

Background: The bioactive metabolites of curcumin are not well defined.
Results: Using [14C]curcumin as tracer, degradation products and unstable reaction intermediates were isolated and identified.
Conclusion: The spontaneous degradation of curcumin is an autoxidation that yields electrophilic and nucleophilic products.
Significance: The unexpected chemical diversity of its metabolites may explain the polypharmacology of curcumin.
Keywords: Analytical Chemistry, Isotopic Tracer, Mass Spectrometry (MS), Metabolism, Natural Product, Nuclear Magnetic Resonance (NMR), Polyphenol, Quinone, Radical
Abstract
Curcumin is a dietary anti-inflammatory and chemopreventive agent consisting of two methoxyphenol rings connected by a conjugated heptadienedione chain. Curcumin is unstable at physiological pH and rapidly degrades in an autoxidation reaction to a major bicyclopentadione product in which the 7-carbon chain has undergone oxygenation and double cyclization. Early degradation products (but not the final bicyclopentadione) mediate topoisomerase poisoning and possibly many other activities of curcumin, but it is not known how many and what autoxidation products are formed, nor their mechanism of formation. Here, using [14C2]curcumin as a tracer, seven novel autoxidation products, including two reaction intermediates, were isolated and identified using one- and two-dimensional NMR and mass spectrometry. The unusual spiroepoxide and vinylether reaction intermediates are precursors to the final bicyclopentadione product. A mechanism for the autoxidation of curcumin is proposed that accounts for the addition and exchange of oxygen that have been determined using 18O2 and H218O. Several of the by-products are formed from an endoperoxide intermediate via reactions that are well precedented in lipid peroxidation. The electrophilic spiroepoxide intermediate formed a stable adduct with N-acetylcysteine, suggesting that oxidative transformation is required for biological effects mediated by covalent adduction to protein thiols. The spontaneous autoxidation distinguishes curcumin among natural polyphenolic compounds of therapeutic interest; the formation of chemically diverse reactive and electrophilic products provides a novel paradigm for understanding the polypharmacological effects of curcumin.
Introduction
The diphenol curcumin, extracted from the rhizomes of the turmeric plant, is used as a coloring agent and preservative in the spice mix curry. In addition to culinary uses, curcumin shows antioxidant, anti-inflammatory, and anti-cancer bioactivities in vitro (1). Curcumin is considered a “polypharmacological” agent because of the plethora of in vitro cellular effects, with review articles listing in excess of 100 distinct targets (2). In animal models, dietary curcumin significantly reduced the incidence and size of colonic tumors and glioblastoma, as well as joint inflammation (3–7). After encouraging and promising in vitro data, animal studies, and apparently safe use as a dietary agent for centuries, curcumin is currently being tested in over 100 clinical trials.
The polypharmacology of curcumin has been linked to its keto-ene moiety acting as a Michael acceptor (8, 9), the β-dicarbonyl as a metal chelator (10), and the phenolic hydroxyl as a H-donor/antioxidant (11, 12). Whether these structural features are sufficient to account for the multitude of diverse biological effects in vitro is debatable. Thus, to rationalize the polypharmacology as well as the discrepancy of low plasma levels of curcumin with the observed in vivo effects, a hypothesis emerged that biological effects are mediated, at least in part, by metabolites (Fig. 1) (13). A similar hypothesis has been suggested for ellagitannins and their urolithin metabolites (14), as well as green tea catechins and their metabolites (15). In contrast to the bioactivity of the ellagitannin and catechin metabolites, however, the known reduced, conjugated, or cleaved metabolites of curcumin are inactive or less active than the parent curcumin (16–19).
FIGURE 1.

Pathways of metabolism of curcumin.
Recently, a novel transformation of curcumin was discovered and identified as an autoxidation reaction leading to oxygen incorporation and the formation of a bicyclopentadione derivative of curcumin (20). Autoxidative transformation is the major pathway of degradation of curcumin at physiological pH in vitro (21). Biological relevance for this transformation was found in the topoisomerase poisoning activity of curcumin (22, 23). Topoisomerase poisoning is the therapeutic mechanism of many anticancer drugs in clinical use (24, 25). Topoisomerase poisoning by curcumin required oxidative transformation to an active compound with short half-life (26). These studies indicated that unstable oxidative transformation products are an active principle rather than the parent curcumin.
The oxidative transformation to the bicyclopentadione is intriguing, but crucial mechanistic questions about the double cyclization and oxygenation reaction remain to be elucidated (20). Furthermore, for a complete evaluation of the biological relevance of oxidative transformation, it is necessary to identify all reaction products and to develop methods for the isolation of unstable reaction intermediates. Here we describe the isolation and identification of unstable spiroepoxide and vinylether intermediates, as well as additional novel products of the autoxidation of curcumin. Isotopic labeling studies were performed to determine their mechanism of formation. The by-products, intermediates, and the final bicyclopentadione comprise a number of diverse structural elements that are likely contributors to the polypharmacology of curcumin.
EXPERIMENTAL PROCEDURES
Materials
Curcumin was synthesized as previously described (27). A 5 mm stock solution of curcumin in ethanol was prepared on the day of the experiments. 18O-Labeled water (97 atom % 18O) was obtained from Isotec. Oxygen-18O2 (99 atom % 18O) was obtained from Sigma. [14C]Methyl iodide (2 mCi/mmol) was from American Radiolabeled Chemicals, Inc. (St. Louis, MO). [d3]Methyl iodide (99.5%) and all other reagents were from Sigma. [14C2]Curcumin and 3′-OCD3,4″-O-methylcurcumin were prepared as described with appropriate modifications (28).
Autoxidation Reactions
Autoxidation of [14C2]curcumin (30 μm; 4 mCi/mmol) was conducted in 500 μl of 10 mm NH4OAc buffer, pH 7.4, at room temperature. After 45 min, 250 μl of the reaction mixture were analyzed without extraction using RP-HPLC.2
For preparative isolation of the autoxidation products, four reactions (50 ml of 10 mm NH4OAc buffer, pH 7.4, 50 μm curcumin) were conducted in parallel. For isolation of 6 and 7, the reaction was allowed to proceed for 20 or 60 min, respectively, followed by extraction without acidification. For all other products a reaction time of 3 h was used. Samples were extracted using a preconditioned 2 g Waters HLB cartridge and eluted with two aliquots of 2 ml of acetonitrile. The solvent was evaporated under a stream of nitrogen, and the sample was dissolved in acetonitrile/water 1:1 for RP-HPLC analysis.
Dihydroxycyclopentadione Isomer 1 (2a)
1H NMR (CD3OD, 600 MHz): δH 6.59 (2H, dd, J = 8.1, 1.9 Hz, H6′ and H6″), 6.55 (2H, d, J = 8.1 Hz, H5′ and H5″), 6.41 (2H, d, J = 1.8 Hz, H2′ and H2″), 4.7 (1H, H4), 3.76 (2H, d, J = 3.2 Hz, H1 and H7), 3.62 (6H, s, OCH3), 3.34 (2H, d, J = 3.2 Hz, H2 and H6). 13C NMR (CD3OD, 150 MHz): δC 206 (2, C3 and C5), 147 (2, C3′ and C3″), 144 (2, C4′ and C4″), 132 (2, C1′ and C1″), 120 (2, C2′ and C2″), 114 (2, C6′ and C6″), 112 (2, C5′ and C5″), 54.8 (2, OCH3), 47 (2, C2 and C6), 46 (2, C1 and C7).
Dihydroxycyclopentadione Isomer 2 (2b)
1H NMR (CD3OD, 600 MHz): δH 6.69 (1H, d, J = 1.2 Hz, H2′), 6.67 (1H, d, J = 7.9 Hz, H5′), 6.65 (1H, dd, J = 8.1, 1.6 Hz, H6′), 6.61 (1H, d, J = 8.0 Hz, H5″), 6.59 (1H, d, J = 1.6 Hz, H2″), 6.52 (1H, dd, J = 8.1, 1.7 Hz, H6″), 4.51 (1H, d, J = 9.0 Hz, H1), 4.34 (1H, d, J = 3.4 Hz, H7), 3.84 (1H, s, H4), 3.79 (3H, s, OCH3), 3.74 (3H, s, OCH3), 2.52, (1H, dd, J = 3.9, 2.8 Hz, H6), 2.30 (1H, dd, J = 8.9, 2.9 Hz, H2). 13C NMR (CD3OD, 150 MHz): δC 207 (C3), 119 (C6′), 118 (C6″), 114 (2, C5′ and C5″), 109 (2, C2′ and C2″), 77 (C1), 73, C7), 55 (2, OCH3), 52 (C6), 51 (C2); expected signals for carbons 4, 5, 1′, 3′, 4′, 1″, 3″, and 4″ were not seen.
Hemiacetalcyclopentadione (3)
1H NMR (CD3OD, 600 MHz): δH 6.95 (1H, d, J = 1.6 Hz, H2′), 6.85 (1H, dd, J = 8.1, 1.6 Hz, H6′), 6.72 (1H, d, J = 8.1 Hz, H5′), 6.52 (1H, d, J = 8.1 Hz, H5″), 6.29 (1H, dd, J = 8.2, 1.9 Hz, H6″), 6.01 (1H, d, J = 1.6 Hz, H2″), 5.23 (1H, d, J = 2.4 Hz, H1), 5.07 (1H, s, H4), 3.75 (3H, s, OCH3), 3.58 (3H, s, OCH3), 3.51 (1H, d, J = 2.2 Hz, H6), 2.57 (1H, s, H2). 13C NMR (CD3OD, 150 MHz): δC 147 (C3′), 146.5 (C3″), 145 (C4′), 143.6 (C4″), 135.4 (C1′), 134.8 (C1″), 119 (C6″), 118 (C6′), 112 (2, C5′ and C5″), 111 (C2″), 108 (C2′), 70 (C1), 64 (C2), 55.4 (2, OCH3), 50 (C6); expected signals for carbons 3, 5, and 7 were not seen.
Hydroxyketocyclopentadione (4)
1H NMR (CD3OD, 600 MHz): δH 7.39 (1H, dd, J = 8.4, 1.6 Hz, H6″), 7.31 (1H, d, J = 1.4 Hz, H2″) 6.80 (1H, d, J = 1.2 Hz, H2′), 6.75 (1H, d, J = 8.3 Hz, H5″), 6.70 (1H, dd, J = 8.0, 1.3 Hz, H6′), 6.54 (1H, d, J = 8.1 Hz, H5′), 4.66 (1H, s, H6), 5.26 (1H, s, H1), 3.86 (3H, s, OCH3), 3.57 (3H, s, OCH3), 3.23 (1H, s, H2); expected signals for H4 and H7 were not seen. 13C NMR (CD3OD, 150 MHz): δC 151 (C4″), 147 (C3″), 146.5 (C3′), 135.7 (C1′), 134.5 (C1″), 124 (C6″), 117.5 (C6′), 114 (2, C5′ and C5″), 111 (C2″), 108 (C2′), 69.6 (C1), 58 (C2), 54 (2, OCH3), 53 (C6); expected signals for carbons 3, 4, 5, and 7 were not seen.
Cyclobutylcyclopentadione (5)
1H NMR (CD3OD, 600 MHz): δH 6.71 (2H, d, J = 1.7 Hz, H2′ and H2″), 6.66 (2H, d, J = 8.1 Hz, H5′ and H5″), 6.54 (2H, dd, J = 8.0, 1.7 Hz, H6′ and H6″), 4.55 (2H, s, H1 and H7), 3.79 (6H, s, OCH3), 2.64 (2H, d, J = 2.5 Hz, H2 and H6); expected signal for H4 was not seen. 13C NMR (CD3OD, 150 MHz): δC 206 (2, C3 and C5), 146.7 (2, C3′ and C3″), 145 (2, C4′ and C4″), 134 (2, C1′ and C1″), 72 (2C, C1 and C7), 54.7 (2C, OCH3), 51 (2C, C2 and C6); expected signal for C4 was not seen.
Spiroepoxidecyclopentadione Isomer 1 (6a)
1H NMR (CD3OD, 600 MHz): δH 7.06 (1H, d, J = 1.9 Hz, H2″), 6.89 (1H, dd, J = 8.1, 2.0 Hz, H6″), 6.74 (1H, d, J = 8.2 Hz, H5″), 6.52 (1H, dd, J = 10.0, 2.5 Hz, H6′), 6.32, d, J = 9.9 Hz, H5′), 5.25 (1H, d, J = 2.3 Hz, H2′), 5.20 (1H, d, J = 2.7 Hz, H7), 4.90 (1H, s, H4), 3.80 (3H, s, OCH3″), 3.60 (3H, s, OCH3′), 3.28 (1H, d, J = 9.0 Hz, H1), 2.91 (1H, dd, J = 2.7, 2.2 Hz, H6), 2.63 (1H, dd, J = 8.9, 1.9 Hz, H2). 13C NMR (CD3OD, 150 MHz): δC 204 (C5), 203 (C3), 184 (C4′), 153.5 (C3′), 147.3 (C3″), 144.7 (C4″), 135.5 (C1″), 130.2 (C5′), 118.0 (C6″), 114.0 (C5″), 111.5 (C2′), 109.5 (C2″), 103.5 (C4), 71.5 (C7), 68.2 (C1), 59.2 (C1′), 57.0 (C6), 55.0 (OCH3″), 54.0 (OCH3′), 44.5 (C2).
Spiroepoxidecyclopentadione Isomer 2 (6b)
1H NMR (CD3OD, 600 MHz): δH 7.01 (1H, d, J = 1.7 Hz, H2″), 6.84 (1H, dd, J = 8.1, 1.9 Hz, H6″), 6.72 (1H, d, J = 8.1 Hz, H5″), 6.59 (1H, dd, J = 9.9, 2.5 Hz, H6′), 6.36, d, J = 9.9 Hz, H5′), 5.28 (1H, d, J = 2.4 Hz, H2′), 4.82 (1H, d, J = 27.6 Hz, H7), 4.76 (1H, s, H4), 3.80 (3H, s, OCH3′), 3.60 (3H, s, OCH3″), 3.40 (1H, d, J = 8.3 Hz, H1), 2.97 (1H, dd, J = 7.7, 2.1 Hz, H6), 2.37 (1H, dd, J = 8.3, 2.0 Hz, H2). 13C NMR (CD3OD, 150 MHz): δC 205 (C5), 202 (C3), 181 (C4′), 153.5 (C3′), 149.8 (C6′), 147 (C3″), 1346 (C4″), 130.0 (C5′), 119.8 (C6″), 114.0 (C5″), 111 (C2′), 110.5 (C2″), 101.7 (C4), 76 (C7), 67.6 (C1), 59.5 (C1′), 55 (C6), 54.0 (2, OCH3), 42.0 (C2).
Vinylethercyclopentadione (7b)
1H NMR (CD3OD, 600 MHz): δH 7.15 (1H, d, J = 1.2 Hz, H1), 6.80 (1H, d, J = 2.0 Hz, H2″), 6.74 (1H, d, J = 8.1 Hz, H5″), 6.70 (1H, d, J = 8.6 Hz, H5′), 6.68 (1H, dd, J = 4.0, 1.4 Hz, H6), 6.65 (1H, dd, J = 81, 1.8 Hz, H6″), 6.43 (1H, d, J = 2.7 Hz, H2′), 6.37 (1H, dd, J = 8.6, 2.7 Hz, H6′), 5.30 (1H, d, J = 4.1 Hz, H7), 4.60, (1H, s, H4), 3.76 (3H, s, OCH3′), 3.73 (3H, s, OCH3″). 13C NMR (CD3OD, 150 MHz): δC 210.7 (C5), 187.6 (C3), 147.3 (2C, C3′ and C3″), 143.5 (C4′), 142.7 (C4″), 142.3 (C1), 118.9 (C6″), 114.2 (C5″), 113.5 (C5′), 111.4 (C6′), 109.8 (C2″), 108.7 (C2), 104.1 (C2′), 103.6 (C4), 73.1 (C7), 55.3 (2, OCH3), 55.2 (C6).
Bicyclopentadione (Major Isomer; 8a)
1H NMR (CD3COCD3, 600 MHz): δH 6.92 (1H, d, J = 2.7 Hz, H2′), 6.87 (1H, d, J = 8.6 Hz, H5′), 6.83 (1H, d, J = 1.8 Hz, H2″), 7.72 (1H, dd, J = 1.9, 8.4 Hz, H6′), 6.71 (1H, dd, J = 2.7, 8.6 Hz, H6″), 6.68 (1H, d, J = 8.1 Hz, H5″), 5.89 (1H, d, J = 3.7 Hz, H1), 5.4 (1H, d, J = 8.2 Hz, H7), 4.88 (1H, d, J = 0.7 Hz, H4), 3.86 (3H, s, OCH3′), 3.75 (3H, s, OCH3″), 3.61 (1H, dd, J = 6.5, 0.8 Hz, H2), 3.25 (1H, dd, J = 8.2, 6.4 Hz, H6). 13C NMR (CD3CN, 150 MHz): δC 201.9 (C5), 188.3 (C3), 148.6 (C3′), 148.3 (C1′), 147.2 (C3″), 146.1 (C4″), 144.9 (C4′), 129.9 (C1″), 120.3 (C6″), 115.5 (C5′), 114.6 (C5″), 112.8 (C6′), 110.9 (C2″), 109.3 (4), 105.2 (C2′), 97.5 (C1), 79.9 (C7), 56.5 (OCH3′), 56.1 (OCH3″), 55.4 (C2), 54.7 (C6).
Bicyclopentadione (Minor Isomers; 8b and 8c)
Isomers were analyzed as a mixture; the signals could not be assigned to an isomer (b/c) and are listed as isomer 1 or 2 using superscript numbers or unassigned): 1H NMR (CD3CN, 600 MHz): δH 7.121 (1H, d, J = 1.3 Hz, H2″), 7.002 (1H, d, J = 1.4 Hz, H2″), 6.881 (1H, dd, J = 7.9, 1.3 Hz, H6″), 6.862 (1H, m, H6″),6.83 (3H, m, H5′, H2′), 6.78 (1H, d, J = 7.9 Hz, H5″), 6.77 (1H, d, J = 7.9 Hz, H5″); 6.75 (1H, d, J = 2.5 Hz, H2′), 6.66 (1H, dd, J = 2.6, 8.5 Hz, H6′), 6.62 (1H, dd, J = 2.6, 8.6 Hz, H6′), 5.741 (1H, s, H1), 5.682 (1H, d, J = 5.8 Hz, H1), 5.021 (1H, d, J = 3.9 Hz, H7), 4.982 (1H, d, J = 7.1 Hz, H7), 3.982 (1H, dd, J = 5.9, 8.3 Hz, H2), 3.85 (3H, s, OCH3), 3.84 (3H, s, OCH3), 3.83 (3H, s, OCH3), 3.82 (3H, s, OCH3), 3.621 (1H, d, J = 7.1 Hz, H2), 3.321 (1H, dd, J = 4.0, 7.0 Hz, H6), 3.152 (1H, dd, J = 7.4, 8.2 Hz, H6).
Diguaiacol (9)
1H NMR (CD3COCD3, 600 MHz): δH 7.66 (1H, s, OH), 7.16 (2H, d, J = 2.0 Hz, H1 and H1′), 7.04 (2H, dd, J = 8.2, 2.0 Hz, H6 and H6′), 6.85 (2H, d, J = 8.2 Hz, H5 and H5′) 3.90 (6H, s, OCH3).
Preparation of 18O-Labeled Products
For LC-MS analyses, reactions were conducted in 1 ml of NH4OAc buffer, pH 7.4, made from a 1:1 mixture of H2O and H218O. For NMR analysis of incorporation of 18O from H218O into 8, two 50-ml reactions were conducted. After 20 min (when 6 is the major product present), the reactions were extracted (2 g HLB cartridge) and eluted with MeOH. The solvent was evaporated under a stream of nitrogen, and the sample was redissolved in 500 μl of H218O buffered at pH 7.4 with 10 mm NH4OAc. After 3 h, the reaction was acidified and combined with a similar scale reaction conducted in H2O in parallel. Product 8 was isolated using RP-HPLC and prepared for 13C NMR analysis.
For autoxidation under 18O2 atmosphere two 50-ml aliquots of buffer were degassed and saturated with 18O2. Curcumin, 500 μl from a 5 mm stock solution in ethanol was added. After 3 h, the reaction mixture was acidified and extracted, and 8a was isolated using RP-HPLC. For 13C NMR analysis, the sample was mixed 1:1 with 8a prepared using O2.
Reaction Progress
Products 6a and 6b were isolated from a 2-ml curcumin autoxidation reaction that was terminated at 20 min by injection on RP-HPLC. The products were dissolved in 0.5 ml of 10 mm NH4OAc buffer, pH 7.4, and aliquots were analyzed by LC-MS using run times of 15 min. Thus, aliquots were analyzed in blocks of three consecutive injections from each of the two reactions for a total of 7 h.
Reaction with N-Acetylcysteine (NAc)
RP-HPLC isolated 6 (a mixture of 6a and 6b; 5 μm final concentration) was added to 1 ml of 20 mm NH4OAc buffer, pH 8.0, containing 10 μm N-acetylcysteine. The reaction was stopped after 2 min by freezing on dry ice followed by LC-MS analysis. In parallel, an autoxidation of curcumin (50 μm) was conducted in the presence of 500 μm NAc in 1 ml of 20 mm NH4OAc buffer, pH 8.0. After 30 min, 5 milliunits of horseradish peroxidase and H2O2 (10 μm) were added. The sample was analyzed at 1 min before the addition of horseradish peroxidase/H2O2 and 10 min after the addition of horseradish peroxidase/H2O2.
Synthesis of Diguaiacol 9
Guaiacol (2 mg, 0.02 mmol) was dissolved to a final concentration of 16 mm in 1 ml 10 mm pyrophosphate buffer containing H2O2 (0.35 mm), GSH (1.6 mg, 5 mm), and manganese acetate (0.04 mg, 1 mm) (29). The reaction was allowed to proceed at room temperature for 12 h before extraction on a preconditioned 50-mg Waters HLB cartridge eluted with MeOH. The sample was concentrated and analyzed using RP-HPLC and LC-MS.
HPLC Analyses
Reactions were analyzed using a Waters Atlantis T3 5-μm column (4.6 × 250 mm) eluted with 10% acetonitrile in 10 mm NH4OAc, pH 7.4, for 10 min. After 10 min, a linear gradient to 80% acetonitrile within 20 min was used. For isolation of products 1-4, the solvent was 2% acetonitrile in 10 mm NH4OAc, pH 7.4. Samples were eluted at a flow rate of 1 ml/min, and the products were monitored using an Agilent 1200 diode array detector coupled to a Radiomatic A100 radioactive flow detector.
LC-MS Analyses
LC-MS analysis was conducted using a Thermo Finnigan LTQ ion trap mass spectrometer equipped with an ESI interface. The instrument was operated in the negative ion mode, and mass spectra were acquired at a rate of 2 s/scan. The settings for the heated capillary (300 °C), spray voltage (4.0 kV), spray current (0.22 μA), auxiliary (37 mTorr), and sheath gas (16 mTorr) were optimized using direct infusion of a solution of curcumin (20 ng/μl) in acetonitrile/water 95/5 (v/v), containing 10 mm NH4OAc. Samples were introduced into the instrument using a Waters Symmetry Shield C18 3.5-μm column (2.1 × 100 mm) eluted with acetonitrile containing 10 mm NH4OAc (3/97 isocratic and then a linear gradient to 95/5 from 5 to 10 min) at 0.2 ml/min.
Thiol adducts were analyzed using a Thermo Vantage triple quadrupole mass spectrometer equipped with a heated ESI source. The settings were: heated capillary (300 °C), vaporizer (275 °C), spray current (0.22 μA), auxiliary (10 mTorr), and sheath gas (60 mTorr). Samples were introduced using a Waters Symmetry Shield C18 3.5-μm column (2.1 × 100 mm) eluted with acetonitrile containing 10 mm NH4OAc (3/97 isocratic and then a linear gradient to 95/5 from 2 to 6 min) at 0.25 ml/min.
NMR
Samples were dissolved in 150 μl of methanol-d4 or acetonitrile-d3 in 3-mm-inner diameter sample tubes. NMR spectra were recorded using a Bruker DRX 600-MHz spectrometer equipped with a cryoprobe at 284 K. Chemical shifts are reported in ppm relative to residual nondeuterated solvent (δ 3.30 for methanol-d4 and 1.94 ppm for acetonitrile-d3). Pulse frequencies for the H,H-COSY, heteronuclear single quantum coherence, and heteronuclear multiple bond correlation experiments were taken from the Bruker library. Carbon chemical shifts were determined from the heteronuclear single quantum coherence and heteronuclear multiple bond correlation experiments.
RESULTS
Isolation and Characterization of Curcumin Autoxidation Products
Curcumin (λmax 425 nm) degraded in phosphate buffer (10 mm, pH 7.4) within a 10-min reaction time (Fig. 2A). Products with λmax at 263 nm were formed in parallel. The absorbance at 263 nm reached a maximum at 10 min and decreased slowly over the next 30 min (Fig. 2B). Thus, the spectrophotometric analyses confirmed that curcumin degrades rapidly at physiological pH (20, 30) and, in addition, suggested formation of intermediate products that transform more slowly.
FIGURE 2.
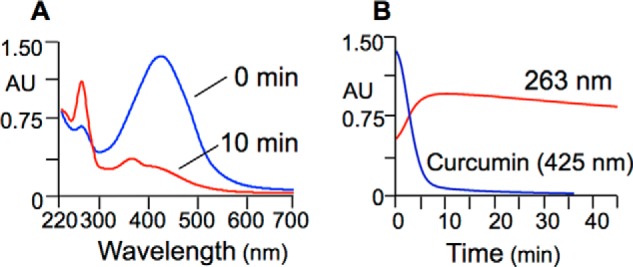
Spectrophotometric analysis of the autoxidation of curcumin. Curcumin (50 μm) was dissolved in phosphate buffer, pH 7.4, and analyzed by scanning from 700 to 220 nm at 0 and 10 min of reaction time (A) and by continuous monitoring at 263 nm or 425 nm for 45 min (B).
To aid chromatographic isolation of products and reaction intermediates, [14C2]curcumin (28) was used as tracer in autoxidation reactions. At 45-min reaction time, an aliquot of the reaction was directly injected on RP-HPLC with diode array and in-line radioactivity detection. The chromatogram in Fig. 3A shows elution of products 1-9 followed by unreacted curcumin as the last peak. The most polar of the products (1-4) were collected as a group and further resolved into five distinct peaks (Fig. 3B). The UV-visible spectra recorded during the HPLC analyses (Fig. 3C) showed that the increase in absorbance at 263 nm during autoxidation (Fig. 2) was caused by formation of 6; the local maximum at ∼360 nm was due to 7.
FIGURE 3.
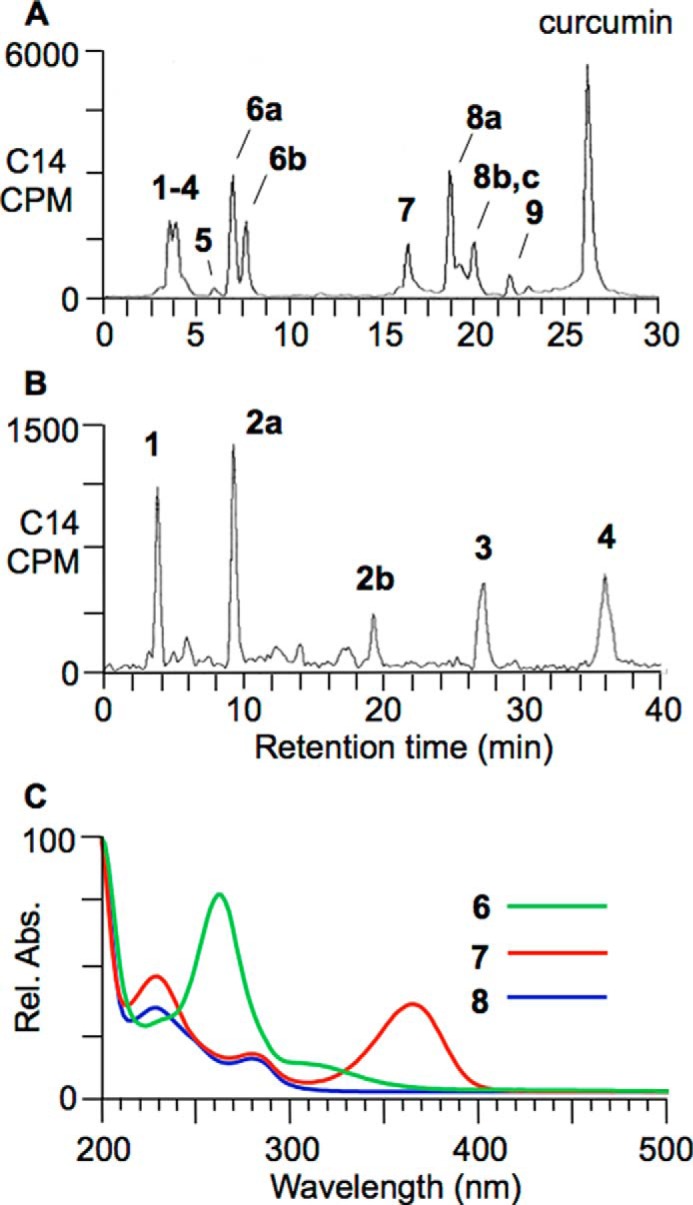
Reversed phase radio-HPLC analysis of an autoxidation reaction of [14C2]curcumin. A, the products were resolved using isocratic elution with a solvent of 10% acetonitrile in 50 mm NH4OAc, pH 7.4, for 10 min followed by a gradient to 80% acetonitrile over 20 min. B, products 1-4 were analyzed using isocratic elution with a solvent of 2% acetonitrile in 50 mm NH4OAc, pH 7.4. C, UV-visible spectra were recorded during RP-HPLC using a diode array detector. Rel. Abs., relative absorbance.
Compounds 1-7 and 9 are novel products of the autoxidation of curcumin that are formed in addition to the previously identified bicyclopentadione isomers 8a–8c (20, 21, 31, 32). The newly identified products are highly polar (1-4) and/or unstable at acidic pH (6 and 7), which might explain why they have not been described as products before. Extraction and chromatographic purification of the products required use of C18 solid phase cartridges without acidification and polar RP-HPLC solvents (e.g. 10 and 2% acetonitrile, respectively, in Fig. 3) in combination with a RP-HPLC column optimized for polar analytes (e.g. Waters Atlantis T3). To prevent further transformation of unstable reaction intermediates, it was necessary to maintain a pH of 7.4 at all steps, including chromatography and preparation for NMR analysis.
The structures of products 2-7 and 9 were elucidated using one- and two-dimensional homo- and heteronuclear NMR, LC-ESI-MS, and ESI high resolution MS analyses (Fig. 4 and Table 1). Products 2-7 contained the characteristic cyclopentadione ring found in the bicyclopentadione product 8. The cyclopentadione ring is formed of carbons C-2 through C-6 of the heptadienedione chain of curcumin. 2-7 are dioxygenated products that incorporate oxygen atoms at C-1 and C-7, respectively, with the exception of 5, which instead has a carbon-carbon bond connecting C-1 and C-7. 2 is a reduced analog (molecular weight 402) of the hydroxy-keto product 4 (molecular weight 400) that is in equilibrium with its hemiketal 3. 9 is diguaiacol. Although 1 chromatographed as a single peak in HPLC-UV analyses, subsequent LC-MS analyses suggested the presence of multiple cleavage products that were not sufficiently abundant for structural identification.
FIGURE 4.
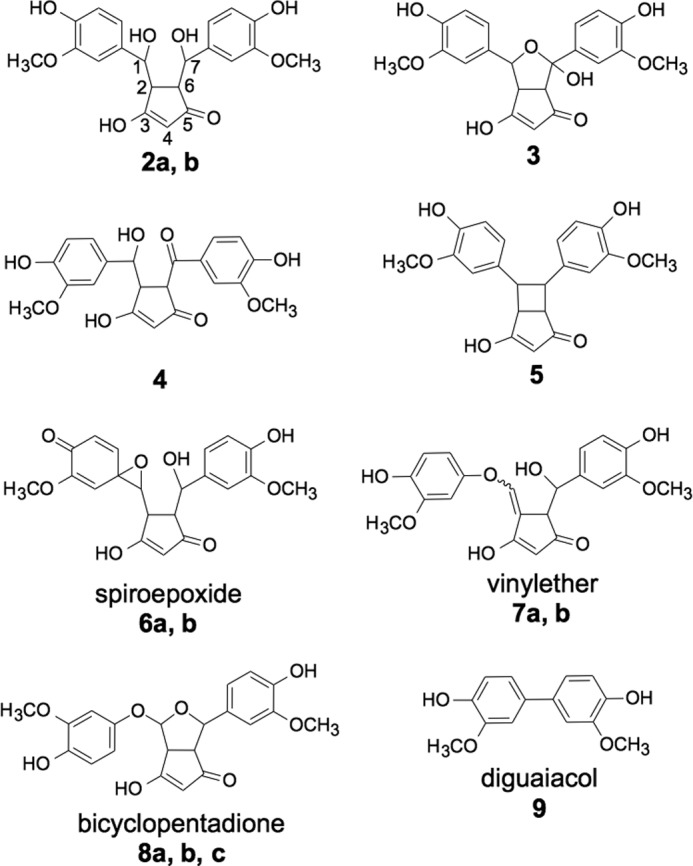
Products isolated from the autoxidation of curcumin.
TABLE 1.
LC-ESI-MS2 and high resolution MS analysis of curcumin oxidations products 2–7
The analyses were conducted in the negative ion mode. HR, high resolution.
| Product | Major MS2 product ions m/z | HR-MS [M-H]− |
||
|---|---|---|---|---|
| Found | Calculated | Error | ||
| ppm | ||||
| 2a | 249; 151 | 401.1249 | 401.1242 | 1.7 |
| 2b | 249; 151 | 401.1261 | 401.1242 | 4.7 |
| 3 | 247; 151; 355; 313 | 399.1099 | 399.1085 | 3.5 |
| 4 | 247; 151; 355; 313 | 399.1084 | 399.1085 | 0.3 |
| 5 | 173; 217; 271 | 367.1195 | 367.1187 | 2.2 |
| 6a | 259; 315; 355 | 399.1088 | 399.1085 | 0.8 |
| 6b | 259; 313; 355 | 399.1086 | 399.1085 | 0.3 |
| 7a | 247; 151 | 399.1092 | 399.1085 | 1.8 |
Identification of Spiroepoxide 6 and Vinylether 7
The spectroscopic data used for identification of the unusual compounds 6 and 7 are presented in detail. 6a and 6b are diastereomeric spiroepoxy cyclopentadiones. 7a and 7b are diastereomeric vinylether cyclopentadiones. Only one of the isomers of product 7 (7b) was sufficiently stable for complete structural characterization including NMR. 7a was identical to 7b with respect to its HPLC retention time, MS, MS-MS, and UV spectra. The UV-visible spectra of 6 and 7 showed λmax at 263 nm and 360 nm, respectively (Fig. 3C). LC-MS analyses showed incorporation of two oxygen atoms in 6 and 7 (m/z 399 in negative ion mode). The keto-enol moiety of the cyclopentadione ring was defined by a sp2 carbon signal (C-4) at 103.5 ppm in 6 and 103.6 ppm in 7, respectively, and two signals each for the C-3 and C-5 carbonyls (203 and 204 ppm in 6; 187 and 210 ppm in 7). Cyclization was evident from the 3J coupling of H-2 and H-6 as well as from the correlations of H-1 with C-6 and of H-7 with C-2, respectively, in the heteronuclear multiple bond correlation spectra of 6 and 7 (Fig. 5).
FIGURE 5.
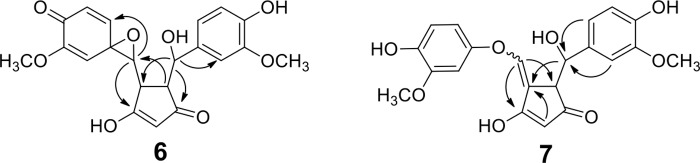
Structurally diagnostic heteronuclear multiple bond correlations for spiroepoxide (6) and vinylether (7).
The spiroepoxide moiety of 6 was established by the carbon chemical shifts of C-1 (68.2 ppm) and C-1′ (59.2 ppm), showing that both were connected to oxygen, and the proton chemical shift of H1 at 3.28 ppm (33). The ring attached to the epoxide was oxidized to a quinone (C-4′ as a carbonyl at 184.0 ppm), resulting in chemical shifts of H-2′, H-5′, and H-6′ compatible with double bonds rather than an aromatic ring. The second oxygen was present as a hydroxyl attached to C-7 (73.1 ppm).
The vinylether moiety in 7 was evident from the chemical shifts of C-1 (142.3 ppm) and H-1 (7.15 ppm), both compatible with a sp2 carbon connected to an oxygen. The ether linkage into the aromatic ring was also evident from a carbon chemical shift of 147 ppm for C-1′. The C-1/C-2 double bond is further corroborated by the sp2 carbon chemical shift of C-2 (108.7 ppm), and because there is no H-2, H-1 appears as a singlet and H-6 appears as a doublet. The second oxygen was present as a hydroxyl at C-7. The extended conjugation between the aromatic and cyclopentadione rings of 7 was also reflected in the UV-visible spectrum with a bathochromic shift to a λmax at 360 nm (Fig. 3C) compared with 6 and the other products that show a local maximum at ∼230 nm.
The side chains in 8a were attached in cis configuration to the cyclopentadione ring, which was inferred from the coupling constant 3J2,6 = 6.3 Hz for H-2 and H-6. The cis configuration is compatible with 5-exo cyclization to the cyclopentadione ring (see “Discussion”). The 3J2,6 value, however, was considerably smaller in all other products: 3.2 Hz in 2, 2.5 Hz in 3, 0 Hz in 4, 2.5 Hz in 5, and 2.0 Hz in 6. Whether the small coupling constant truly indicates a trans configuration of the side chains in 2-6 is difficult to decide in the absence of authentic reference compounds for these highly substituted cyclopentadione rings.
Reaction Progress
A spiroepoxide has been proposed previously as intermediate in the autoxidation of curcumin to bicyclopentadione 8 (20). The successful isolation and identification of 6 allowed us to test whether it is a true intermediate in the formation of 8. RP-HPLC isolated 6a and 6b were incubated in 10 mm NH4OAc pH 7.4, and aliquots of the reactions were analyzed using LC-MS analyses over the course of 7 h. As expected, 6a disappeared within the first 3 h and transiently formed 7a, which gave rise to bicyclopentadiones 8b and 8c (Fig. 6, A and B). Likewise, 6b was transformed via intermediate 7b to 8a (Fig. 6, C and D). Thus, 6 and 7 are reaction intermediates in the autoxidation of curcumin.
FIGURE 6.
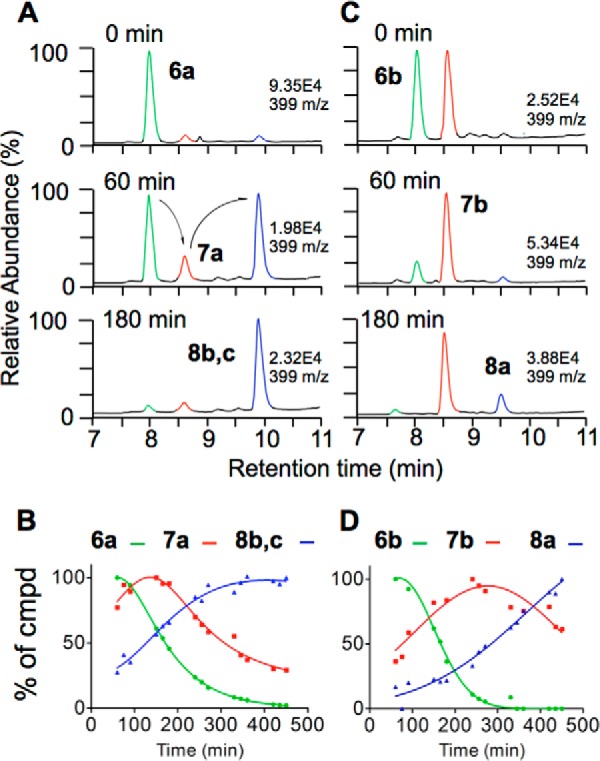
Time course of the transformation of 6a and 6b in 50 mm NH4OAc, pH 7.4. A and C, ion chromatograms for m/z 399 from LC-ESI-MS analyses at 0, 60, and 180 min. B and D, relative abundance of the diastereomers of 6, 7, and 8 over 7 h of reaction time.
Transformation of the diastereomers of 6 and 7 occurred at different rates, implicating differences in their stability. Neither 6 nor 7 gave products 2-5 upon further incubation in buffer pH 7.4. These analyses placed the spiroepoxide 6 and vinylether 7 into the reaction sequence as shown in Reaction 1.
6 was the earliest intermediate we were able to isolate from autoxidation reactions of curcumin, suggesting that prior intermediates are radicals. As shown below, oxygen incorporation studies unexpectedly revealed that 6 and 8 are formed by different mechanisms than previously proposed (20).
Oxygen Incorporation in Bicyclopentadione 8
Preliminary studies have shown that one of the two oxygen atoms incorporated into bicyclopentadione 8 was derived from water (20). Although it had not been shown directly, it was concluded that the other oxygen was derived from O2. Thus, O2 is split during the transformation such that one oxygen atom remains in the final product 8, whereas the other is exchanged with H2O. Here, we examined the sites of incorporation of 18O from 18O2 and H218O in 8. The sites were identified using the differences in chemical shift of carbon atoms bound to 18O versus 16O in the 13C NMR spectra (34) and by LC-ESI-MSn analyses.
When 8 was formed using 18O2 gas, the 13C NMR signals for both C-1 (96.9 ppm) and C-7 (79.3 ppm) were shifted by 0.02 ppm upfield, indicating that the oxygen of the tetrahydrofuran ring was derived from O2 (marked red in Fig. 7A). When 8 was formed in H218O, only the signal for C-1 was shifted, but no shift was observed for C-7 (Fig. 7B). This indicated that the oxygen connecting C-1 to the methoxyphenol ring was derived from H2O. The expected corresponding shift of the C-1′ signal of the methoxyphenol ring of 8 formed in H218O, however, was not observed.
FIGURE 7.
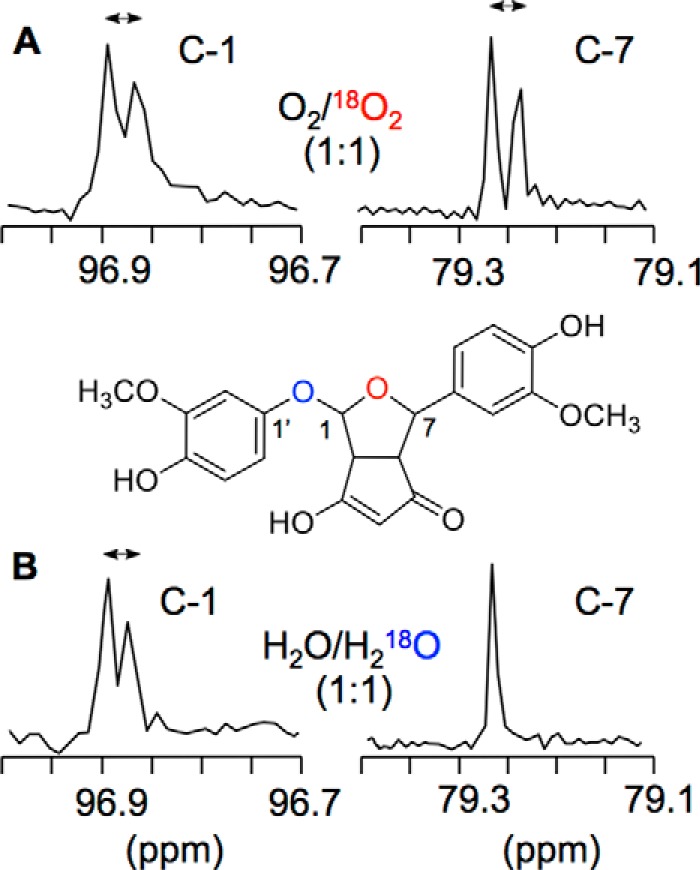
13C NMR analysis of 1:1 mixtures of 8 formed in O2 and 18O2 (A) and H2O and H218O (B). Expanded spectra showing the signals for C-1 and C-7 are shown.
LC-MSn analyses were conducted to corroborate the results for 18O incorporation deduced from the NMR studies. Because the fragment ions obtained in MSn experiments of 8 could not be assigned unequivocally, we prepared an unsymmetrical curcumin analog (3′-OCD3,4″-O-methylcurcumin) that contained OCD3 in place of the methoxy group on one of the aromatic rings and on the other aromatic ring a methoxy group in place of the para hydroxyl. K3[Fe(CN)6]-mediated oxidation of 3′-OCD3,4″-O-methylcurcumin gave the corresponding 3′-OCD3,4″-O-substituted 8 (structure established by NMR). Negative ion LC-MS-MS fragmentation data were compared for 8 formed in H2O (MS2 of m/z 399; Fig. 8A), 8 in H218O (MS2 of m/z 401; Fig. 8B), to those of 3′-OCD3,4″-O-methyl-8 in H2O (MS2 of m/z 416; Fig. 8C), and 3′-OCD3,4″-O-methyl-8 formed in H218O (MS2 of m/z 418; Fig. 8D). The fragment ion m/z 247 of 8 (formed in H2O) was increased to m/z 249 in 18O-8 (formed in H218O), indicating that this fragment contained the oxygen from water. In 3′-OCD3,4″-O-methyl-8 (H2O), the fragment was shifted to m/z 250, indicating that this fragment ion also contained the ring carrying the d3-methoxy group. As expected, the fragment ion was shifted to m/z 252 in 3′-OCD3,4″-O-methyl-8 when formed in H218O. This fragmentation pattern is best rationalized if the water-derived oxygen in 8 forms the ether bridge linking the bicycle to the methoxyphenol ring (Fig. 8E).
FIGURE 8.

Negative ion LC-MS2 spectra of 8 and 3′-OCD3,4″-O-methyl-8, respectively, formed in H2O (A and C) and H218O (B and D). E, proposed fragmentation of 18O-3′-OCD3,4″-O-methyl-8.
LC-MS fragmentation data of 3′-OCD3,4″-O-methyl substituted 8 formed in H218O were consistent with the results on oxygen incorporation obtained in the NMR analyses. Thus, the oxygen atom from O2 is inserted into the tetrahydrofuran ring of 8, whereas the ether bridge to the methoxyphenol ring is formed by an oxygen atom that has been exchanged from water. This is the opposite result from what was deduced previously from less detailed analyses (20).
Oxygen Exchange
We next determined the step at which the exchange of oxygen occurred. An autoxidation reaction of curcumin was conducted in phosphate buffer, pH 7.4, prepared using a 1:1 mixture of H2O and H218O. Negative ion LC-MS analyses showed that only vinylether 7 and bicyclopentadione 8 contained 18O from H218O, whereas spiroepoxide 6 and products 2-4 did not contain or exchange 18O from H218O (Fig. 9). When isolated 7 (formed in H2O) was incubated in 92% H218O in buffer pH 7.4 for 1 h, we found that neither recovered 7 nor formed 8 had incorporated 18O from H218O. Likewise, 8 does not exchange 18O when incubated in H218O buffer. Thus, the exchange of oxygen occurred during transformation of 6 to 7.
FIGURE 9.
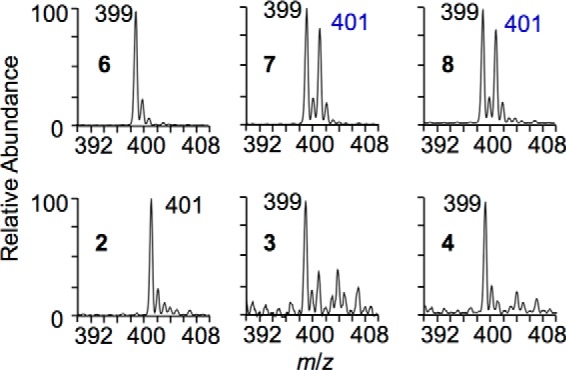
Partial LC-ESI mass spectra of curcumin autoxidation products formed in a 1:1 mixture of H2O and H218O (negative ion mode). Note that m/z 401 for 2 is not due to incorporation of 18O but to its molecular weight (402).
Additional Mechanistic Studies
RP-HPLC purified 6 was incubated in D2O (10 mm NH4OAc, pH 7.4) for 3 h followed by isolation of 8. NMR analysis showed loss of the proton signal at C-2, suggesting incorporation of deuterium at that position. This was also evident from the change in the splitting pattern of H-6 from a dd to a d signal.
Formation of minor amounts of diguaiacol 9 suggested that one of the methoxyphenol rings is released as a side reaction. Likewise, temporary release of a methoxyphenol ring is proposed during the transformation of 6 to 7 (see “Discussion”). To test whether the released methoxyphenol ring can undergo exchange before reattaching, a 4:1 mixture of d6-curcumin and curcumin was autoxidized, and the products were analyzed using LC-ESI-MS. Products 6, 7, and 8 had the same 4:1 isotopic ratio for d6 to d0 as the starting material, and there was no evidence for formation of a d3-analog of 6, 7, or 8, indicating that isotopic scrambling had not occurred.
Thiol Adduction of Spiroepoxide 6
We tested whether the spiroepoxide 6 was a functional electrophile by analyzing its ability to form covalent adducts with the thiol reagent, NAc. RP-HPLC isolated 6 reacted rapidly (2-min reaction time) with NAc to an adduct with a molecular ion at m/z 562 (negative ion LC-MS), equivalent to the sum of the molecular weight of 6 and NAc (Fig. 10, A and B). When curcumin (50 μm) was autoxidized in the presence of a 10-fold excess of NAc, curcumin was stabilized, and there was negligible formation of the m/z 562 adduct after 30 min, implying an efficient antioxidative effect of NAc. The addition of horseradish peroxidase/H2O2 resulted in transformation of curcumin and formation of products with m/z 562 (NAc adducts) and 399 (autoxidation products) (Fig. 10C). Adducts with m/z 530 representing a Michael addition of curcumin to NAc were not detected in these reactions. These analyses showed that oxidative transformation of curcumin was required for the formation of thiol adducts.
FIGURE 10.
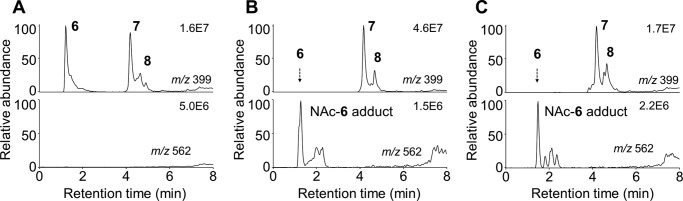
The spiroepoxide 6 forms a covalent adduct with NAc. RP-HPLC isolated 6 was placed in acetonitrile (A) or incubated with a 2-fold molar excess of N-acetylcysteine in 10 mm NH4OAc, pH 8 (B), for 2 min and analyzed using LC-ESI-MS. A, elution of 6, 7, and 8 in the m/z 399 ion trace. B, 6 is ablated in the m/z 399 ion trace, and instead a NAc-6 adduct is detected with a molecular ion of m/z 562. C, oxidative transformation of curcumin (50 μm) by horseradish peroxidase/H2O2 in the presence of NAc (500 μm) resulted in the formation of the same NAc-6 adduct as in A. Ion traces for m/z 399 (oxidative metabolites of curcumin) and m/z 562 (N-acetylcysteine adducts of oxidative metabolites) from negative ion LC-ESI-MS analyses are shown.
DISCUSSION
Mechanism of Autoxidation
Isolation and structural identification of novel products and reaction intermediates in conjunction with detailed isotopic incorporation studies have enabled us to propose a mechanism for the autoxidation of curcumin (Fig. 11). The initiating step is H-abstraction from one of the phenolic hydroxyls of curcumin. Delocalization of the radical to C-2 of the heptadienedione chain leaves the ring as a quinone methide and enables 5-exo cyclization to the cyclopentadione ring present in all isolated products (2-8) except for diguaiacol 9. Cyclization places the radical at C-7 followed by addition of O2 to form a peroxyl radical. Addition of the peroxyl radical to the quinone methide gives an endoperoxide and a tertiary radical that performs SHi attack (35, 36) at the endoperoxide to give spiroepoxide 6. 6 undergoes opening of the epoxide and exchange of oxygen such that the vinylether oxygen in 7 is derived from water rather than retained from the epoxide. The addition of the hydroxyl of 7 across the 1,2 double bond completes formation of 8 as the major autoxidation product.
FIGURE 11.

Proposed mechanism of autoxidation of curcumin. Products that have been isolated and identified are boxed.
The transformation of 6 to 8 is considerably more complex than previously recognized and involves vinylether 7 as an unexpected intermediate. The incorporation of 18O from H218O during the transformation of 6 to 7 can be explained by SN1 opening of the epoxide triggered by the electron-donating methoxy group and H-bonding by the cyclopentadione enol (Fig. 11). The addition of water (H218O) at C-1′ of the ring is followed by heterolytic C–C bond cleavage of vicinal diol 15, which restores aromaticity of the methoxyphenol ring. Identical carbon bond cleavage of substituted cyclohexa-2,5-dienones has been observed before in an analogous system (37). Carbon bond cleavage likely occurs in a solvent cage because isotopic scrambling was not observed under the reaction conditions used. Even if the reaction partners separate from the cage, hemiacetal formation at the 1′-OH would be preferred because of steric factors. Instant addition of the released methoxyphenol to the aldehyde gives hemiacetal 16 (not isolated) that loses water (containing the original epoxide oxygen) en route to vinylether 7.
Formation of the minor products 5 and 9 can be rationalized from reactions of early radical intermediates in the autoxidation of curcumin. The cyclobutyl ring of 5 is formed by the addition of the carbon-centered radical of 11 to the quinone methide as a competing reaction to oxygenation and peroxyl radical formation (Fig. 11). Diguaiacol 9 is recognized as one of the oxidation products when guaiacol is used as a co-substrate in peroxidase catalysis (38, 39). Like in peroxidase catalysis, 9 is the net result of combination of two methoxyphenol radicals. Their formation by carbon bond cleavage may require additional oxygenation reactions. Cleavage likely occurs during alternate transformation of one or more of radical intermediates 11-14. Consistent with its formation from 14 (or a prior radical intermediate), 9 was not formed from isolated 6 upon further incubation.
Four prior mechanistic assumptions (20) can now be ruled out, namely (i) addition of water to the quinone methide, (ii) formation of a hydroperoxide intermediate, (iii) its Hock cleavage, and (iv) direct transformation of spiroepoxide 6 to bicyclopentadione 8 by intramolecular hydrolysis of the epoxide. The previously proposed addition of water to the quinone methide implied that 6 contains 18O from H218O. We found this was not the case (Fig. 9). Instead of adding water, the quinone methide reacts with the peroxyl radical to form an endoperoxide. A hydroperoxide intermediate is not formed. Thus, 6 is not formed by Hock cleavage of a putative hydroperoxide intermediate but instead by intramolecular homolytic attack (SHi) of tertiary radical 13 on the endoperoxide.
Mechanistic Analogies to Lipid Peroxidation
Products 2-4 are formed from proposed endoperoxide intermediate 17 in transformations that are well precedented in lipid peroxidation chemistry (Fig. 11) (40–43). Opening of the endoperoxide moiety in a Kornblum-DeLaMare rearrangement (44) gives 4, similar to the formation of prostaglandins E2 and D2 from the prostaglandin endoperoxide PGH2 (45). 4 is in equilibrium with its hemiketal 3. Reductive opening of the endoperoxide leads to 2, similar to the formation of PGF2α from PGH2 that can occur as a nonenzymatic reduction. Both oxygen atoms introduced in 2, 3, and 4, respectively, were derived from O2, consistent with their origin from an endoperoxide intermediate (Fig. 9).
Like other naturally occurring (poly)phenolic compounds, curcumin can act as an inhibitor of lipid peroxidation (11, 12). The antioxidant reaction is the transfer of a hydrogen atom from curcumin to a fatty acyl peroxyl radical, thereby interrupting the lipid peroxidation chain reaction (46). The site of hydrogen abstraction on curcumin (β-diketo methylene versus phenolic hydroxyl) has been the topic of a controversial debate (47–50) that was resolved when the specific effects of polar and nonpolar solvents on hydrogen atom transfer versus a sequential proton loss-electron transfer process were uncovered (12, 51). The latter transfer process is relevant in aqueous solution: deprotonation of the phenolic hydroxyl at basic pH precedes the transfer of an electron to O2 acting as the initial electron acceptor and being reduced to superoxide (20). Hydrogen abstraction from the phenolic hydroxyl can also be achieved enzymatically in a peroxidase reaction, e.g. catalyzed by horseradish peroxidase or the peroxidase activity of cyclooxygenase (20).
Our studies answer a question about the antioxidant mechanism of curcumin that has received little attention so far: what is the fate of the curcumin phenoxyl radical formed in the antioxidant reaction? Curcumin dimers have been isolated from the early phase of radical-initiated degradation conducted in organic solvent (11). Dimerization of curcumin is reminiscent of the dimerization of resveratrol as a consequence of cyclooxygenase catalyzed hydrogen abstraction (52, 53). Analogous reactions conducted in the presence of linoleic acid or its hydroperoxides resulted in the formation of curcumin-linoleate adducts (54). In aqueous solution at physiological pH, however, the ultimate consequence of phenoxyl radical formation is the stable incorporation of oxygen into the heptadienedione moiety of curcumin leading to the dioxygenated bicyclopentadione 8.
The Cyclopentadione as a Carboxylic Acid Isostere
The elution times of 2-6 in RP-HPLC analyses were much earlier than expected from comparison with structurally related 7 and 8. The increase in polarity is due to the cyclopentadione moiety acting as a carboxylic acid isostere, such that products 2-6 chromatograph as polar anions in the aqueous RP-HPLC solvent. Cyclopentadiones with pKa values of ∼3–4 have been employed as carboxylic acid isosteres in drug development (55). It appears that the functional groups attached to the cyclopentadione rings of 2-8 have a major influence on the apparent acidity of the methylenic hydrogen of the β-diketo moiety.
Implications for the Biological Activity of Curcumin
Biochemical studies have shown that the initially formed quinone methides, e.g. 10, 11, and 12, or a later intermediate like spiroepoxide 6 are the active species responsible for the inactivation (poisoning) of human topoisomerase, rather than the parent curcumin or the final bicyclopentadione (26). Although not shown directly, it was inferred that electrophilic reaction intermediates form covalent adducts with cysteine residues in the topoisomerase active site that triggered DNA cleavage. In support of this hypothesis, we have shown that spiroepoxide 6 is a reactive electrophile forming a stable covalent adduct with NAc (Fig. 10). The same adduct was formed when curcumin was oxidized in the presence of NAc by horseradish peroxidase. Notably, curcumin did not form a stable Michael-type adduct with NAc, underscoring the requirement for oxidative transformation as the mechanism of adduct formation. These preliminary studies underscore the relevance of oxidative transformation, resulting in reactive intermediates that can alter cellular function by forming covalent adducts with redox regulated thiols (cysteines) in proteins.
The transformation of curcumin into reactive electrophiles as the biologically active species is reminiscent of the action of other small molecule electrophiles like 4-hydroxy-nonenal (56), cyclopentenone prostaglandins (57), and oleanane triterpenoids (58) that covalently adduct to regulatory cysteines in the active sites of target proteins, e.g. KEAP-1/Nrf2 or IKKβ/NF-κB. It can be speculated that, given its instability at physiological pH, many of the bioactivities of curcumin are mediated by its electrophilic transformation products forming covalent adducts with cellular proteins.
Curcumin autoxidizes at physiological pH to a spectrum of dioxygenated metabolites with electrophilic, as well as nucleophilic moieties. The previously unrecognized diversity of chemical entities formed in the autoxidation (quinone methide, peroxyl radical, endoperoxide, spiroepoxide, vinylether, cyclopentadione, etc.) is remarkable. Undoubtedly, this provides a novel paradigm to understand the vast cellular effects of curcumin (polypharmacology) based on the number and diversity of its spontaneous transformation products. The work described here is the first example for isolation and direct structural characterization of fleeting reaction intermediates formed during oxidative transformation of a natural polyphenolic agent of high medicinal interest. We have determined conditions under which the reaction intermediates are sufficiently stable for chromatographic and spectroscopic analysis. The methods developed here are suitable to investigate metabolic transformation of related polyphenols and enable preparation and isolation of reaction intermediates and products to investigate their particular roles within the spectrum of biological activities of curcumin.
Acknowledgments
We thank Drs. Brian Bachman, Jin Cha, Ned Porter, and Gary Sulikowski for helpful discussion.
This work was supported, in whole or in part, by National Institutes of Health Grants CA159382, AT006896, and GM076592 (to C. S.) and NIGMS Training Grant 2T32GM07628 and NCCAM Predoctoral Fellowship Award F31AT007287 (to O. N. G.). This work was also supported by the Camille Dreyfus Foundation (to H. O. S.), Pilot Award P30DK058404 from the Vanderbilt Institute in Chemical Biology, the Vanderbilt Digestive Diseases Research Center, and the NCI SPORE in GI Cancer Pilot Award 5P50CA095103.
- RP
- reversed phase
- ESI
- electrospray ionization
- NAc
- N-acetylcysteine.
REFERENCES
- 1. Esatbeyoglu T., Huebbe P., Ernst I. M., Chin D., Wagner A. E., Rimbach G. (2012) Curcumin-from molecule to biological function. Angew. Chem. Int. Ed. Engl. 51, 5308–5332 [DOI] [PubMed] [Google Scholar]
- 2. Anand P., Sundaram C., Jhurani S., Kunnumakkara A. B., Aggarwal B. B. (2008) Curcumin and cancer: an “old-age” disease with an “age-old” solution. Cancer Lett. 267, 133–164 [DOI] [PubMed] [Google Scholar]
- 3. Huang M. T., Lou Y. R., Ma W., Newmark H. L., Reuhl K. R., Conney A. H. (1994) Inhibitory effects of dietary curcumin on forestomach, duodenal, and colon carcinogenesis in mice. Cancer Res. 54, 5841–5847 [PubMed] [Google Scholar]
- 4. Pereira M. A., Grubbs C. J., Barnes L. H., Li H., Olson G. R., Eto I., Juliana M., Whitaker L. M., Kelloff G. J., Steele V. E., Lubet R. A. (1996) Effects of the phytochemicals, curcumin and quercetin, upon azoxymethane-induced colon cancer and 7,12-dimethylbenz[a]anthracene-induced mammary cancer in rats. Carcinogenesis 17, 1305–1311 [DOI] [PubMed] [Google Scholar]
- 5. Funk J. L., Frye J. B., Oyarzo J. N., Zhang H., Timmermann B. N. (2010) Anti-arthritic effects and toxicity of the essential oils of turmeric (Curcuma longa L.). J. Agric. Food Chem. 58, 842–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Funk J. L., Oyarzo J. N., Frye J. B., Chen G., Lantz R. C., Jolad S. D., Sólyom A. M., Timmermann B. N. (2006) Turmeric extracts containing curcuminoids prevent experimental rheumatoid arthritis. J. Nat. Prod. 69, 351–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Coban D., Milenkovic D., Chanet A., Khallou-Laschet J., Sabbe L., Palagani A., Vanden Berghe W., Mazur A., Morand C. (2012) Dietary curcumin inhibits atherosclerosis by affecting the expression of genes involved in leukocyte adhesion and transendothelial migration. Mol. Nutr. Food Res. 56, 1270–1281 [DOI] [PubMed] [Google Scholar]
- 8. Balogun E., Hoque M., Gong P., Killeen E., Green C. J., Foresti R., Alam J., Motterlini R. (2003) Curcumin activates the haem oxygenase-1 gene via regulation of Nrf2 and the antioxidant-responsive element. Biochem. J. 371, 887–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dinkova-Kostova A. T., Talalay P. (2008) Direct and indirect antioxidant properties of inducers of cytoprotective proteins. Mol. Nutr. Food Res. 52, S128–S138 [DOI] [PubMed] [Google Scholar]
- 10. Jiao Y., Wilkinson J., 4th, Di X., Wang W., Hatcher H., Kock N. D., D'Agostino R., Jr., Knovich M. A., Torti F. M., Torti S. V. (2009) Curcumin, a cancer chemopreventive and chemotherapeutic agent, is a biologically active iron chelator. Blood 113, 462–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Masuda T., Hidaka K., Shinohara A., Maekawa T., Takeda Y., Yamaguchi H. (1999) Chemical studies on antioxidant mechanism of curcuminoid: analysis of radical reaction products from curcumin. J. Agric. Food Chem. 47, 71–77 [DOI] [PubMed] [Google Scholar]
- 12. Foti M. C. (2007) Antioxidant properties of phenols. J. Pharm. Pharmacol. 59, 1673–1685 [DOI] [PubMed] [Google Scholar]
- 13. Shen L., Ji H. F. (2012) The pharmacology of curcumin: is it the degradation products? Trends Mol. Med. 18, 138–144 [DOI] [PubMed] [Google Scholar]
- 14. Giménez-Bastida J. A., González-Sarrías A., Larrosa M., Tomás-Barberán F., Espín J. C., García-Conesa M. T. (2012) Ellagitannin metabolites, urolithin A glucuronide and its aglycone urolithin A, ameliorate TNF-α-induced inflammation and associated molecular markers in human aortic endothelial cells. Mol. Nutr. Food Res. 56, 784–796 [DOI] [PubMed] [Google Scholar]
- 15. Lambert J. D., Sang S., Yang C. S. (2007) Biotransformation of green tea polyphenols and the biological activities of those metabolites. Mol. Pharm. 4, 819–825 [DOI] [PubMed] [Google Scholar]
- 16. Ireson C., Orr S., Jones D. J., Verschoyle R., Lim C. K., Luo J. L., Howells L., Plummer S., Jukes R., Williams M., Steward W. P., Gescher A. (2001) Characterization of metabolites of the chemopreventive agent curcumin in human and rat hepatocytes and in the rat in vivo, and evaluation of their ability to inhibit phorbol ester-induced prostaglandin E2 production. Cancer Res. 61, 1058–1064 [PubMed] [Google Scholar]
- 17. Anand P., Thomas S. G., Kunnumakkara A. B., Sundaram C., Harikumar K. B., Sung B., Tharakan S. T., Misra K., Priyadarsini I. K., Rajasekharan K. N., Aggarwal B. B. (2008) Biological activities of curcumin and its analogues (Congeners) made by man and Mother Nature. Biochem. Pharmacol. 76, 1590–1611 [DOI] [PubMed] [Google Scholar]
- 18. Dhillon N., Sung B., Kurzrock R., Aggarwal B. B. (2009) Could antitumor activity of curcumin in patients be due to its metabolites? A response. Clin. Cancer Res. 15, 7108–710919903776 [Google Scholar]
- 19. Wu J. C., Lai C. S., Badmaev V., Nagabhushanam K., Ho C. T., Pan M. H. (2011) Tetrahydrocurcumin, a major metabolite of curcumin, induced autophagic cell death through coordinative modulation of PI3K/Akt-mTOR and MAPK signaling pathways in human leukemia HL-60 cells. Mol. Nutr. Food Res. 55, 1646–1654 [DOI] [PubMed] [Google Scholar]
- 20. Griesser M., Pistis V., Suzuki T., Tejera N., Pratt D. A., Schneider C. (2011) Autoxidative and cyclooxygenase-2 catalyzed transformation of the dietary chemopreventive agent curcumin. J. Biol. Chem. 286, 1114–1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gordon O. N., Schneider C. (2012) Vanillin and ferulic acid: not the major degradation products of curcumin. Trends Mol. Med. 18, 361–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Martín-Cordero C., López-Lázaro M., Gálvez M., Ayuso M. J. (2003) Curcumin as a DNA topoisomerase II poison. J. Enzyme Inhib. Med. Chem. 18, 505–509 [DOI] [PubMed] [Google Scholar]
- 23. López-Lázaro M., Willmore E., Jobson A., Gilroy K. L., Curtis H., Padget K., Austin C. A. (2007) Curcumin induces high levels of topoisomerase I- and II-DNA complexes in K562 leukemia cells. J. Nat. Prod. 70, 1884–1888 [DOI] [PubMed] [Google Scholar]
- 24. Deweese J. E., Osheroff N. (2009) The DNA cleavage reaction of topoisomerase II: wolf in sheep's clothing. Nucleic Acids Res. 37, 738–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nitiss J. L. (2009) Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 9, 338–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ketron A. C., Gordon O. N., Schneider C., Osheroff N. (2013) Oxidative metabolites of curcumin poison human type II topoisomerases. Biochemistry 52, 221–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pabon H. J. J. (1964) A synthesis of curcumin and related compounds. Recl. Trav. Chim. Pays Bas 83, 379–386 [Google Scholar]
- 28. Gordon O. N., Graham L. A., Schneider C. (2013) Facile synthesis of deuterated and [14C]labeled analogs of vanillin and curcumin for use as mechanistic and analytical tools. J. Labelled Comp. Radiopharm. 56, 696–699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Forrester I. T., Grabski A. C., Burgess R. R., Leatham G. F. (1988) Manganese, Mn-dependent peroxidases, and the biodegradation of lignin. Biochem. Biophys. Res. Commun. 157, 992–999 [DOI] [PubMed] [Google Scholar]
- 30. Wang Y. J., Pan M. H., Cheng A. L., Lin L. I., Ho Y. S., Hsieh C. Y., Lin J. K. (1997) Stability of curcumin in buffer solutions and characterization of its degradation products. J. Pharm. Biomed. Anal. 15, 1867–1876 [DOI] [PubMed] [Google Scholar]
- 31. Schneider C., Amberg A., Feurle J., Ross A., Roth M., Tóth G., Schreier P. (1998) 2-[(4″-Hydroxy-3′-methoxy)-phenoxyl]-4-(4″-hydroxy-3″-methoxyphenyl)-8-hydroxy-6-oxo-3-oxabicyclo[3.3.0]-7-octene: unusual product of the soybean lipoxygenase-catalyzed oxygenation of curcumin. J. Mol. Catalysis B Enzymatic 4, 219–227 [Google Scholar]
- 32. Toth G., Roth M., Weckerle B., Schreier P. (2000) Structural elucidation of two novel products from the soybean lipoxygenase-catalysed dioxygenation of curcumin. Magn. Res. Chem. 38, 51–54 [Google Scholar]
- 33. Gierer J., Imsgard F., Noren I. (1977) Studies on the degradation of phenolic lignin units of β-aryl ether type with oxygen in alkaline media. Acta Chem. Scand. B 31, 561–572 [Google Scholar]
- 34. Vederas J. C. (1980) Structural dependence of O-18-isotope shifts in C-13-NMR spectra. J. Am. Chem. Soc. 102, 374–376 [Google Scholar]
- 35. Porter N. A., Zuraw P. J., Sullivan J. A. (1984) Peroxymercuration-demercuration of lipid hydroperoxides. Tetrahed. Lett. 25, 807–810 [Google Scholar]
- 36. Schneider C., Boeglin W. E., Brash A. R. (2004) Identification of two cyclooxygenase active site residues, leucine-384 and glycine-526, that control carbon ring cyclization in prostaglandin biosynthesis. J. Biol. Chem. 279, 4404–4414 [DOI] [PubMed] [Google Scholar]
- 37. Karhu M. (1981) Formation of diphenyl ethers from cyclohexa-2,5-dienones via 4-phenoxy-4-(1-alkoxy)cyclohexa-2,5-dienones as probable intermediates. J. Chem. Soc. Perkin Trans. 1, 303–306 [Google Scholar]
- 38. Taurog A., Dorris M. L., Guziec F. S., Jr. (1992) An unexpected side reaction in the guaiacol assay for peroxidase. Anal. Biochem. 205, 271–277 [DOI] [PubMed] [Google Scholar]
- 39. Doerge D. R., Divi R. L., Churchwell M. I. (1997) Identification of the colored guaiacol oxidation product produced by peroxidases. Anal. Biochem. 250, 10–17 [DOI] [PubMed] [Google Scholar]
- 40. Porter N. A., Caldwell S. E., Mills K. A. (1995) Mechanisms of free radical oxidation of unsaturated lipids. Lipids 30, 277–290 [DOI] [PubMed] [Google Scholar]
- 41. Rouzer C. A., Marnett L. J. (2003) Mechanism of free radical oxygenation of polyunsaturated fatty acids by cyclooxygenases. Chem. Rev. 103, 2239–2304 [DOI] [PubMed] [Google Scholar]
- 42. Jahn U., Galano J. M., Durand T. (2008) Beyond prostaglandins: chemistry and biology of cyclic oxygenated metabolites formed by free-radical pathways from polyunsaturated fatty acids. Angew. Chem. Int. Ed. Engl. 47, 5894–5955 [DOI] [PubMed] [Google Scholar]
- 43. Yin H., Xu L., Porter N. A. (2011) Free radical lipid peroxidation: mechanisms and analysis. Chem. Rev. 111, 5944–5972 [DOI] [PubMed] [Google Scholar]
- 44. Kornblum N., DeLaMare H. E. (1951) The base catalyzed decomposition of a dialkyl peroxide. J. Am. Chem. Soc. 73, 880–881 [Google Scholar]
- 45. Hamberg M., Samuelsson B. (1967) Oxygenation of unsaturated fatty acids by the vesicular gland of sheep. J. Biol. Chem. 242, 5344–5354 [PubMed] [Google Scholar]
- 46. Litwinienko G., Ingold K. U. (2007) Solvent effects on the rates and mechanisms of reaction of phenols with free radicals. Acc. Chem. Res. 40, 222–230 [DOI] [PubMed] [Google Scholar]
- 47. Jovanovic S. V., Steenken S., Boone C. W., Simic M. G. (1999) H-atom transfer is a preferred antioxidant mechanism of curcumin. J. Am. Chem. Soc. 121, 9677–9681 [Google Scholar]
- 48. Barclay L. R., Vinqvist M. R., Mukai K., Goto H., Hashimoto Y., Tokunaga A., Uno H. (2000) On the antioxidant mechanism of curcumin: classical methods are needed to determine antioxidant mechanism and activity. Org. Lett. 2, 2841–2843 [DOI] [PubMed] [Google Scholar]
- 49. Priyadarsini K. I., Maity D. K., Naik G. H., Kumar M. S., Unnikrishnan M. K., Satav J. G., Mohan H. (2003) Role of phenolic O-H and methylene hydrogen on the free radical reactions and antioxidant activity of curcumin. Free Radic. Biol. Med. 35, 475–484 [DOI] [PubMed] [Google Scholar]
- 50. Galano A., Alvarez-Diduk R., Ramirez-Silva M. T., Alarcon-Angeles G., Rojas-Hernandez A. (2009) Role of the reacting free radicals on the antioxidant mechanism of curcumin. Chem. Phys. 363, 13–23 [Google Scholar]
- 51. Litwinienko G., Ingold K. U. (2004) Abnormal solvent effects on hydrogen atom abstraction. 2. Resolution of the curcumin antioxidant controversy: the role of sequential proton loss electron transfer. J. Org. Chem. 69, 5888–5896 [DOI] [PubMed] [Google Scholar]
- 52. Szewczuk L. M., Forti L., Stivala L. A., Penning T. M. (2004) Resveratrol is a peroxidase-mediated inactivator of COX-1 but not COX-2: a mechanistic approach to the design of COX-1 selective agents. J. Biol. Chem. 279, 22727–22737 [DOI] [PubMed] [Google Scholar]
- 53. Szewczuk L. M., Lee S. H., Blair I. A., Penning T. M. (2005) Viniferin formation by COX-1: evidence for radical intermediates during co-oxidation of resveratrol. J. Nat. Prod. 68, 36–42 [DOI] [PubMed] [Google Scholar]
- 54. Masuda T., Maekawa T., Hidaka K., Bando H., Takeda Y., Yamaguchi H. (2001) Chemical studies on antioxidant mechanism of curcumin: analysis of oxidative coupling products from curcumin and linoleate. J. Agric. Food Chem. 49, 2539–2547 [DOI] [PubMed] [Google Scholar]
- 55. Ballatore C., Soper J. H., Piscitelli F., James M., Huang L., Atasoylu O., Huryn D. M., Trojanowski J. Q., Lee V. M., Brunden K. R., Smith A. B. (2011) Cyclopentane-1,3-dione: a novel isostere for the carboxylic acid functional group. Application to the design of potent thromboxane (A2) receptor antagonists. J. Med. Chem. 54, 6969–6983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ji C., Kozak K. R., Marnett L. J. (2001) IκB kinase, a molecular target for inhibition by 4-hydroxy-2-nonenal. J. Biol. Chem. 276, 18223–18228 [DOI] [PubMed] [Google Scholar]
- 57. Rossi A., Kapahi P., Natoli G., Takahashi T., Chen Y., Karin M., Santoro M. G. (2000) Anti-inflammatory cyclopentenone prostaglandins are direct inhibitors of IκB kinase. Nature 403, 103–108 [DOI] [PubMed] [Google Scholar]
- 58. Liby K., Hock T., Yore M. M., Suh N., Place A. E., Risingsong R., Williams C. R., Royce D. B., Honda T., Honda Y., Gribble G. W., Hill-Kapturczak N., Agarwal A., Sporn M. B. (2005) The synthetic triterpenoids, CDDO and CDDO-imidazolide, are potent inducers of heme oxygenase-1 and Nrf2/ARE signaling. Cancer Res. 65, 4789–4798 [DOI] [PubMed] [Google Scholar]