

Background: A mutation of the extended X loop of MDR3 caused hereditary liver cholestasis.
Results: Wild type MDR3 exhibited PC-induced ATPase activity, but the Q1174E mutant displayed no stimulation.
Conclusion: The glutamine preceding the ABC signature motif communicates substrate binding within the TMD to the extended X loop of the NBD.
Significance: This study provides evidence for a transmission interface coupling ATP hydrolysis to substrate transport.
Keywords: ABC Transporter, ATPase, Lipid Transport, Liver Injury, Multidrug Transporter, MDR1, MDR3, PFIC-3, Transmission Interface, X Loop
Abstract
The human multidrug resistance protein 3 (MDR3/ABCB4) belongs to the ubiquitous family of ATP-binding cassette (ABC) transporters and is located in the canalicular membrane of hepatocytes. There it flops the phospholipids of the phosphatidylcholine (PC) family from the inner to the outer leaflet. Here, we report the characterization of wild type MDR3 and the Q1174E mutant, which was identified previously in a patient with progressive familial intrahepatic cholestasis type 3 (PFIC-3). We expressed different variants of MDR3 in the yeast Pichia pastoris, purified the proteins via tandem affinity chromatography, and determined MDR3-specific ATPase activity in the presence or absence of phospholipids. The ATPase activity of wild type MDR3 was stimulated 2-fold by liver PC or 1,2-dioleoyl-sn-glycero-3-phosphatidylethanolamine lipids. Furthermore, the cross-linking of MDR3 with a thiol-reactive fluorophore blocked ATP hydrolysis and exhibited no PC stimulation. Similarly, phosphatidylethanolamine, phosphatidylserine, and sphingomyelin lipids did not induce an increase of wild type MDR3 ATPase activity. The phosphate analogues beryllium fluoride and aluminum fluoride led to complete inhibition of ATPase activity, whereas orthovanadate inhibited exclusively the PC-stimulated ATPase activity of MDR3. The Q1174E mutation is located in the nucleotide-binding domain in direct proximity of the leucine of the ABC signature motif and extended the X loop, which is found in ABC exporters. Our data on the Q1174E mutant demonstrated basal ATPase activity, but PC lipids were incapable of stimulating ATPase activity highlighting the role of the extended X loop in the cross-talk of the nucleotide-binding domain and the transmembrane domain.
Introduction
The human multidrug resistance protein 3 (MDR3/ABCB4) belongs to the family of ATP-binding cassette (ABC)2 transporters and is highly expressed in the canalicular membrane of hepatocytes. In the canaliculus, phosphatidylcholine (PC) lipids form mixed micelles with bile salts and cholesterol to reduce the destructive detergent activity of bile salts and to protect the biliary ducts (1). Because the flop of PC lipids from the inner leaflet of the lipid bilayer to the outer leaflet is a very slow process, PC lipids are translocated by the PC floppase MDR3 energized by ATP binding and hydrolysis (2–7).
The first evidence that MDR3 flops phospholipids (PLs) was obtained by the generation of homozygous knock-out mice for the murine Mdr2 gene (Mdr2−/− mice), which is homologous to human MDR3 (2). These mice showed a complete absence of PLs from bile. The function of Mdr2 could be substituted in these mice by expressing the human MDR3 gene, indicating that MDR3 acts as a PL floppase (3). Further studies used the lower hydrophobicity of short chain lipids (C5-C6 chain) to determine Mdr2/MDR3-mediated lipid translocation in yeast and cultured mammalian cells, respectively. Direct evidence that MDR3 translocates exclusively the PLs of the phosphatidylcholine family was obtained by van Helvoort and co-workers (5). They reported the translocation of fluorescently labeled short chain PC lipids in polarized pig kidney epithelial cells transfected with MDR3. Subsequently, it was demonstrated in HEK293 cells stably expressing MDR3 that PC lipids are excreted in a bile salt-dependent manner (6–8).
MDR3 is a 1279-amino acid glycoprotein and is composed of two nucleotide-binding domains (NBDs) and two transmembrane domains (TMDs), which are encoded on a single gene forming a so-called full-size ABC transporter (9). MDR3 shares up to 76% identity and 86% similarity in the amino acid sequence with the human multidrug resistance protein 1 (MDR1/ABCB1) while fulfilling a different physiological function (9–15). As of yet, no specific ATPase activity of MDR3 could be determined when the protein was expressed at high levels in insect (Sf9) cells or HEK293 cells, although MDR1 exhibited high ATPase activity in these systems (16, 17). Smith et al. (16) demonstrated vanadate-dependent nucleotide trapping of MDR3 in Sf9 plasma membranes, which could be inhibited by the MDR1 reversal agents verapamil and cyclosporin A. In addition, Ishigami et al. (17) ascertained the drug-stimulated ATPase activity of a chimera protein containing the TMDs of MDR1 and the NBDs of MDR3. They demonstrated that the purified chimera protein exhibited 10-fold lower drug-stimulated ATPase activity compared with MDR1. These findings confirmed that the NBDs of MDR3 bind ATP but that ATP hydrolysis takes place with an apparent low turnover number.
Previously, we demonstrated that human wild type MDR3 exhibited PC-induced ATPase activity, whereas the ATPase-deficient mutant of both Walker B motifs did not show stimulation (18). In this study, we characterized the ATPase activity of wild type MDR3 in terms of kinetic parameters, substrate spectrum, and the effect of phosphate analogues.
Furthermore, we analyzed the ATPase activity of the MDR3 Q1174E mutant in vitro, which was identified in a patient with progressive familial intrahepatic cholestasis type 3 (PFIC-3) and described previously in Kubitz et al. (19). This mutation is located in the extended X loop of NBD2 (TRVGDKGTQ). Based on structural and biochemical data, the NBDs dimerize in a head-to-tail orientation in the presence of ATP (20–22). Each ATP-binding site harbors highly conserved motifs, the Walker A (GXXGXGK(S/T), where X can be any amino acid residue), and the Walker B motif (ΦΦΦΦD, where Φ can be any hydrophobic residue) of one NBD, and the ABC signature motif (C loop, LSGGQ) of the opposing NBD (23). A highly conserved motif, the X loop (TEVGERG), which is located in close proximity to the ABC signature motif, was identified by Dawson and Locher (22) in ABC exporters. The X loop contacts the first intracellular loop (ICL1) of the TMD and likely transmits conformational changes generated by ATP binding and hydrolysis to the TMD (22, 24, 25). To date, the molecular function of this transmission interface with respect to coupling of the ATP hydrolysis cycle with substrate translocation is still not entirely clear. We expressed the Q1174E mutant in Pichia pastoris and purified the mutant via tandem affinity purification (TAP). The detergent-solubilized Q1174E mutant exhibited basal ATPase activity, which was demonstrated by modifying this mutant with a thiol-reactive fluorophore, but no substrate-stimulated ATPase activity in contrast to the wild type floppase. Thus, we suggest that glutamine 1174 is involved in the cross-talk of NBD and TMD.
EXPERIMENTAL PROCEDURES
Chemicals and Routine Procedures
Fos-choline 16 (FC-16) was obtained from Anatrace, and lipids were purchased from Avanti Polar Lipids. All other chemicals were from Sigma. The protein concentration was determined by a Bradford assay using the Coomassie Plus Assay (Pierce). The Mini-Protean 3 system (Bio-Rad) was used for SDS-PAGE on 7% gels. Immunoblotting was performed with a Tank blot system (Bio-Rad) using the monoclonal anti-P-gp C219 antibody (Merck) employing standard procedures.
Cloning of Human MDR3 and Site-directed Mutagenesis
We cloned human wild type MDR3 (NM_000443.3) as described previously (18). Site-directed mutagenesis was carried out with the QuikChange® XL (Agilent Technologies) and the Phusion site-directed mutagenesis kit (Thermo Scientific), respectively. To generate the ATPase-deficient mutant, we exchanged Glu-558 and Glu-1207 of the conserved Walker B motif against Gln with the primer pair as described before (18). The Q1174E mutant was introduced into MDR3 with the Phusion site-directed mutagenesis kit (Thermo Scientific) using forward primer 5′-GATAAGGGGACTGAGCTCTCAGGAGGTCAAAAAC-3′ and the reverse primer 5′-CCCACTCTTGTTTCATATTTGTGGGGTAACG-3′. The sequences of all constructs were verified by DNA sequencing (GATC Biotech).
Transformation of P. pastoris and Expression Screening
MDR3 expression constructs were transformed into electro-competent P. pastoris X33 cells (Invitrogen) using standard procedures, and the expression level was analyzed as described in Ellinger et al. (18).
Fermentation of MDR3 Transformed P. pastoris Cells
For large-scale expression, P. pastoris cells containing the chromosomally integrated wild type MDR3, the E558Q/E1702Q double mutant, or the Q1174E mutant gene were fermented in a 15-liter table-top glass fermenter (Applikon Biotechnology) according to the P. pastoris fermentation guidelines from Invitrogen. Usually, a volume of 7 liters of basal salt media was inoculated with 1 liter of an overnight culture grown in MGY (1.34% yeast nitrogen base, 1% glycerol, and 4 × 10−5% biotin) media. During the glycerol-fed batch phase, ∼500 ml of 50% (v/v) glycerol added with 12 ml/liter PTM1 salts was fed for 4 h. Protein expression was induced by addition of 3.6 ml/h/liter of methanol for 48 h. During the complete fermentation, the temperature was set at 30 °C; oxygenation was kept above 20% O2 saturation, and the mixer was set at 1000 rpm. Cells were harvested by centrifugation (5000 × g, 10 min, 4 °C), flash-frozen in liquid nitrogen, and stored at −80 °C until further use. Under these conditions, 1.4 kg of wet cell mass were routinely obtained.
Preparation of Crude Membrane Vesicles for Protein Purification
Generally, 100 g of frozen P. pastoris cells expressing the respective MDR3 protein (wild type or mutant) were thawed on ice, washed with 500 ml of ice-cold 50 mm Tris-HCl, pH 8.0, and resuspended at a concentration of 0.5 g of cells/ml in homogenization buffer (50 mm Tris-HCl, pH 8.0, 0.33 m sucrose, 75 mm NaCl, 1 mm EDTA, 1 mm EGTA, 100 mm 6-aminocaproic acid, 1 mm DTT) containing a protease inhibitor mixture (Roche Applied Science). Cells were disrupted by two passages through a pre-cooled TS Series Cell Disrupter (Constant Systems) at 2.7 kbar. After cell debris was removed by three centrifugation steps (10 min at 5000 × g, 4 °C, and two times for 30 min at 15,000 × g, 4 °C), crude membrane vesicles were prepared by ultracentrifugation for 1 h at 125,000 × g at 4 °C. Membrane vesicles were resuspended in buffer A (50 mm Tris-HCl, pH 8.0, 50 mm NaCl, 30% (v/v), glycerol) and flash-frozen in liquid N2.
Solubilization and Purification of MDR3
The purification of wild type MDR3 or mutant was performed as described previously with a few modifications (18). All procedures were carried out at 4 °C. Crude membrane vesicles equivalent to 100 g of wet cells were diluted to a final concentration of 15 mg/ml total protein with buffer A and solubilized with 1% (w/v) of FC-16 for 1 h at 4 °C. Nonsolubilized membrane vesicles were removed by centrifugation at 125,000 × g at 4 °C for 1 h. All buffers typically contained 2× critical micelle concentration of FC-16 (0.0011% (w/v) FC-16) and were cooled to 4 °C. The supernatant supplemented with 20 mm imidazole was loaded onto a Ni2+-loaded HiTrap chelating column (5 ml, GE Healthcare) and washed with 10 column volumes of buffer A supplemented with 20 mm imidazole. Proteins were eluted in one step with buffer B (50 mm Tris-HCl, pH 8.0, 50 mm NaCl, 200 mm imidazole, 20% (v/v) glycerol). The immobilized metal ion affinity chromatography eluate was diluted five times with CaCl2 binding buffer (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1 mm MgCl2, 2 mm CaCl2, and 20% (v/v) glycerol), applied to 4 ml of calmodulin affinity resin equilibrated in CaCl2 binding buffer and incubated with the calmodulin resin overnight at 4 °C on a rotator. The resin was transferred into a gravity flow column and washed with 10 column volumes of CaCl2 binding buffer. Protein was eluted with 5 column volumes of EGTA elution buffer (2 mm EGTA, 50 mm Tris-HCl, pH 7.4, 150 mm NaCl, and 20% (v/v) glycerol). The purified protein was directly used for ATPase activity measurements or aliquoted, snap-frozen in liquid nitrogen, and stored at −80 °C. Aliquots of the samples were analyzed by Coomassie Blue-stained SDS-PAGE and immunoblotting.
Labeling of MDR3 with BODIPY® FL Maleimide
Purified wild type MDR3 and mutants were incubated with 10-fold molar excess of BODIPY® FL maleimide (BODIPY® FL N-(2-aminoethyl))maleimide, Molecular Probes) at room temperature for 20 min. BODIPY®FL maleimide was added from a stock solution prepared in DMSO. The concentration of DMSO in the mixture did not exceed 0.2% (v/v). The reaction was terminated by the addition of a 20-fold molar excess of dithiothreitol, and samples were stored on ice until ATPase activity measurements. The labeling was analyzed by SDS-PAGE and fluorescence imaging at 488 nm excitation and 520 nm emission wavelength.
ATPase Activity Measurements of MDR3
The ATPase activity of MDR3 was examined with the malachite green assay by determination of released free inorganic orthophosphate as described previously with a few changes in the experimental procedures (18, 26, 27). Reactions were performed in a total volume of 100 μl in 50 mm Tris-HCl, pH 7.5 (at 37 °C), containing 2× critical micelle concentration of FC-16 and 10 mm MgCl2. 5–10 μg of purified detergent-solubilized MDR3 proteins were used, and the reaction was started by adding 2 mm ATP at 37 °C and stopped after 0 and 40 min by the addition of 25 μl of the reaction mixture into 175 μl of 20 mm ice-cold H2SO4. Subsequently, 50 μl of dye solution (0.096% (w/v) malachite green, 1.48% (w/v) ammonium molybdate, and 0.173% (w/v) Tween 20 in 2.36 m H2SO4) was added. After 15 min, the amount of free phosphate was quantified by measuring the absorption at 595 nm. For substrate-stimulated ATPase activity, purified MDR3 was incubated at room temperature for 20 min with a defined volume taken from a 5 mm lipid stock solution and sonicated for 30 s to facilitate the incorporation of lipids into the detergent-protein micelles. The lipid-protein sample was directly used for ATPase activity measurement. For determination of kinetic parameters, ATP concentration was varied. The kinetic data were analyzed according to Michaelis-Menten kinetics as shown in Equation 1,
 |
Here, v describes the reaction velocity; vmax is the maximal reaction velocity; S the substrate concentration, and Km is the Michaelis-Menten constant.
Inhibition by phosphate analogues was assayed with stock solutions containing 100 mm BeCl2 complemented with 500 mm NaF (100 mm BeFx) and 100 mm AlCl3 complemented with 500 mm NaF (100 mm AlFx), respectively. Orthovanadate solutions (100 and 10 mm) were prepared from Na3VO4 at pH 10 and boiled for 2 min prior to use (28). To determine the IC50 values, the ATPase activity was plotted against the log of inhibitor concentration. The data were analyzed according to Equation 2,
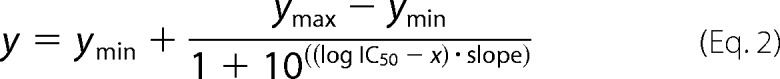 |
Here, ymax denotes the ATPase activity in the starting plateau, and ymin denotes the ATPase activity of the final inhibited plateau. y describes the ATPase activity value, and x represents the logarithmic concentration of the inhibitor. The IC50 value is calculated as the value of the inhibitor concentration used at an ATPase activity inhibition of 50%. This corresponds to the inflection point of the resulting curves.
For subsequent data evaluation, a reaction with EDTA (final concentration of 20 mm) was performed, and the autohydrolysis of ATP was subtracted. For calibration of free phosphate concentrations, a Na2HPO4 standard curve was used. All experiments were generally performed three times if not otherwise stated. Fitting was carried out using the GraphPad Prism Software (version 5.0a).
Immunofluorescence of Liver Tissue and MDR3-EYFP-transfected HEK293 Cells
Immunofluorescence staining and confocal microscopy were performed as described recently (29, 30). Briefly, snap-frozen sample liver tissue was cryo-sectioned and incubated with the transporter-specific antibodies K24 for bile salt export pump and P3II26 for MDR3 (Thermo Scientific) (31). Staining was then performed with secondary antibodies conjugated to Alexa Fluor 488 (green) and Alexa Fluor 546 (red). HEK293 cells were transiently transfected with either wild type MDR3-EYFP or MDR3Q1174E-EYFP for 48 h using the pEYFP-N1 vector and polyethyleneimine ((average mass of 25 kDa; Sigma) as a transfection agent. After fixation for 30 s with ice-cold methanol, plasma membranes were immunostained with a monoclonal Na+/K+-ATPase antibody (clone M7-PB-E9, Sigma). Goat anti-mouse Cy3 (Dianova) was used as the secondary antibody. Tissue and cells were visualized with an LSM 510 Meta confocal laser scanning microscope (Zeiss) using excitation wavelengths of 488 nm for EYFP and Alexa Fluor 488 or 543 nm for Cy3 and Alexa Fluor 546, respectively. 505–530-nm (green) and 560–615-nm band pass filters (red) were used for signal detection.
RESULTS
Expression and Purification of the Human ABC Transporter MDR3 in P. pastoris
Previously, we described the expression of wild type MDR3 and the ATP hydrolysis-deficient mutant (E558Q/E1207Q, later called the EQ/EQ mutant) in the methylotrophic yeast P. pastoris. To determine the influence of the Q1174E mutation located in the extended X loop on the ATPase activity, we mutated the Gln to Glu at position 1174 of MDR3 and expressed the mutant in P. pastoris. The fermentation of each P. pastoris strain, containing MDR3 chromosomally integrated, yielded about 1.4 kg of wet cell weight. We found that the detergent Fos-choline 16 (FC-16) solubilized MDR3 in large quantities, and fluorescence detection size exclusion chromatography analysis demonstrated an appropriate quality in terms of monodispersity of MDR3 solubilized in FC-16 (18). Therefore, we solubilized and purified all three MDR3 variants in FC-16 by means of tandem-affinity chromatography (TAP), which was established by Rigaut et al. (32). The purity of wild type MDR3, EQ/EQ, and Q1174E mutants was analyzed by Coomassie Brilliant Blue-stained SDS gels, and MDR3 was identified by immunoblot analysis using the C219 antibody, which is specific for the human ABC transporters MDR1 and MDR3 (Fig. 1A, left and middle panels). For wild type MDR3, we obtained 6.3 mg of highly purified (>90%) protein from 100 g wet cell weight in eight independent purifications compared with MDR3 EQ/EQ mutant with 3.4 mg of protein and the MDR3 Q1174E mutant with 2.1 mg of protein per 100 g wet cell weight (Table 1). The lower yield and purity of the mutants compared with the wild type were likely due to the reduced expression levels of the mutants in P. pastoris (data not shown).
FIGURE 1.

A, human wild type MDR3, the E558Q/E1207Q double mutant, and the Q1174E mutant purified from P. pastoris. The MDR3 variants containing a C-terminal His6 tag and a calmodulin-binding peptide were expressed in the yeast P. pastoris and purified as described under “Experimental Procedures.” 10 μg of purified MDR3 was resolved on a 7% SDS-PAGE and either stained with Coomassie Brilliant Blue (left panel) or detected by immunoblotting (middle panel) using the monoclonal anti-P-gp C219 antibody. MDR3 was cross-linked by the thiol-reactive maleimide-BODIPY fluorophore (MDR3-BODIPY) and analyzed by fluorescence imaging using excitation wavelength at 488 nm and emission wavelength at 520 nm (right panel). Molecular mass markers are shown on the left. MDR3 is indicated with an arrow, and degradation products of MDR3 are shown with asterisks. B, ATPase activity of purified wild type MDR3 (black triangle) and MDR3-BODIPY (blue circles) in the absence (left panel) or presence (right panel) of DOPC lipids. C, ATPase activity of purified wild type MDR3 (blue), the ATPase-deficient MDR3 EQ/EQ mutant (green), and the Q1174E mutant (magenta) and the corresponding cross-linked BODIPY derivatives in the presence of 2 mm ATP and 300 μm DOPC (PC).
TABLE 1.
Purification and ATPase activity of wild type MDR3, the EQ/EQ, and the Q1174E mutant
| Purified protein | Yield | No. of purifications | Km (MgATP) | vmax | kcat |
|---|---|---|---|---|---|
| mg/100 g wet cell weight | mm | nmol min−1 mg−1 | s−1 | ||
| Wild type | 6.3 ± 1.2 | 8 | 2.17 ± 0.20 | 354 ± 13 | 0.83 ± 0.03 |
| 1.78 ± 0.10a | 536 ± 11a | 1.26 ± 0.03a | |||
| Wild type-BODIPY | 1.26 ± 0.10 | 186 ± 6 | 0.44 ± 0.01 | ||
| 1.43 ± 0.19a | 175 ± 10a | 0.41 ± 0.02a | |||
| E558Q/E1207Q | 3.4 ± 0.6 | 5 | b | b | b |
| Q1174E | 2.0 ± 0.2 | 5 | 1.04 ± 0.15 | 286 ± 16 | 0.64 ± 0.04 |
| Q1174E-BODIPY | 0.74 ± 0.10 | 198 ± 5 | 0.46 ± 0.01 |
a ATPase activity was in the presence of 300 μm DOPC lipids. The subtraction of the ATPase activity in the absence and presence of DOPC lipids revealed a Δvmax of 182 nmol min−1 mg−1 and Δkcat of 0.43 s−1 for wild type, a Δvmax of −9 nmol min−1 mg−1, and a Δkcat of ≤ 0.03 s−1 for labeled wild type MDR3-BODIPY.
b The observed ATPase activity of the EQ/EQ mutant results from co-purified, contaminating ATPase. Please see text for further details.
ATPase Activity of Isolated Wild Type MDR3, EQ/EQ Mutant, and Maleimide-BODIPY-labeled MDR3
In a previous study, we demonstrated that MDR3 exhibits a PC-induced ATPase activity of 15 nmol min−1 per mg of purified MDR3 in comparison with the ATPase activity of the ATP hydrolysis-deficient Walker B (EQ/EQ) mutant in the presence of 2 mm ATP and 10 mm Mg2+ (18). We used mass spectrometry to analyze an FC-16 solubilized and purified sample of wild type MDR3. Here, we observed that high amounts of detergent molecules were attached to MDR3 (data not shown). We increased the ratio of MDR3 containing crude membranes to FC-16 during solubilization and reduced the amount of FC-16 during the purification protocol as described under “Experimental Procedures.” Thus, we obtained a 6-fold higher stimulation of 105 ± 30 nmol min−1 mg−1 in the presence of DOPC lipids and 2 mm ATP (Fig. 1B). A similar effect was reported for the multidrug resistance protein LmrA from Lactococcus lactis (27).
In a first step of analyzing MDR3-related ATPase activity in detergent solution in the presence or absence of lipids, we determined the maximal reaction velocity (vmax) and the Michaelis-Menten constant (Km) of ATP hydrolysis of wild type MDR3 and the EQ/EQ mutant in the absence and presence of DOPC lipids, respectively. Purified protein was first activated with 300 μm DOPC lipids, and the amount of released inorganic phosphate was measured at various ATP concentrations at 37 °C for 40 min.
The ATP hydrolysis-deficient EQ/EQ mutant exhibited an ATPase activity with a vmax of 240 ± 5 nmol min−1 mg−1, but it could not be stimulated by adding DOPC lipids (vmax = 245 ± 5 nmol min−1 mg−1) (Table 1). The exchange of the highly conserved Glu to Gln in the Walker B motif of ABC transporter prevents in general the hydrolysis of ATP in both nucleotide-binding sites (21). Groen et al. (7) reported an important cytotoxicity caused by expression of wild type MDR3 in HEK293T cells, which was counteracted by the single mutation E558Q of the Walker B motif of the first NBD resulting in an inactive floppase. This suggests that the observed ATPase activity EQ/EQ mutant is likely derived from co-purified contaminating ATPases. For wild type MDR3, we obtained an increased vmax value of 354 ± 13 nmol min−1 mg−1 in the absence of DOPC lipids corresponding to a kcat value of 0.83 ± 0.03 s−1 (Fig. 1B, left panel; Table 1). ATPase activity of wild type MDR3 as well as the corresponding kcat value were increased in the presence of DOPC lipids. We observed a vmax value of 536 ± 11 nmol min−1 mg−1 corresponding to a kcat value of 1.26 ± 0.03 s−1 (Fig. 1B, right panel; Table 1). Furthermore, the activity of wild type MDR3 displayed a relatively high Km value of 2.17 ± 0.20 mm in the absence and 1.78 ± 0.10 mm in the presence of DOPC lipids. Within experimental error, these values are more or less identical. The subtraction of the maximal ATPase activity of ATP-deficient EQ/EQ mutant from the measured vmax value of wild type MDR3 resulted in the maximal basal ATPase activity of 114 nmol min−1 mg−1 for wild type MDR3. However, a higher ATPase activity of wild type MDR3 cannot be ruled out, because of the differences in quality of the purified proteins. To get rid of contaminating ATPases, we performed additional purification steps such as ion exchange chromatography or size exclusion chromatography, but both purification steps resulted in no improvement (data not shown). This might be caused by the incorporation of contaminating proteins and MDR3 within the same detergent-lipid micelles. In addition, we varied ion strength, exchanged FC-16 with a number of commonly used detergents such as dodecyl β-d-maltopyranoside or octyl glucoside during the purification procedure, or elongated the washing step during immobilized metal ion affinity chromatography resulting in loss of ATPase activity or aggregation of protein, which might be caused by a de-lipidation of MDR3, for example.
Cross-linking of Cys in the Walker A motif of MDR1 with maleimide derivatives was demonstrated to block ATP hydrolysis, and the covalent modification was used to investigate the function of MDR1 (33–35). Because the Cys residues 433 and 1073 in the Walker A motifs are conserved in MDR3 and MDR1 and both proteins share high amino acid sequence identity, we reasoned that cross-linking of Cys of MDR3 using a maleimide-fluorophore might block ATP hydrolysis of MDR3. Thus, the difference in activity of wild type and modified MDR3 should reflect the basal ATPase activity. We labeled wild type MDR3 and mutants with the thiol-reactive fluorophore maleimide-BODIPY (further called MDR3-BODIPY). Full-length MDR3 was predominantly labeled beside degradation products of MDR3 as determined by comparison of the fluorescence image and the immunoblot (Fig. 1A, middle and right panels). To exclude that labeling also inhibited the ATP hydrolysis of co-purified ATPases, we compared the ATPase activity of the EQ/EQ mutant with the corresponding labeled protein in the presence of 2 mm ATP and observed a slight decrease of the ATPase activity of 17 nmol min−1 mg−1 (Fig. 1C), whereas the ATPase activity of wild type MDR3-BODIPY was decreased by 67 nmol min−1 mg−1 in the presence of 2 mm ATP. This difference therefore reflects the basal activity of MDR3. To examine whether co-purified ATPases exhibited PC-induced stimulation, we determined the ATPase activity of MDR3-BODIPY in the presence of DOPC lipids and observed no stimulation (Fig. 1, B and C). For MDR3-BODIPY, we determined a vmax value of 186 ± 6 nmol min−1 mg−1 and a corresponding Km value of 1.26 ± 0.10 mm in the absence of DOPC lipids, which was identical in the presence of DOPC lipids within the experimental error (vmax = 175 ± 10 nmol min−1 mg−1 and Km = 1.43 ± 0.19 mm) (Fig. 1B and Table 1). These data demonstrated that MDR3 exhibited a basal ATPase activity between 114 and 168 nmol min−1 mg−1 and that the PC-induced ATPase activity exclusively reflected MDR3-specific ATP hydrolysis.
Headgroup of the Lipid Moiety Determines MDR3 Substrate Specificity
The physiological function of MDR3 is the flop of PC lipids into the canaliculus of hepatocytes. Transport experiments with MDR3-transfected polarized pig kidney epithelial cells demonstrated that MDR3 flops fluorescently labeled short chain PC lipids but not the corresponding PE variants. Furthermore, van Helvoort et al. (5) ascertained that MDR3 distinguished between PC and sphingomyelin (SM) lipids. These findings led to the conclusion that MDR3 binds predominantly phospholipids with a choline headgroup and a diacyl backbone.
To exclude that the increased ATPase activity in the presence of PC lipids is caused by an unspecific effect of these lipids on MDR3, we determined the ATPase activity of wild type MDR3 in the presence of brain PE, DOPE, PS, and SM lipids. Data derived for other human ABC transporters demonstrated that lipids might display a stabilizing effect on the conformation of the protein, which would result in an increased ATPase activity (36–39). We found that neither the presence of brain PE nor DOPE lipids increased MDR3 ATPase activity (Fig. 2, A and B). Furthermore, the addition of PS or SM lipids resulted in no significant stimulation (Fig. 2, C and D). To further investigate the influence of liver PC and DOPC lipids on the ATPase activity of wild type MDR3 in the solubilized state, Km and vmax values were determined by Michaelis-Menten kinetic analysis (Fig. 2, E and F). Liver PC and DOPC lipids stimulated the ATPase activity in a concentration-dependent manner. The maximal velocity vmax value was slightly increased for liver PC (109.8 ± 5.9 nmol min−1 mg−1) as compared with DOPC lipids (84.0 ± 3.4 nmol min−1 mg−1) (Table 2). Assuming that 100% homogeneous and active MDR3 is present in the ATPase assay, approximately 4 s are required to catalyze the flop of one lipid molecule by one molecule of MDR3 in the case of DOPC and liver PC lipids. The Km values were 16.6 ± 2.6 μm for liver PC lipids and 12.0 ± 1.6 μm for DOPC lipids.
FIGURE 2.

Concentration dependence of the ATPase activity of MDR3 in the presence of different kinds of lipids: A, brain PE lipids; B, DOPE lipids; C, PS lipids; D, SM lipids; E, liver PC lipids; and F, DOPC lipids, respectively. The ATPase activity was started by addition of 2 mm ATP and assayed for 40 min at 37 °C. The ATPase activity in the absence of lipids (159 ± 14 nmol min−1 mg−1) was subtracted from the ATPase activity in the presence of lipids. The data represent the average of at least six independent experiments (mean ± S.D.).
TABLE 2.
Kinetic parameters of MDR3 ATPase activity in the presence of different kinds of lipids
ATPase activity in the absence of lipids was determined and subtracted from the ATPase activity of MDR3 in the presence of liver PC, DOPC, brain PE, and DOPE lipids, respectively.
| Lipids | Km | vmax | kcat |
|---|---|---|---|
| μm | nmol min−1 mg−1 | s−1 | |
| Liver PC | 16.6 ± 2.6 | 109.8 ± 5.9 | 0.26 ± 0.01 |
| DOPC | 12.0 ± 1.6 | 84.0 ± 3.4 | 0.20 ± 0.01 |
Inhibition of MDR3 ATPase by Transition and Ground State Analogues
Next, we examined whether MDR3-specific ATP hydrolysis could be inhibited by transition and ground state analogues of phosphate. Phosphate analogues have been extensively used to analyze, for example, the catalytic mechanism of MDR1 and other human ABC transporters (36, 40–42). We determined the ATPase activity of wild type MDR3 after adding BeFx, AlFx, and orthovanadate in the presence and absence of DOPC lipids, respectively (Fig. 3A). BeFx and AlFx led to complete inhibition of ATP hydrolysis at a concentration of 1 mm and abolished the ATPase activity of MDR3 as well as the hydrolysis activity of the co-purified ATPases. No inhibition of the ATPase activity by orthovanadate was observed in the absence of DOPC lipids even at concentrations up to 3 mm. However, ATP hydrolysis in the presence of DOPC lipids is more strongly inhibited than in the absence of DOPC lipids (∼20%) at a concentration of 3 mm orthovanadate. Additionally, the Q1174E mutant was not susceptible to vanadate inhibition (Fig. 3B). Hence, orthovanadate inhibited exclusively the PC-stimulated ATPase activity of MDR3.
FIGURE 3.
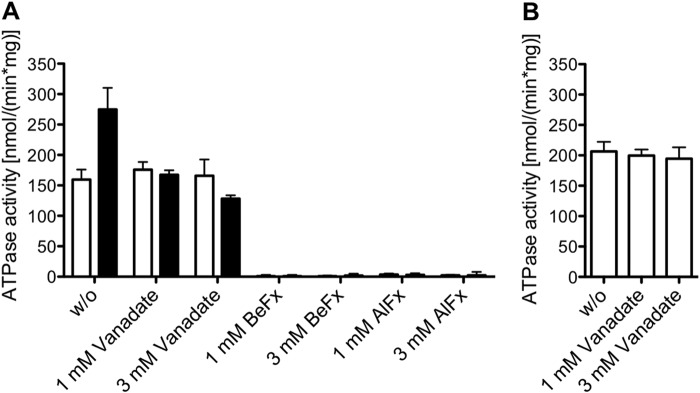
Inhibition of MDR3 ATPase activity by phosphate analogues. A, ATPase activity of purified wild type MDR3 was measured without (w/o, white column) and with 300 μm DOPC lipids (black column) in the presence of orthovanadate, BeFx, and AlFx. The reaction mixture contained 2 mm ATP and inhibitors at a final concentration of 1 or 3 mm. The data are means ± S.D. of at least four independent experiments. B, ATPase activity of Q1174E mutant in the presence of orthovanadate. The data represent the average of at least two independent experiments (mean ± S.D.).
Furthermore, we investigated the half-maximal inhibitory concentration (IC50) of BeFx, AlFx, and orthovanadate in the absence and presence of DOPC lipids. The calculated IC50 values are summarized in Table 3. The IC50 value for BeFx is slightly decreased in the presence of DOPC lipids (28.6 ± 1.0 μm) compared with the IC50 value of 38.0 ± 1.1 μm in the absence of DOPC lipids. In good agreement with this, the half-maximal ATPase inhibition of the close homologue MDR1 was achieved at a concentration of 23 μm BeFx (43). AlFx exhibited similar IC50 values of MDR3 ATPase activity of 199.8 ± 1.0 μm in the absence and 183.8 ± 1.0 μm in the presence of DOPC lipids, respectively. Interestingly, orthovanadate inhibited the MDR3-specific DOPC-stimulated ATPase activity, and we determined an IC50 value of 397.5 ± 2.0 μm. In contrast, purified human MDR1, which shares more than 86% amino acid similarity to MDR3, exhibited an IC50 of 2.3 μm (41, 44).
TABLE 3.
IC50 values of MDR3 ATPase activity inhibited by phosphate analogues in the absence or presence of 300 μm DOPC lipids
| Phosphate analogue | IC50 |
|
|---|---|---|
| −DOPC | +DOPC | |
| μm | ||
| Orthovanadate | 397.5 ± 2.0 | |
| BeFx | 38.0 ± 1.1 | 28.6 ± 1.0 |
| AlFx | 199.8 ± 1.0 | 183.8 ± 1.0 |
MDR3 Expression in a Liver Biopsy of a Patient with PFIC-3 and in Transfected HEK293 Cells
A 3-year-old girl with a PFIC-3 phenotype was compound heterozygous for a nucleotide exchange (c.3520C>G) in coding exon 26, resulting in the missense mutation Q1174E, as well as for the intronic donor splice site mutation c.286 + 1G>A of ABCB4 (Gene ID, 5244; mRNA reference NM_000443.3), which likely disrupts MDR3-mRNA expression from the related allele (19). Immunofluorescence staining of the patient's liver revealed an apparently normal immunoreactivity for MDR3 and the bile salt export pump (used here as a canalicular marker protein) at the canalicular membrane as compared with a normal liver (Fig. 4A). HEK293 cells were transiently transfected with wild type MDR3-EYFP or MDR3 Q1174E-EYFP (Fig. 4B). Both MDR3 variants were equally targeted to the plasma membrane. This indicates that the amino acid exchange allows normal protein folding and trafficking and that the mutation more likely results in a functional defect of MDR3.
FIGURE 4.
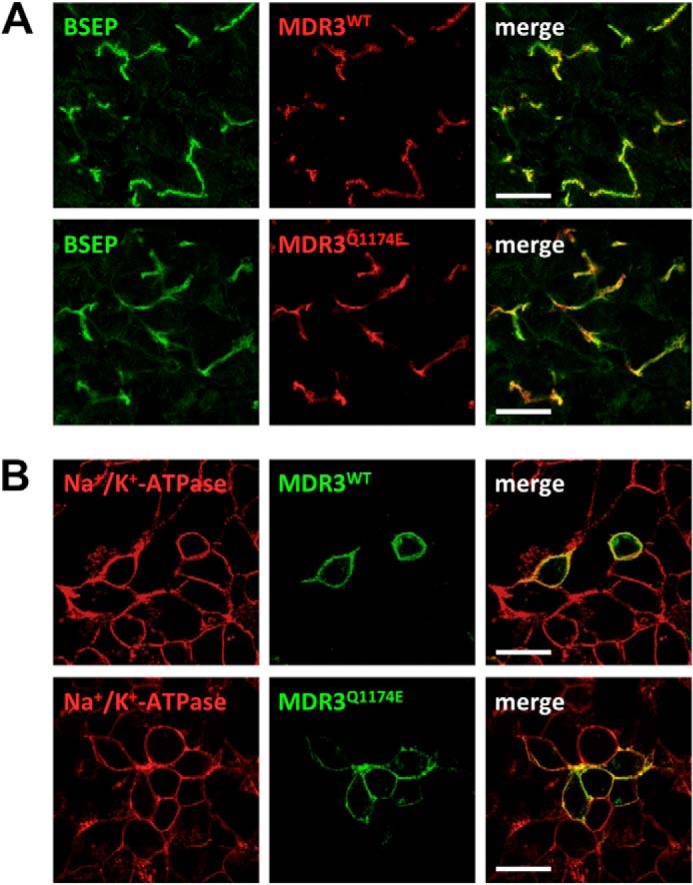
MDR3 mutation Q1174E does not affect protein localization in vivo or in vitro. A, sample from a liver with normal MDR3 expression (upper panel) and from the patient carrying the heterozygous Q1174E mutation (lower panel). The green fluorescence corresponds to bile salt export pump (BSEP), which acts as a canalicular marker, and the red fluorescence represents MDR3. B, transient expression of human wild type (upper panel) and Q1174E (lower panel) MDR3-eYFP (green) after transfection into HEK293 cells. Cells were fixed after 48 h and stained for the Na+/K+-ATPase as a plasma membrane marker (red). Scale bars, 20 μm.
Extended X Loop Mutation Q1174E Abolished PC-induced ATPase Activity
In ABC exporters, the highly conserved X loop (TEVGERG) is localized in the helical subdomain of the NBD in direct proximity of the ABC signature motif. X-ray structures of two bacterial ABC exporters, Sav1866 from Staphylococcus aureus and MsbA from Salmonella typhimurium, and five eukaryotic ABC transporters, MDR1 from Mus musculus, Cyanidioschyzon merolae, and Caenorhabditis elegans, ABCB10 from Homo sapiens, and Atm1 from Saccharomyces cerevisiae, revealed an enlarged transmission interface of the NBD and the TMD (22, 45–50). This transmission interface includes the X loop, which contacts the intracellular loop 1 (ICL1) of the opposing TMD and transmits signals of ATP binding and hydrolysis by the nearby ABC signature motif to the TMD (22). Oancea et al. (24) substituted the conserved glutamate (Glu-602) of the X loop of the transporter associated with antigen processing (TAP1/2) and demonstrated that peptide binding was not affected; however, transport activity was reduced from 20% for the E602D mutant to complete disruption for the E602R mutant, suggesting a pivotal role in transmitting conformational changes generated by ATP hydrolysis and substrate translocation. However, the molecular function of this transmission interface in terms of the coupling of the ATP hydrolysis cycle and substrate translocation is still not entirely clear.
We focused on the interaction between the glutamine at position 1174 of the NBD2, which is located in the extended X loop (TRVGDKGTQ) next to the leucine of the ABC signature motif and conserved in human ABC transporters such as MDR1, TAP1/2, and the cystic fibrosis transmembrane conductance regulator (CFTR) (Fig. 5A) (24, 25, 46) We hypothesized that the glutamine (Gln-1174) is indispensable for the transmission of the conformational change of the NBD2 to the ICL1 of the TMD.
FIGURE 5.

A, alignment of the amino acid sequence of MDR3 with selected human ABC transporters. The X loop motif is shaded in blue and the ABC signature motif in orange. Gln-1174 is colored in red. B, homology model of human MDR3 based on the structure of Sav1866 (Protein Data Bank code 2HYD) and the amino acid sequence of isoform B of MDR3 (51). One transporter half, consisting of transmembrane domain (TMD) and nucleotide-binding domain (NBD), is shown in blue and the other in yellow. The coupling helices are highlighted in red, and the X loop is colored in cyan. C, overlay of the MDR3 NBD2 of the isoform A (green) and isoform B (blue). Gln-1174 of the isoform A is a rotamer of Gln-1181 of the isoform B. D, close-up view of the interface between TMD and NBD2. Gln-1174 of the X loop is shown in stick representation (cyan).
Previously, we generated the homology model of human wild type MDR3 based on the crystal structure of the homologue of multidrug resistance ABC transporters Sav1866 (Protein Data Bank code 2HYD) as a structural template and the amino acid sequence of the isoform B of MDR3 (Fig. 5B) (51). The isoform B of MDR3 contains seven additional amino acids within the NBD2 compared with the major isoform A, which was used in our experiments. Nevertheless, the Gln of isoform A at position 1174 and of isoform B at position 1181 did not differ in the homology model of NBD2, because the orientation of the side chain of Gln-1174 represents a rotamer of Gln-1181 (Fig. 5C). The NBD structures of both isoforms are well aligned and had a root mean square deviation value of 1.6 Å over 229 Cα atoms. Thus, we used this structural model of MDR3 to generate the structure of the MDR3 Q1181E mutant (Q1174E isoform A). The close-up view of the transmission interface clearly indicates that Gln-1181 of human MDR3 can be replaced by a Glu without large conformational rearrangement, but it prohibited hydrogen bonding between Gln-1181 (Q1174 of isoform A) and Asp-166 of the coupling helix of ICL1 (Fig. 5D).
To ascertain whether the mutation of Gln-1174 to Glu abrogates ATP hydrolysis in vitro, we purified MDR3 Q1174E via TAP and assayed ATPase stimulation with DOPC, 1,2-dipalmitoyl-phosphatidylcholine, and liver PC lipids (Fig. 6). These data clearly demonstrated that all tested PC lipids were incapable of stimulating ATPase activity of MDR3, a situation comparable with the ATPase-deficient EQ/EQ mutant and the labeled wild type MDR3-BODIPY sample (Fig. 1). To clarify whether the Q1174E mutant exhibits basal ATPase activity or abrogates the ATPase activity completely, we determined ATPase activity of the Q1174E mutant and the corresponding Cys-labeled Q1174E-BODIPY sample in the presence of 2 mm ATP (Fig. 1C). The ATPase activity of Q1174E was higher as compared with wild type MDR3 likely due to a higher degree of protein impurities in the purified sample. Notably, the inhibition of ATPase activity of Q1174E-BODIPY (69 nmol min−1 mg−1) was comparable with wild type MDR3-BODIPY (67 nmol min−1 mg−1) with 2 mm ATP indicating that the Q1174E mutant exhibited basal ATPase activity. In addition, we determined the kinetic parameters of Q1174E and Q1174E-BODIPY according to Michaelis-Menten (Table 1). We obtained a maximal velocity of 286 ± 16 and 198 ± 5 nmol min−1 mg−1 for Q1174E-BODIPY. The Km values were similar for Q1174E (Km = 1.04 ± 0.15 mm) and the corresponding labeled sample (Km = 0.74 ± 0.1 mm) within experimental error. Taken together, the Q1174E mutant exhibited a higher affinity to ATP and a reduced basal activity of ∼90 nmol min−1 mg−1 compared with wild type floppase (vmax = 168 nmol min−1 mg−1; Km = 2.17 ± 0.2 mm). On the basis of these data, we suggest that the signals of substrate binding and substrate transport are not transmitted properly in this mutant.
FIGURE 6.
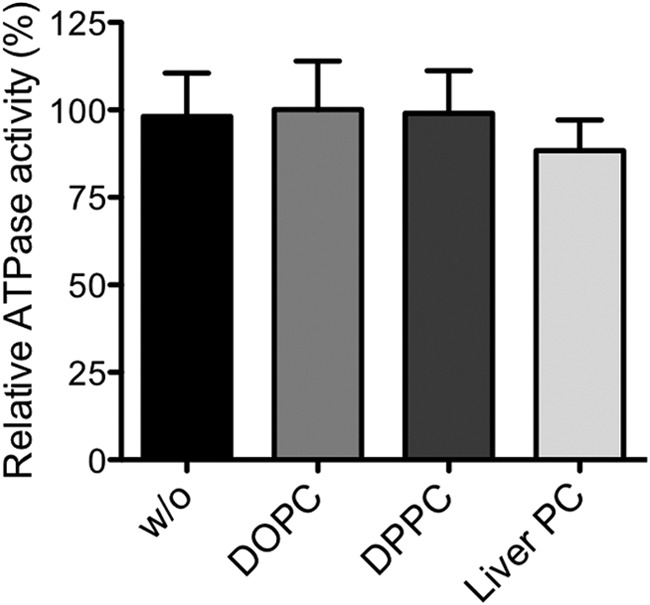
ATPase activity of the purified Q1174E mutant was measured without (w/o) lipids and with DOPC, DPPC, or liver PC lipids as described under “Experimental Procedures.” None of these lipids stimulated the ATPase activity of the Q1174E mutant. One hundred percent activity represents 223 ± 25 nmol min−1 mg−1.
DISCUSSION
Recently, we established the expression of human MDR3 in the yeast P. pastoris and purified the protein in amounts suitable for a detailed functional analysis with a purity of more than 90% in the case of wild type MDR3 (Fig. 1). In ABC transporters, binding of the substrate to the TMD typically stimulates ATP hydrolysis at the NBDs, and the energy from the hydrolysis is used to translocate the substrate across the membrane (52). The modulation of ATP hydrolysis upon substrate binding and translocation has been reported for several ABC transporters and is frequently used as a readout of transporter function and activity (36, 40, 44, 53, 54). However, no specific ATPase activity of MDR3 has been reported so far (16, 17). We demonstrated previously that wild type MDR3 exhibited a PC-stimulated ATPase activity (18). In this study, we characterized the ATPase activity of wild type MDR3 in terms of kinetic parameters, substrate spectrum, and the effect of phosphate analogues. We demonstrated that lower amounts of FC-16 increased the ATPase activity of MDR3. Former studies have shown that especially these zwitterionic detergents tend to deactivate proteins (55). The influence of the concentration of detergent on the ATPase activity was also demonstrated for MDR1 (37, 56). We observed a basal ATPase activity of wild type MDR3, but due to co-purified contaminating impurities that might act as ATPases, the observed basal ATPase activity is derived from MDR3 and impurities.
Cross-linking of wild type MDR3 with maleimide-BODIPY blocks basal and PC-induced ATPase activity as demonstrated for MDR1 (33–35), whereas the ATP hydrolysis-deficient EQ double mutant (E558Q/E1207Q) showed no PC stimulation, and ATPase activity of the labeled EQ/EQ mutant was only marginally reduced (Table 1 and Fig. 1). The PC-induced ATPase activity of wild type MDR3 was about 9-fold lower than the described ATPase activity of detergent-soluble mouse (1.5 μmol min−1 mg−1) and human MDR1 (1.8 μmol min−1 mg−1) in the presence of verapamil and lipids, but it was comparable with the substrate-induced ATPase activity of other members of the human liver ABC transporter family such as ABCC3 (vmax = 170 nmol min−1 mg−1 in the presence of 1 mm taurocholate) and ABCG5/G8 (vmax = 256 nmol min−1 mg−1 in the presence of taurocholate or taurodeoxycholate) (36, 37, 40, 44). Furthermore, purified chimera protein composed of MDR1 TMDs and MDR3 NBDs expressed in HEK293 cells exhibited drug-stimulated ATPase in the presence of vinblastine (vmax = 170 nmol min−1 mg−1) and verapamil (vmax = 450 nmol min−1mg−1), respectively, but it did not mediate bile salt-dependent PC efflux (17). These findings confirm that MDR3 NBDs are able to accomplish substrate-stimulated ATP hydrolysis in the detergent-solubilized state. Despite the high degree of amino acid sequence identity between MDR1 and MDR3 (>85% homology to human MDR1 and 80% to mouse Mdr1a (previously called mouse MDR3)), they exhibited different maximal ATPase activities in the presence of the transport substrate but similar and relatively high Km values for ATP (MDR3, Km = 1.90 ± 0.27 mm; MDR1, Km = 1.5 mm (44)), which were in a range typical for ABC transporters (36, 40, 44, 57). Because PC lipids are present in high concentrations in the plasma membrane, Ishigami et al. (17) suggested that MDR3 is a low affinity transporter optimized for PC translocation. Our data confirm such a suggestion. Nevertheless, bile salts were shown to be the driving force for PC secretion, and currently, we cannot rule out that bile salts influence the ATPase activity of MDR3 (6, 7, 58).
Furthermore, we demonstrated that PC lipids specifically stimulated MDR3 ATPase activity, whereas brain PE, DOPE, PS, and SM lipids did not induce any stimulation (Fig. 2). This is consistent with previously reported data (3–6, 16). All experiments showed that MDR3 translocates fluorescently labeled short chain PC lipids or long chain derivatives but not PE, SM, or ceramides. In conclusion, MDR3 binds exclusively phospholipids with a choline headgroup. However, we cannot exclude that MDR3 translocates MDR1 substrates as well. There is evidence that MDR3 is able to transport digoxin, paclitaxel, and vinblastine and that this transport is inhibited by either verapamil, cyclosporin A, or PSC833 (16). Furthermore, MDR3 gene products were found in paclitaxel-, doxorubicin-, and vincristine-resistant cell lines, indicating that MDR3 might be involved in multidrug resistance (59). Further investigations are required to address this issue.
The ATPase activity of many ABC proteins, such as MDR1, is efficiently inhibited by phosphate analogues such as orthovanadate, aluminum, and beryllium fluoride (41, 44). For MDR3, it was shown that nucleotide trapping by orthovanadate is indeed possible (16). Here, we have demonstrated that aluminum fluoride and beryllium fluoride inhibited ATPase activity of MDR3 and co-purified contaminating NTPases. Importantly, orthovanadate inhibited exclusively PC-induced MDR3-specific ATPase activity at high concentrations (Fig. 3). In contrast, mouse Mdr1a was completely inhibited at a concentration of 200 μm orthovanadate, and human MDR1 showed an IC50 of 2.3 μm, ∼165-fold lower than the IC50 value found for MDR3 (Table 3) (41, 44). Nevertheless, our data are in agreement with other studies on human ABC transporters. Orthovanadate did not inhibit ABCA1, and bile acid-stimulated ATPase hydrolysis of ABCG5/G8 was also only inhibited at high orthovanadate concentrations (40, 54).
Structural studies on the full-length ABC transporter Sav1866 have revealed that the NBDs of ABC exporters contain a conserved X loop motif, which transmits the conformational changes of ATP binding and hydrolysis to the ICLs of the TMD (22). The molecular function of this transmission interface relating coupling of the ATP hydrolysis cycle and substrate transport has not been extensively studied yet. To date, only a few studies dealt with the X loop of ABC transporters (22, 24, 25). Previously, the X loop Q1174E mutant of MDR3 in a young patient showing symptoms of PFIC-3 was described in Kubitz et al. (19). Thus, we substituted the conserved Gln in the extended X loop of MDR3 to Glu, expressed the Q1174E mutant in P. pastoris, and purified the protein employing the same procedure as described for wild type MDR3. We observed that the Q1174E mutant was purified with smaller yields and homogeneity, which is likely caused by the lower expression levels in P. pastoris. However, lower integration numbers of the gene expression cassette into the target chromosome cannot be ruled out. Here, we demonstrated that the Q1174E mutant was properly located in the plasma membrane of hepatocytes and MDR3-transfected HEK293 cells indicating a functional defect of MDR3 (Fig. 4). Cross-linking of the Q1174E mutant demonstrated that the Q1174E exhibited basal ATPase activity comparable with wild type in the presence of 2 mm ATP (Fig. 1). In contrast, the Q1174E exhibited a slightly increased affinity to ATP and a reduced maximal ATPase activity (Table 1) as the wild type floppase. Furthermore, all tested PC lipids were not capable of stimulating ATPase activity (Fig. 6). Based on these data and the structural model of MDR3, the exchange of the Gln at position 1174 of NBD2 prohibited the interaction with Asp-166 of the ICL1, and the conformational changes of the ICL as a result of PC lipid binding were not transmitted to the extended X loop of the NBD (Fig. 5).
This is in agreement with mutational and biochemical analysis of the ABC transporter TAP1/2 and CFTR and molecular dynamics simulation of human MDR1 (24, 25, 60). The mutational analysis of the conserved Glu within the X loop of TAP1/2 (TEVDEA(G/T)DVGEKG) demonstrated that substrate binding was not affected; however, substrate transport was drastically reduced (24). Moreover, Cys pair cross-linking experiments of the chloride channel CFTR showed that the X loops are in close proximity to ICLs connecting TMDs and also to the ATP-binding sites (25). He et al. (25) demonstrated that the interface between NBDs and ICLs of CFTR was involved in the stabilizing of interdomain contacts and regulation of the channel gating. Furthermore, Chang et al. (60) modeled the structure of human MDR1 based on mouse Mdr1a and investigated the transmission interface between NBDs and TMDs. They demonstrated that the amino acid residue Gln-1175 of NBD2, which is identical to Gln-1174 in MDR3, hydrogen bonds with Asp-164 (Asp-166 in MDR3) of the ICL1 and identified this residue pair Asp-164–Gln-1175 as key residue pair in the transmission interface. Furthermore, they suggested that the X loop plays an important role in formation of the outward-facing conformation of human MDR1 (60). Based on the biochemical data, we suggest that the conserved Gln next to the Leu of the ABC signature motif is crucial for the cross-talk between the extended X loop of the NBD and the coupling helices of the TMDs similar to the role of this region in MDR1. Further investigations are required to clarify the role of the individual amino acids, which are involved in signal transmission between the extended X loop and ICL1 of the TMDs, and should aid our understanding of how mutations relating to cholestatic diseases disrupt interdomain interactions of ATP binding and hydrolysis on a molecular level.
Taken together, we demonstrated exclusively that PC lipids stimulate the ATPase activity of detergent-solubilized human MDR3, which was exclusively inhibited by orthovanadate. For the first time, this study provides evidence that the glutamine next to the leucine of the ABC signature motif participates in the transmission of the substrate binding at the ICLs of the TMD to the extended X loop of the NBD.
Acknowledgments
We are indebted to Dr. E. Dumont (University of Rochester Medical Center, Rochester, NY) for the pSGP18 plasmid. We thank Nathalie Walter for technical assistance and Prof. Dr. Patrick Gerner for support. We thank Philipp Ellinger and Nils Hanekop for support and stimulating discussions and Dr. Sander Smits for critically reading the manuscript.
This work was supported by the Deutsche Forschungsgemeinschaft through the Clinical Research Group 217 TP 3 (to L. S.) and TP 1 (to R. K.) and the Collaborative Research Center 974 TP B3 (to R. K. and L. S.).
- ABC
- ATP-binding cassette
- CFTR
- cystic fibrosis transmembrane conductance regulator
- DOPC
- 1,2-dioleoyl-sn-glycero-3-phosphocholine
- DOPE
- 1,2-dioleoyl-sn-glycero-3-phosphatidylethanolamine
- MDR3/1
- multidrug resistance protein 3/1
- NBD
- nucleotide-binding domain
- PC
- phosphatidylcholine
- PFIC
- progressive familial intrahepatic cholestasis
- TAP
- tandem affinity purification
- TAP1/2
- transporter associated with antigen processing
- TMD
- transmembrane domain
- ICL
- intracellular loop
- EYFP
- enhanced YFP
- PL
- phospholipid
- BeFx
- beryllium fluoride
- AlFx
- aluminum fluoride.
REFERENCES
- 1. Oude Elferink R. P., Paulusma C. C. (2007) Function and pathophysiological importance of ABCB4 (MDR3 P-glycoprotein). Pflugers Arch. 453, 601–610 [DOI] [PubMed] [Google Scholar]
- 2. Smit J. J., Schinkel A. H., Oude Elferink R. P., Groen A. K., Wagenaar E., van Deemter L., Mol C. A., Ottenhoff R., van der Lugt N. M., van Roon M. A. (1993) Homozygous disruption of the murine mdr2 P-glycoprotein gene leads to a complete absence of phospholipid from bile and to liver disease. Cell 75, 451–462 [DOI] [PubMed] [Google Scholar]
- 3. Smith A. J., de Vree J. M., Ottenhoff R., Oude Elferink R. P., Schinkel A. H., Borst P. (1998) Hepatocyte-specific expression of the human MDR3 P-glycoprotein gene restores the biliary phosphatidylcholine excretion absent in Mdr2(−/−) mice. Hepatology 28, 530–536 [DOI] [PubMed] [Google Scholar]
- 4. Smith A. J., Timmermans-Hereijgers J. L., Roelofsen B., Wirtz K. W., van Blitterswijk W. J., Smit J. J., Schinkel A. H., Borst P. (1994) The human MDR3 P-glycoprotein promotes translocation of phosphatidylcholine through the plasma membrane of fibroblasts from transgenic mice. FEBS Lett. 354, 263–266 [DOI] [PubMed] [Google Scholar]
- 5. van Helvoort A., Smith A. J., Sprong H., Fritzsche I., Schinkel A. H., Borst P., van Meer G. (1996) MDR1 P-glycoprotein is a lipid translocase of broad specificity, while MDR3 P-glycoprotein specifically translocates phosphatidylcholine. Cell 87, 507–517 [DOI] [PubMed] [Google Scholar]
- 6. Morita S. Y., Kobayashi A., Takanezawa Y., Kioka N., Handa T., Arai H., Matsuo M., Ueda K. (2007) Bile salt-dependent efflux of cellular phospholipids mediated by ATP-binding cassette protein B4. Hepatology 46, 188–199 [DOI] [PubMed] [Google Scholar]
- 7. Groen A., Romero M. R., Kunne C., Hoosdally S. J., Dixon P. H., Wooding C., Williamson C., Seppen J., Van den Oever K., Mok K. S., Paulusma C. C., Linton K. J., Oude Elferink R. P. (2011) Complementary functions of the flippase ATP8B1 and the floppase ABCB4 in maintaining canalicular membrane integrity. Gastroenterology 141, 1927–1937 [DOI] [PubMed] [Google Scholar]
- 8. Oude Elferink R. P., Groen A. K. (2000) Mechanisms of biliary lipid secretion and their role in lipid homeostasis. Semin. Liver Dis. 20, 293–305 [DOI] [PubMed] [Google Scholar]
- 9. van der Bliek A. M., Kooiman P. M., Schneider C., Borst P. (1988) Sequence of mdr3 cDNA encoding a human P-glycoprotein. Gene 71, 401–411 [DOI] [PubMed] [Google Scholar]
- 10. Gottesman M. M., Fojo T., Bates S. E. (2002) Multidrug resistance in cancer: role of ATP-dependent transporters. Nat. Rev. Cancer 2, 48–58 [DOI] [PubMed] [Google Scholar]
- 11. Ueda K., Cardarelli C., Gottesman M. M., Pastan I. (1987) Expression of a full-length cDNA for the human “MDR1” gene confers resistance to colchicine, doxorubicin, and vinblastine. Proc. Natl. Acad. Sci. U.S.A. 84, 3004–3008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gottesman M. M., Pastan I. (1993) Biochemistry of multidrug resistance mediated by the multidrug transporter. Annu. Rev. Biochem. 62, 385–427 [DOI] [PubMed] [Google Scholar]
- 13. Buschman E., Gros P. (1991) Functional analysis of chimeric genes obtained by exchanging homologous domains of the mouse mdr1 and mdr2 genes. Mol. Cell. Biol. 11, 595–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Buschman E., Gros P. (1994) The inability of the mouse mdr2 gene to confer multidrug resistance is linked to reduced drug binding to the protein. Cancer Res. 54, 4892–4898 [PubMed] [Google Scholar]
- 15. Gros P., Buschman E. (1993) The mouse multidrug resistance gene family: structural and functional analysis. Int. Rev. Cytol. 137, 169–197 [PubMed] [Google Scholar]
- 16. Smith A. J., van Helvoort A., van Meer G., Szabo K., Welker E., Szakacs G., Varadi A., Sarkadi B., Borst P. (2000) MDR3 P-glycoprotein, a phosphatidylcholine translocase, transports several cytotoxic drugs and directly interacts with drugs as judged by interference with nucleotide trapping. J. Biol. Chem. 275, 23530–23539 [DOI] [PubMed] [Google Scholar]
- 17. Ishigami M., Tominaga Y., Nagao K., Kimura Y., Matsuo M., Kioka N., Ueda K. (2013) ATPase activity of nucleotide binding domains of human MDR3 in the context of MDR1. Biochim. Biophys. Acta 1831, 683–690 [DOI] [PubMed] [Google Scholar]
- 18. Ellinger P., Kluth M., Stindt J., Smits S. H., Schmitt L. (2013) Detergent screening and purification of the human liver ABC transporters BSEP (ABCB11) and MDR3 (ABCB4) expressed in the yeast Pichia pastoris. PLoS One 8, e60620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kubitz R., Bode J., Erhardt A., Graf D., Kircheis G., Müller-Stöver I., Reinehr R., Reuter S., Richter J., Sagir A., Schmitt M., Donner M. (2011) Cholestatic liver diseases from child to adult: the diversity of MDR3 disease. Z. Gastroenterol. 49, 728–736 [DOI] [PubMed] [Google Scholar]
- 20. Zaitseva J., Jenewein S., Wiedenmann A., Benabdelhak H., Holland I. B., Schmitt L. (2005) Functional characterization and ATP-induced dimerization of the isolated ABC-domain of the haemolysin B transporter. Biochemistry 44, 9680–9690 [DOI] [PubMed] [Google Scholar]
- 21. Zaitseva J., Jenewein S., Oswald C., Jumpertz T., Holland I. B., Schmitt L. (2005) A molecular understanding of the catalytic cycle of the nucleotide-binding domain of the ABC transporter HlyB. Biochem. Soc. Trans. 33, 990–995 [DOI] [PubMed] [Google Scholar]
- 22. Dawson R. J., Locher K. P. (2006) Structure of a bacterial multidrug ABC transporter. Nature 443, 180–185 [DOI] [PubMed] [Google Scholar]
- 23. Schmitt L., Tampé R. (2002) Structure and mechanism of ABC transporters. Curr. Opin. Struct. Biol. 12, 754–760 [DOI] [PubMed] [Google Scholar]
- 24. Oancea G., O'Mara M. L., Bennett W. F., Tieleman D. P., Abele R., Tampé R. (2009) Structural arrangement of the transmission interface in the antigen ABC transport complex TAP. Proc. Natl. Acad. Sci. U.S.A. 106, 5551–5556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. He L., Aleksandrov A. A., Serohijos A. W., Hegedus T., Aleksandrov L. A., Cui L., Dokholyan N. V., Riordan J. R. (2008) Multiple membrane-cytoplasmic domain contacts in the cystic fibrosis transmembrane conductance regulator (CFTR) mediate regulation of channel gating. J. Biol. Chem. 283, 26383–26390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Baykov A. A., Evtushenko O. A., Avaeva S. M. (1988) A malachite green procedure for orthophosphate determination and its use in alkaline phosphatase-based enzyme immunoassay. Anal. Biochem. 171, 266–270 [DOI] [PubMed] [Google Scholar]
- 27. Infed N., Hanekop N., Driessen A. J., Smits S. H., Schmitt L. (2011) Influence of detergents on the activity of the ABC transporter LmrA. Biochim. Biophys. Acta 1808, 2313–2321 [DOI] [PubMed] [Google Scholar]
- 28. Gordon J. A. (1991) Use of vanadate as protein-phosphotyrosine phosphatase inhibitor. Methods Enzymol. 201, 477–482 [DOI] [PubMed] [Google Scholar]
- 29. Kubitz R., Sütfels G., Kühlkamp T., Kölling R., Häussinger D. (2004) Trafficking of the bile salt export pump from the Golgi to the canalicular membrane is regulated by the p38 MAP kinase. Gastroenterology 126, 541–553 [DOI] [PubMed] [Google Scholar]
- 30. Keitel V., Burdelski M., Warskulat U., Kühlkamp T., Keppler D., Häussinger D., Kubitz R. (2005) Expression and localization of hepatobiliary transport proteins in progressive familial intrahepatic cholestasis. Hepatology 41, 1160–1172 [DOI] [PubMed] [Google Scholar]
- 31. Noé J., Stieger B., Meier P. J. (2002) Functional expression of the canalicular bile salt export pump of human liver. Gastroenterology 123, 1659–1666 [DOI] [PubMed] [Google Scholar]
- 32. Rigaut G., Shevchenko A., Rutz B., Wilm M., Mann M., Séraphin B. (1999) A generic protein purification method for protein complex characterization and proteome exploration. Nat. Biotechnol. 17, 1030–1032 [DOI] [PubMed] [Google Scholar]
- 33. Loo T. W., Clarke D. M. (1995) Covalent modification of human P-glycoprotein mutants containing a single cysteine in either nucleotide-binding fold abolishes drug-stimulated ATPase activity. J. Biol. Chem. 270, 22957–22961 [DOI] [PubMed] [Google Scholar]
- 34. Loo T. W., Clarke D. M. (1999) Determining the structure and mechanism of the human multidrug resistance P-glycoprotein using cysteine-scanning mutagenesis and thiol-modification techniques. Biochim. Biophys. Acta 1461, 315–325 [DOI] [PubMed] [Google Scholar]
- 35. Gabriel M. P., Storm J., Rothnie A., Taylor A. M., Linton K. J., Kerr I. D., Callaghan R. (2003) Communication between the nucleotide binding domains of P-glycoprotein occurs via conformational changes that involve residue 508. Biochemistry 42, 7780–7789 [DOI] [PubMed] [Google Scholar]
- 36. Chloupková M., Pickert A., Lee J. Y., Souza S., Trinh Y. T., Connelly S. M., Dumont M. E., Dean M., Urbatsch I. L. (2007) Expression of 25 human ABC transporters in the yeast Pichia pastoris and characterization of the purified ABCC3 ATPase activity. Biochemistry 46, 7992–8003 [DOI] [PubMed] [Google Scholar]
- 37. Lerner-Marmarosh N., Gimi K., Urbatsch I. L., Gros P., Senior A. E. (1999) Large scale purification of detergent-soluble P-glycoprotein from Pichia pastoris cells and characterization of nucleotide binding properties of wild-type, Walker A, and Walker B mutant proteins. J. Biol. Chem. 274, 34711–34718 [DOI] [PubMed] [Google Scholar]
- 38. Urbatsch I. L., Gimi K., Wilke-Mounts S., Lerner-Marmarosh N., Rousseau M. E., Gros P., Senior A. E. (2001) Cysteines 431 and 1074 are responsible for inhibitory disulfide cross-linking between the two nucleotide-binding sites in human P-glycoprotein. J. Biol. Chem. 276, 26980–26987 [DOI] [PubMed] [Google Scholar]
- 39. Schölz C., Parcej D., Ejsing C. S., Robenek H., Urbatsch I. L., Tampé R. (2011) Specific lipids modulate the transporter associated with antigen processing (TAP). J. Biol. Chem. 286, 13346–13356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Johnson B. J., Lee J. Y., Pickert A., Urbatsch I. L. (2010) Bile acids stimulate ATP hydrolysis in the purified cholesterol transporter ABCG5/G8. Biochemistry 49, 3403–3411 [DOI] [PubMed] [Google Scholar]
- 41. Urbatsch I. L., Tyndall G. A., Tombline G., Senior A. E. (2003) P-glycoprotein catalytic mechanism: studies of the ADP-vanadate inhibited state. J. Biol. Chem. 278, 23171–23179 [DOI] [PubMed] [Google Scholar]
- 42. Russell P. L., Sharom F. J. (2006) Conformational and functional characterization of trapped complexes of the P-glycoprotein multidrug transporter. Biochem. J. 399, 315–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sankaran B., Bhagat S., Senior A. E. (1997) Inhibition of P-glycoprotein ATPase activity by beryllium fluoride. Biochemistry 36, 6847–6853 [DOI] [PubMed] [Google Scholar]
- 44. Urbatsch I. L., Wilke-Mounts S., Gimi K., Senior A. E. (2001) Purification and characterization of N-glycosylation mutant mouse and human P-glycoproteins expressed in Pichia pastoris cells. Arch. Biochem. Biophys. 388, 171–177 [DOI] [PubMed] [Google Scholar]
- 45. Ward A., Reyes C. L., Yu J., Roth C. B., Chang G. (2007) Flexibility in the ABC transporter MsbA: Alternating access with a twist. Proc. Natl. Acad. Sci. U.S.A. 104, 19005–19010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Aller S. G., Yu J., Ward A., Weng Y., Chittaboina S., Zhuo R., Harrell P. M., Trinh Y. T., Zhang Q., Urbatsch I. L., Chang G. (2009) Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 323, 1718–1722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kodan A., Yamaguchi T., Nakatsu T., Sakiyama K., Hipolito C. J., Fujioka A., Hirokane R., Ikeguchi K., Watanabe B., Hiratake J., Kimura Y., Suga H., Ueda K., Kato H. (2014) Structural basis for gating mechanisms of a eukaryotic P-glycoprotein homolog. Proc. Natl. Acad. Sci. U.S.A. 111, 4049–4054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jin M. S., Oldham M. L., Zhang Q., Chen J. (2012) Crystal structure of the multidrug transporter P-glycoprotein from Caenorhabditis elegans. Nature 490, 566–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shintre C. A., Pike A. C., Li Q., Kim J. I., Barr A. J., Goubin S., Shrestha L., Yang J., Berridge G., Ross J., Stansfeld P. J., Sansom M. S., Edwards A. M., Bountra C., Marsden B. D., von Delft F., Bullock A. N., Gileadi O., Burgess-Brown N. A., Carpenter E. P. (2013) Structures of ABCB10, a human ATP-binding cassette transporter in apo- and nucleotide-bound states. Proc. Natl. Acad. Sci. U.S.A. 110, 9710–9715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Srinivasan V., Pierik A. J., Lill R. (2014) Crystal structures of nucleotide-free and glutathione-bound mitochondrial ABC transporter Atm1. Science 343, 1137–1140 [DOI] [PubMed] [Google Scholar]
- 51. Dzagania T., Engelmann G., Häussinger D., Schmitt L., Flechtenmacher C., Rtskhiladze I., Kubitz R. (2012) The histidine-loop is essential for transport activity of human MDR3. A novel mutation of MDR3 in a patient with progressive familial intrahepatic cholestasis type 3. Gene 506, 141–145 [DOI] [PubMed] [Google Scholar]
- 52. Higgins C. F., Linton K. J. (2004) The ATP switch model for ABC transporters. Nat. Struct. Mol. Biol. 11, 918–926 [DOI] [PubMed] [Google Scholar]
- 53. McDevitt C. A., Collins R., Kerr I. D., Callaghan R. (2009) Purification and structural analyses of ABCG2. Adv. Drug Deliv. Rev. 61, 57–65 [DOI] [PubMed] [Google Scholar]
- 54. Takahashi K., Kimura Y., Kioka N., Matsuo M., Ueda K. (2006) Purification and ATPase activity of human ABCA1. J. Biol. Chem. 281, 10760–10768 [DOI] [PubMed] [Google Scholar]
- 55. Seddon A. M., Curnow P., Booth P. J. (2004) Membrane proteins, lipids and detergents: not just a soap opera. Biochim. Biophys. Acta 1666, 105–117 [DOI] [PubMed] [Google Scholar]
- 56. Doige C. A., Yu X., Sharom F. J. (1993) The effects of lipids and detergents on ATPase-active P-glycoprotein. Biochim. Biophys. Acta 1146, 65–72 [DOI] [PubMed] [Google Scholar]
- 57. Pozza A., Perez-Victoria J. M., Sardo A., Ahmed-Belkacem A., Di Pietro A. (2006) Purification of breast cancer resistance protein ABCG2 and role of arginine-482. Cell. Mol. Life Sci. 63, 1912–1922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pohl A., Devaux P. F., Herrmann A. (2005) Function of prokaryotic and eukaryotic ABC proteins in lipid transport. Biochim. Biophys. Acta 1733, 29–52 [DOI] [PubMed] [Google Scholar]
- 59. Januchowski R., Wojtowicz K., Andrzejewska M., Zabel M. (2014) Expression of MDR1 and MDR3 gene products in paclitaxel-, doxorubicin- and vincristine-resistant cell lines. Biomed. Pharmacother. 68, 111–117 [DOI] [PubMed] [Google Scholar]
- 60. Chang S. Y., Liu F. F., Dong X. Y., Sun Y. (2013) Molecular insight into conformational transmission of human P-glycoprotein. J. Chem. Phys. 139, 225102. [DOI] [PubMed] [Google Scholar]