDB
- Protein Data Bank.
REFERENCES
- 1. Bode W., Gomis-Rüth F. X., Stöcker W. (1993) Astacins, serralysins, snake venom and matrix metalloproteinases exhibit identical zinc-binding environments (HEXXHXXGXXH and Met-turn) and topologies and should be grouped into a common family, the “metzincins”. FEBS Lett. 331, 134–140 [DOI] [PubMed] [Google Scholar]
- 2. Gomis-Rüth F. X. (2003) Structural aspects of the metzincin clan of metalloendopeptidases. Mol. Biotechnol. 24, 157–202 [DOI] [PubMed] [Google Scholar]
- 3. Gomis-Rüth F. X. (2009) Catalytic domain architecture of metzincin metalloproteases. J. Biol. Chem. 284, 15353–15357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Stöcker W., Grams F., Baumann U., Reinemer P., Gomis-Rüth F. X., McKay D. B., Bode W. (1995) The metzincins: topological and sequential relations between the astacins, adamalysins, serralysins, and matrixins (collagenases) define a superfamily of zinc-peptidases. Protein Sci. 4, 823–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cerdà-Costa N., Gomis-Rüth F. X. (2014) Architecture and function of metallopeptidase catalytic domains. Protein Sci. 23, 123–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Balaban N. P., Rudakova N. L., Sharipova M. R. (2012) Structural and functional characteristics and properties of metzincins. Biochemistry 77, 119–127 [DOI] [PubMed] [Google Scholar]
- 7. Stöcker W., Gomis-Rüth F. X. (2013) Astacins:proteases in development and tissue differentiation. in Proteases: Structure and Function (Brix K., Stöcker W., eds) pp. 235–263, Springer Verlag, Vienna [Google Scholar]
- 8. Gomis-Rüth F. X. (2013) Zinc adamalysins. in Encyclopedia of Metalloproteins (Uversky V. N., Kretsinger R. H., Permyakov E. A., eds) pp. 2345–2349, Springer Verlag, Heidelberg [Google Scholar]
- 9. Cerdà-Costa N., Guevara T., Karim A. Y., Ksiazek M., Nguyen K. A., Arolas J. L., Potempa J., Gomis-Rüth F. X. (2011) The structure of the catalytic domain of Tannerella forsythia karilysin reveals it is a bacterial xenologue of animal matrix metalloproteinases. Mol. Microbiol. 79, 119–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sang Q. A., Douglas D. A. (1996) Computational sequence analysis of matrix metalloproteinases. J. Protein Chem. 15, 137–160 [DOI] [PubMed] [Google Scholar]
- 11. Fanjul-Fernández M., Folgueras A. R., Cabrera S., López-Otín C. (2010) Matrix metalloproteinases: evolution, gene regulation and functional analysis in mouse models. Biochim. Biophys. Acta 1803, 3–19 [DOI] [PubMed] [Google Scholar]
- 12. Marino G., Funk C. (2012) Matrix metalloproteinases in plants: a brief overview. Physiol. Plant 145, 196–202 [DOI] [PubMed] [Google Scholar]
- 13. de Souza S. J., Brentani R. (1993) Sequence homology between a bacterial metalloproteinase and eukaryotic matrix metalloproteinases. J. Mol. Evol. 36, 596–598 [DOI] [PubMed] [Google Scholar]
- 14. Murphy G. J., Murphy G., Reynolds J. J. (1991) The origin of matrix metalloproteinases and their familial relationships. FEBS Lett. 289, 4–7 [DOI] [PubMed] [Google Scholar]
- 15. Jackson B. C., Carpenter C., Nebert D. W., Vasiliou V. (2010) Update of human and mouse forkhead box (FOX) gene families. Hum. Genomics 4, 345–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Massova I., Kotra L. P., Fridman R., Mobashery S. (1998) Matrix metalloproeinases: structures, evolution, and diversification. FASEB J. 12, 1075–1095 [PubMed] [Google Scholar]
- 17. Angerer L., Hussain S., Wei Z., Livingston B. T. (2006) Sea urchin metalloproteases: a genomic survey of the BMP-1/tolloid-like, MMP and ADAM families. Dev. Biol. 300, 267–281 [DOI] [PubMed] [Google Scholar]
- 18. Das S., Mandal M., Chakraborti T., Mandal A., Chakraborti S. (2003) Structure and evolutionary aspects of matrix metalloproteinases: a brief overview. Mol. Cell. Biochem. 253, 31–40 [DOI] [PubMed] [Google Scholar]
- 19. Keeling P. J. (2009) Functional and ecological impacts of horizontal gene transfer in eukaryotes. Curr. Opin. Genet. Dev. 19, 613–619 [DOI] [PubMed] [Google Scholar]
- 20. Anderson M. T., Seifert H. S. (2011) Opportunity and means: horizontal gene transfer from the human host to a bacterial pathogen. mBio 2, e00005–00011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Moncrief J. S., Duncan A. J., Wright R. L., Barroso L. A., Wilkins T. D. (1998) Molecular characterization of the fragilysin pathogenicity islet of enterotoxigenic Bacteroides fragilis. Infect. Immun. 66, 1735–1739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Klein T., Bischoff R. (2011) Active metalloproteases of the A Disintegrin and Metalloprotease (ADAM) family: biological function and structure. J. Proteome Res. 10, 17–33 [DOI] [PubMed] [Google Scholar]
- 23. Takeda S., Takeya H., Iwanaga S. (2012) Snake venom metalloproteinases: structure, function and relevance to the mammalian ADAM/ADAMTS family proteins. Biochim. Biophys. Acta 1824, 164–176 [DOI] [PubMed] [Google Scholar]
- 24. Edwards D. R., Handsley M. M., Pennington C. J. (2008) The ADAM metalloproteinases. Mol. Aspects Med. 29, 258–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gomis-Rüth F. X., Meyer E. F., Kress L. F., Politi V. (1998) Structures of adamalysin II with peptidic inhibitors: implications for the design of tumor necrosis factor α convertase inhibitors. Protein Sci. 7, 283–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Goulas T., Arolas J. L., Gomis-Rüth F. X. (2011) Structure, function and latency regulation of a bacterial enterotoxin potentially derived from a mammalian adamalysin/ADAM xenolog. Proc. Natl. Acad. Sci. U.S.A. 108, 1856–1861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Goulas T., Gomis-Rüth F. X. (2013) 186 Fragilysin. in Handbook of Proteolytic Enzymes (Rawlings N. D., Salvesen G. S., eds) 3rd Ed., pp. 887–891, Academic Press, Oxford [Google Scholar]
- 28. Guevara T., Ksiazek M., Skottrup P. D., Cerdà-Costa N., Trillo-Muyo S., de Diego I., Riise E., Potempa J., Gomis-Rüth F. X. (2013) Structure of the catalytic domain of the Tannerella forsythia matrix metallopeptidase karilysin in complex with a tetrapeptidic inhibitor. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 69, 472–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jusko M., Potempa J., Karim A. Y., Ksiazek M., Riesbeck K., Garred P., Eick S., Blom A. M. (2012) A metalloproteinase karilysin present in the majority of Tannerella forsythia isolates inhibits all pathways of the complement system. J. Immunol. 188, 2338–2349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Karim A. Y., Kulczycka M., Kantyka T., Dubin G., Jabaiah A., Daugherty P. S., Thogersen I. B., Enghild J. J., Nguyen K. A., Potempa J. (2010) A novel matrix metalloprotease-like enzyme (karilysin) of the periodontal pathogen Tannerella forsythia ATCC 43037. Biol. Chem. 391, 105–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Koziel J., Karim A. Y., Przybyszewska K., Ksiazek M., Rapala-Kozik M., Nguyen K. A., Potempa J. (2010) Proteolytic inactivation of LL-37 by karilysin, a novel virulence mechanism of Tannerella forsythia. J. Innate Immun. 2, 288–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Potempa J., Gomis-Rüth F. X., Karim A. Y. (2013) Karilysin in Handbook of Proteolytic Enzymes (Rawlings N. D., Salvesen G. S., eds) 3rd Ed., pp. 883–886, Academic Press, Oxford [Google Scholar]
- 33. Skottrup P. D., Sørensen G., Ksiazek M., Potempa J., Riise E. (2012) A phage display selected 7-mer peptide inhibitor of the Tannerella forsythia metalloprotease-like enzyme karilysin can be truncated to Ser-Trp-Phe-Pro. PloS One 7, e48537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pomerantsev A. P., Pomerantseva O. M., Moayeri M., Fattah R., Tallant C., Leppla S. H. (2011) A Bacillus anthracis strain deleted for six proteases serves as an effective host for production of recombinant proteins. Protein Expr. Purif. 80, 80–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gomis-Rüth F. X., Botelho T. O., Bode W. (2012) A standard orientation for metallopeptidases. Biochim. Biophys. Acta 1824, 157–163 [DOI] [PubMed] [Google Scholar]
- 36. Stöcker W., Bode W. (1995) Structural features of a superfamily of zinc-endopeptidases: the metzincins. Curr. Opin. Struct. Biol. 5, 383–390 [DOI] [PubMed] [Google Scholar]
- 37. Stroud R. M., Kossiakoff A. A., Chambers J. L. (1977) Mechanisms of zymogen activation. Annu. Rev. Biophys. Bioeng. 6, 177–193 [DOI] [PubMed] [Google Scholar]
- 38. Bryan P. N. (2002) Prodomains and protein folding catalysis. Chem. Rev. 102, 4805–4816 [DOI] [PubMed] [Google Scholar]
- 39. Khan A. R., James M. N. (1998) Molecular mechanisms for the conversion of zymogens to active proteolytic enzymes. Protein Sci. 7, 815–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Baker D., Shiau A. K., Agard D. A. (1993) The role of proregions in protein folding. Curr. Opin. Cell Biol. 5, 966–970 [DOI] [PubMed] [Google Scholar]
- 41. Graef C., Schacherl M., Waltersperger S., Baumann U. (2012) Crystal structures of archaemetzincin reveal a moldable substrate-binding site. PloS One 7, e43863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Waltersperger S., Widmer C., Wang M., Baumann U. (2010) Crystal structure of archaemetzincin AmzA from Methanopyrus kandleri at 1.5-Å resolution. Proteins 78, 2720–2723 [DOI] [PubMed] [Google Scholar]
- 43. Ng N., Littler D., Le Nours J., Paton A. W., Paton J. C., Rossjohn J., Beddoe T. (2013) Cloning, expression, purification and preliminary x-ray diffraction studies of a novel AB(5) toxin. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 69, 912–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ng N. M., Littler D. R., Paton A. W., Le Nours J., Rossjohn J., Paton J. C., Beddoe T. (2013) EcxAB Is a founding member of a new family of metalloprotease AB toxins with a hybrid cholera-like B subunit. Structure 21, 2003–2013 [DOI] [PubMed] [Google Scholar]
- 45. Gomis-Rüth F. X. (2013) A different look for AB5 toxins. Structure 21, 1909–1910 [DOI] [PubMed] [Google Scholar]
- 46. Yu A. C., Worrall L. J., Strynadka N. C. (2012) Structural insight into the bacterial mucinase StcE essential to adhesion and immune evasion during enterohemorrhagic E. coli infection. Structure 20, 707–717 [DOI] [PubMed] [Google Scholar]
- 47. Tallant C., Marrero A., Gomis-Rüth F. X. (2010) Matrix metalloproteinases: fold and function of their catalytic domains. Biochim. Biophys. Acta 1803, 20–28 [DOI] [PubMed] [Google Scholar]
- 48. Springman E. B., Angleton E. L., Birkedal-Hansen H., Van Wart H. E. (1990) Multiple modes of activation of latent human fibroblast collagenase: evidence for the role of a Cys73 active-site zinc complex in latency and a “cysteine switch” mechanism for activation. Proc. Natl. Acad. Sci. U.S.A. 87, 364–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Van Wart H. E., Birkedal-Hansen H. (1990) The cysteine switch: a principle of regulation of metalloproteinase activity with potential applicability to the entire matrix metalloproteinase gene family. Proc. Natl. Acad. Sci. U.S.A. 87, 5578–5582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Vallee B. L., Auld D. S. (1990) Zinc coordination, function, and structure of zinc enzymes and other proteins. Biochemistry 29, 5647–5659 [DOI] [PubMed] [Google Scholar]
- 51. Rosenblum G., Meroueh S., Toth M., Fisher J. F., Fridman R., Mobashery S., Sagi I. (2007) Molecular structures and dynamics of the stepwise activation mechanism of a matrix metalloproteinase zymogen: challenging the cysteine switch dogma. J. Am. Chem. Soc. 129, 13566–13574 [DOI] [PubMed] [Google Scholar]
- 52. Loechel F., Overgaard M. T., Oxvig C., Albrechtsen R., Wewer U. M. (1999) Regulation of human ADAM 12 protease by the prodomain: evidence for a functional cysteine switch. J. Biol. Chem. 274, 13427–13433 [DOI] [PubMed] [Google Scholar]
- 53. Grams F., Huber R., Kress L. F., Moroder L., Bode W. (1993) Activation of snake venom metalloproteinases by a cysteine switch-like mechanism. FEBS Lett. 335, 76–80 [DOI] [PubMed] [Google Scholar]
- 54. Leonard J. D., Lin F., Milla M. E. (2005) Chaperone-like properties of the prodomain of TNFα-converting enzyme (TACE) and the functional role of its cysteine switch. Biochem. J. 387, 797–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tallant C., García-Castellanos R., Seco J., Baumann U., Gomis-Rüth F. X. (2006) Molecular analysis of ulilysin, the structural prototype of a new family of metzincin metalloproteases. J. Biol. Chem. 281, 17920–17928 [DOI] [PubMed] [Google Scholar]
- 56. Gomis-Rüth F. X., Trillo-Muyo S., Stöcker W. (2012) Functional and structural insights into astacin metallopeptidases. Biol. Chem. 393, 1027–1041 [DOI] [PubMed] [Google Scholar]
- 57. Sterchi E. E., Stöcker W., Bond J. S. (2008) Meprins, membrane-bound and secreted astacin metalloproteinases. Mol. Aspects Med. 29, 309–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bond J. S., Beynon R. J. (1995) The astacin family of metalloendopeptidases. Protein Sci. 4, 1247–1261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Goulas T., Cuppari A., García-Castellanos R., Snipas S., Glockshuber R., Arolas J. L., Gomis-Rüth F. X. (2014) The pCri system: a vector collection for recombinant protein expression and purification. PloS One 9, e112643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ericsson U. B., Hallberg B. M., Detitta G. T., Dekker N., Nordlund P. (2006) Thermofluor-based high-throughput stability optimization of proteins for structural studies. Anal. Biochem. 357, 289–298 [DOI] [PubMed] [Google Scholar]
- 61. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kabsch W. (2010) Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. D Biol. Crystallogr. 66, 133–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Bode W., Gomis-Rüth F. X., Huber R., Zwilling R., Stöcker W. (1992) Structure of astacin and implications for activation of astacins and zinc-ligation of collagenases. Nature 358, 164–167 [DOI] [PubMed] [Google Scholar]
- 65. Gomis-Rüth F. X., Stöcker W., Huber R., Zwilling R., Bode W. (1993) Refined 1.8-Å X-ray crystal structure of astacin, a zinc-endopeptidase from the crayfish Astacus astacus L. Structure determination, refinement, molecular structure and comparison with thermolysin. J. Mol. Biol. 229, 945–968 [DOI] [PubMed] [Google Scholar]
- 66. Cohen S. X., Ben Jelloul M., Long F., Vagin A., Knipscheer P., Lebbink J., Sixma T. K., Lamzin V. S., Murshudov G. N., Perrakis A. (2008) ARP/wARP and molecular replacement: the next generation. Acta Crystallogr. D Biol. Crystallogr. 64, 49–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Debreczeni J. É., Emsley P. (2012) Handling ligands with Coot. Acta Crystallogr. D Biol. Crystallogr. 68, 425–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., Adams P. D. (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Smart O. S., Womack T. O., Flensburg C., Keller P., Paciorek W., Sharff A., Vonrhein C., Bricogne G. (2012) Exploiting structure similarity in refinement: automated NCS and target-structure restraints in BUSTER. Acta Crystallogr. D Biol. Crystallogr. 68, 368–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Blanc E., Roversi P., Vonrhein C., Flensburg C., Lea S. M., Bricogne G. (2004) Refinement of severely incomplete structures with maximum likelihood in BUSTER-TNT. Acta Crystallogr. D Biol. Crystallogr. 60, 2210–2221 [DOI] [PubMed] [Google Scholar]
- 72. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. (2004) UCSF Chimera: a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 73. Krissinel E., Henrick K. (2004) Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 60, 2256–2268 [DOI] [PubMed] [Google Scholar]
- 74. Kabsch W. (1976) A solution for the best rotation to relate two sets of vectors. Acta Crystallogr. A 32, 922–923 [Google Scholar]
- 75. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Chen V. B., Arendall W. B., 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J.-S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., Warren G. L. (1998) Crystallography & NMR System: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 78. Karplus P. A., Diederichs K. (2012) Linking crystallographic model and data quality. Science 336, 1030–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Gomis-Rüth F. X. (2008) Structure and mechanism of metallocarboxypeptidases. Crit. Rev. Biochem. Mol. Biol. 43, 319–345 [DOI] [PubMed] [Google Scholar]
- 80. Matthews B. W. (1988) Structural basis of the action of thermolysin and related zinc peptidases. Acc. Chem. Res. 21, 333–340 [Google Scholar]
- 81. Vendrell J., Querol E., Avilés F. X. (2000) Metallocarboxypeptidases and their protein inhibitors. Structure, function and biomedical properties. Biochim. Biophys. Acta 1477, 284–298 [DOI] [PubMed] [Google Scholar]
- 82. Arolas J. L., Gomis-Rüth F. X. (2013) Zinc metallocarboxypeptidases. in Encyclopedia of Metalloproteins (Uversky V. N., Kretsinger R. H., Permyakov E. A., eds) pp. 2473–2479, Springer Verlag, Heidelberg, Germany [Google Scholar]
- 83. Wetmore D. R., Hardman K. D. (1996) Roles of the propeptide and metal ions in the folding and stability of the catalytic domain of stromelysin (matrix metalloproteinase 3). Biochemistry 35, 6549–6558 [DOI] [PubMed] [Google Scholar]
- 84. Gonzales P. E., Solomon A., Miller A. B., Leesnitzer M. A., Sagi I., Milla M. E. (2004) Inhibition of the tumor necrosis factor-α-converting enzyme by its prodomain. J. Biol. Chem. 279, 31638–31645 [DOI] [PubMed] [Google Scholar]
- 85. Fotouhi N., Lugo A., Visnick M., Lusch L., Walsky R., Coffey J. W., Hanglow A. C. (1994) Potent peptide inhibitors of stromelysin based on the prodomain region of matrix metalloproteinases. J. Biol. Chem. 269, 30227–30231 [PubMed] [Google Scholar]
- 86. Stetler-Stevenson W. G., Talano J. A., Gallagher M. E., Krutzsch H. C., Liotta L. A. (1991) Inhibition of human type IV collagenase by a highly conserved peptide sequence derived from its prosegment. Am. J. Med. Sci. 302, 163–170 [DOI] [PubMed] [Google Scholar]
- 87. Woessner J. F., Jr., Nagase H. (2000) Matrix metalloproteinases and TIMPs (Protein Profile Series) (Sheterline P., ed) Oxford University Press, New York [Google Scholar]
- 88. Guevara T., Yiallouros I., Kappelhoff R., Bissdorf S., Stöcker W., Gomis-Rüth F. X. (2010) Proenzyme structure and activation of astacin metallopeptidase. J. Biol. Chem. 285, 13958–13965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Morgunova E., Tuuttila A., Bergmann U., Isupov M., Lindqvist Y., Schneider G., Tryggvason K. (1999) Structure of human pro-matrix metalloproteinase-2: activation mechanism revealed. Science 284, 1667–1670 [DOI] [PubMed] [Google Scholar]
- 90. Jozic D., Bourenkov G., Lim N. H., Visse R., Nagase H., Bode W., Maskos K. (2005) X-ray structure of human pro-MMP-1: new insights into procollagenase activation and collagen binding. J. Biol. Chem. 280, 9578–9585 [DOI] [PubMed] [Google Scholar]
- 91. Demidyuk I. V., Gromova T. Y., Polyakov K. M., Melik-Adamyan W. R., Kuranova I. P., Kostrov S. V. (2010) Crystal structure of protealysin precursor: insights into propeptide function. J. Biol. Chem. 285, 2003–2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Gao X., Wang J., Yu D. Q., Bian F., Xie B. B., Chen X. L., Zhou B. C., Lai L. H., Wang Z. X., Wu J. W., Zhang Y. Z. (2010) Structural basis for the autoprocessing of zinc metalloproteases in the thermolysin family. Proc. Natl. Acad. Sci. U.S.A. 107, 17569–17574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Bode W., Maskos K. (2003) Structural basis of the matrix metalloproteinases and their physiological inhibitors, the tissue inhibitors of metalloproteinases. Biol. Chem. 384, 863–872 [DOI] [PubMed] [Google Scholar]
- 94. Chothia C., Levitt M., Richardson D. (1977) Structure of proteins: packing of α-helices and pleated sheets. Proc. Natl. Acad. Sci. U.S.A. 74, 4130–4134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Schechter I., Berger A. (1967) On the size of active site in proteases:. I. papain. Biochem. Biophys. Res. Commun. 27, 157–162 [DOI] [PubMed] [Google Scholar]
- 96. Jones S., Marin A., Thornton J. M. (2000) Protein domain interfaces: characterization and comparison with oligomeric protein interfaces. Protein Eng. 13, 77–82 [DOI] [PubMed] [Google Scholar]
- 97. Batra J., Soares A. S., Mehner C., Radisky E. S. (2013) Matrix metalloproteinase-10/TIMP-2 structure and analyses define conserved core interactions and diverse exosite interactions in MMP/TIMP complexes. PloS One 8, e75836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Grams F., Reinemer P., Powers J. C., Kleine T., Pieper M., Tschesche H., Huber R., Bode W. (1995) X-ray structures of human neutrophil collagenase complexed with peptide thiol inhibitors: implications for substrate binding and rational drug design. Eur. J. Biochem. 228, 830–841 [DOI] [PubMed] [Google Scholar]
- 99. Hunter C. A., Sanders J. K. M. (1990) The natutre of π-π interactions. J. Am. Chem. Soc. 112, 5525–5534 [Google Scholar]
- 100. Gomis-Rüth F. X., Kress L. F., Bode W. (1993) First structure of a snake venom metalloproteinase: a prototype for matrix metalloproteinases/collagenases. EMBO J. 12, 4151–4157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Jiang W., Bond J. S. (1992) Families of metalloendopeptidases and their relationships. FEBS Lett. 312, 110–114 [DOI] [PubMed] [Google Scholar]
- 102. Bode W. (1979) Aktivierung, Aktivität und Inhibierung des Rindertrypsins. Die Naturwissenschaften 66, 251–258 [DOI] [PubMed] [Google Scholar]
- 103. Reinemer P., Grams F., Huber R., Kleine T., Schnierer S., Piper M., Tschesche H., Bode W. (1994) Structural implications for the role of the N terminus in the “superactivation” of collagenases: a crystallographic study. FEBS Lett. 338, 227–233 [DOI] [PubMed] [Google Scholar]
- 104. Becker J. W., Marcy A. I., Rokosz L. L., Axel M. G., Burbaum J. J., Fitzgerald P. M., Cameron P. M., Esser C. K., Hagmann W. K., Hermes J. D., Springer J. P. (1995) Stromelysin-1: three-dimensional structure of the inhibited catalytic domain and of the C-truncated proenzyme. Protein Sci. 4, 1966–1976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Elkins P. A., Ho Y. S., Smith W. W., Janson C. A., D'Alessio K. J., McQueney M. S., Cummings M. D., Romanic A. M. (2002) Structure of the C-terminally truncated human ProMMP9, a gelatin-binding matrix metalloproteinase. Acta Crystallogr. D Biol. Crystallogr. 58, 1182–1192 [DOI] [PubMed] [Google Scholar]
- 106. Cha J., Pedersen M. V., Auld D. S. (1996) Metal and pH dependence of heptapeptide catalysis by human matrilysin. Biochemistry 35, 15831–15838 [DOI] [PubMed] [Google Scholar]
- 107. Nagase H. (1997) Activation mechanisms of matrix metalloproteinases. Biol. Chem. 378, 151–160 [PubMed] [Google Scholar]
- 108. Nagase H., Enghild J. J., Suzuki K., Salvesen G. (1990) Stepwise activation mechanisms of the precursor of matrix metalloproteinase 3 (stromelysin) by proteinases and (4-aminophenyl)mercuric acetate. Biochemistry 29, 5783–5789 [DOI] [PubMed] [Google Scholar]
- 109. Harding M. M. (2006) Small revisions to predicted distances around metal sites in proteins. Acta Crystallogr. D Biol. Crystallogr. 62, 678–682 [DOI] [PubMed] [Google Scholar]
- 110. Weiss M. S. (2001) Global indicators of X-ray quality. J. Appl. Crystallogr. 34, 130–135 [Google Scholar]
- 111. Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62, 72–82 [DOI] [PubMed] [Google Scholar]