

Background: Frequent overactivation of c-Myc aggravates a spectrum of cancers. However, cancer-specific inhibition of c-Myc activity poses significant difficulty.
Results: Inhibition of 5-lipoxygenase down-regulates c-Myc expression and triggers proteasomal degradation in cancer cells but not in normal fibroblasts.
Conclusion: Inhibition of 5-lipoxygenase selectively disrupts c-Myc function in cancer cells.
Significance: Inhibition of 5-lipoxygenase may be a novel strategy to control oncogenic c-Myc signaling.
Keywords: Apoptosis, Eicosanoid, Lipoxygenase Pathway, Myc (c-Myc), Prostate Cancer, 5-Lipoxygenase, MK591
Abstract
Myc is up-regulated in almost all cancer types and is the subject of intense investigation because of its pleiotropic effects controlling a broad spectrum of cell functions. However, despite its recognition as a stand-alone molecular target, development of suitable strategies to block its function is hindered because of its nonenzymatic nature. We reported earlier that arachidonate 5-lipoxygenase (5-Lox) plays an important role in the survival and growth of prostate cancer cells, although details of the underlying mechanisms have yet to be characterized. By whole genome gene expression array, we observed that inhibition of 5-Lox severely down-regulates the expression of c-Myc oncogene in prostate cancer cells. Moreover, inhibition of 5-Lox dramatically decreases the protein level, nuclear accumulation, DNA binding, and transcriptional activities of c-Myc. Both the 5-Lox inhibition-induced down-regulation of c-Myc and induction of apoptosis are mitigated when the cells are treated with 5-oxoeicosatetraenoic acid, a metabolite of 5-Lox, confirming a role of 5-Lox in these processes. c-Myc is a transforming oncogene widely expressed in prostate cancer cells and maintains their transformed phenotype. Interestingly, MK591, a specific 5-Lox inhibitor, strongly affects the viability of Myc-overactivated prostate cancer cells and completely blocks their invasive and soft agar colony-forming abilities, but it spares nontransformed cells where expression of 5-Lox is undetectable. These findings indicate that the oncogenic function of c-Myc in prostate cancer cells is regulated by 5-Lox activity, revealing a novel mechanism of 5-Lox action and suggesting that the oncogenic function of c-Myc can be suppressed by suitable inhibitors of 5-Lox.
Introduction
Myc is a basic helix-loop-helix leucine zipper transcription factor that dimerizes with its binding partner MAX and associates with gene promoters containing the E-box motifs (CACGTG or CACATG) to induce gene transcription (1). It is up-regulated in almost all cancer types and is the subject of intense investigation because of its pleiotropic effects controlling a broad spectrum of functions, including cell proliferation, metabolism, differentiation, sensitization to apoptotic stimuli, and genetic instability, which are events intimately associated with initiation, promotion, and progression phases of cancer (2). Myc function is deregulated in most human cancers by a variety of mechanisms, including gene amplification, insertional mutations, or chromosomal translocation of the Myc gene (1, 2). Because of its central role in oncogenesis, Myc has emerged as a promising stand-alone molecular target for therapy of cancers afflicted with cells undergoing oncogene addiction. Recent experimental data suggest that even a brief inhibition of c-Myc expression may be sufficient to permanently stop tumor growth and induce regression of tumors, and Soucek et al. (4) have shown in a preclinical mouse model that c-Myc inhibition of RAS-induced lung adenocarcinoma, using a reversible systemic expression of a Myc mutant that antagonizes Myc activity, regressed lung tumors by triggering apoptosis in cancer cells (3). Myc inhibition also exerted profound growth arrest in normal tissues, although these were well tolerated (4), suggesting that direct targeting Myc could maintain the therapeutic ratio of cancer treatment by preferential killing of tumor cells relative to normal cells. Although Myc has been identified more than 30 years ago and anti-Myc agents such as antisense oligonucleotides, small interfering RNA (siRNA), or phosphorodiamidate morpholino oligomers have been developed, which induce tumor cell growth arrest, differentiation, and trigger apoptosis, direct targeting of Myc has yielded very limited success for clinical use (1–7). Thus, unique upstream or downstream regulator(s) that control Myc functions should be explored that may help to develop additional, more effective measures to modulate deregulation of c-Myc in cancer cells.
Prostate cancer is the most common form of malignancy and the second leading cause of cancer-related deaths in men in the United States (8). Epidemiological studies and experiments with laboratory animals have repeatedly suggested a link between consumption of high fat “Western” diets and clinical prostate cancer (9–11). Recent analysis points toward a role of ω-6 fatty acids, such as arachidonic acid, in the promotion and progression of prostate cancer; however, the underlying mechanisms have yet to be fully characterized. Arachidonic acid, an ω-6 polyunsaturated fatty acid, is metabolized via cyclooxygenase, lipoxygenase, and epoxygenase pathways to generate an array of metabolites that regulate a variety of cell functions, such cell proliferation, survival, motility, invasion, angiogenesis, and metastasis (12, 13). We and others have observed that arachidonic acid promotes growth of prostate cancer cells via metabolic conversion through the 5-Lox2 pathway (14–17). Interestingly, it was observed that prostate cancer cells continuously generate 5-Lox metabolites, from arachidonic acid in serum-free medium without any exogenous stimuli, which deliver signals via the G-protein-coupled receptor (OXER1) and eventual activation of PKC-ϵ (14, 16–20). This feature signifies a deregulated state of 5-Lox in prostate cancer cells because neutrophils, which express 5-Lox under normal culture condition, maintain 5-Lox in an inactive state that does not generate 5-Lox metabolites until activated by phosphorylation and intracellular calcium surge (21–24). Interestingly, we observed that inhibition of 5-Lox blocks production of 5-Lox metabolites and induces apoptosis both in androgen-sensitive as well as androgen-independent prostate cancer cells (16–20). This apoptosis is prevented by exogenous 5-hydroxyeicosatetraenoic acid and its dehydrogenated derivative 5-oxoeicosatetraenoic acid (5-oxoETE), suggesting that the 5-Lox activity plays an essential role in the survival of prostate cancer cells. Recently, we observed that 5-Lox is not expressed in normal prostate epithelium but is highly expressed both in human and mouse prostate tumor tissues as well as in prostate cancer cell lines,3 suggesting that 5-Lox is specifically expressed in prostate tumors, which together with its critical role in the survival of prostate cancer cells makes 5-Lox a promising target for therapy. However, details of the signaling mechanisms mediating the effects of 5-Lox metabolites in the regulation of survival and growth characteristics of prostate cancer cells are yet to be fully understood.
To gain an insight into downstream signaling mechanisms of 5-Lox, we performed a global gene expression array and observed that MK591, a specific inhibitor of 5-Lox activity (25–29), remarkably inhibits the expression of c-Myc mRNA. Moreover, inhibition of 5-Lox dramatically reduced the protein level, nuclear accumulation, DNA binding, transcriptional activity, and oncogenic function of c-Myc in prostate cancer cells. Interestingly, MK591 does not alter the basal level of c-Myc and its target proteins in normal fibroblasts, which do not express 5-Lox, suggesting that the oncogenic c-Myc function in transformed cells is specifically suppressed by inhibition of the 5-Lox activity. We also observed that Myc-driven prostate tumors express high levels of 5-Lox, and Myc-transformed prostate cancer cells depend on 5-Lox activity for their survival as well as metastatic abilities. Altogether, our findings indicate that the activity of 5-Lox is required for the oncogenic functions of c-Myc in prostate cancer cells and suggest that prostate cancer cells promote their survival and metastasis using arachidonic acid, a common fatty acid in “modern diets” via metabolic conversion through the 5-Lox pathway. Because deregulation of Myc is one of the most common abnormalities in human malignancy and is frequently observed in aggressive cancer cells, our findings open up a new avenue to effectively regulate c-Myc function through inhibition of 5-Lox activity by specific chemical inhibitors, such as MK591.
EXPERIMENTAL PROCEDURES
Cell Culture and Reagents
LNCaP human prostate cancer cells and human foreskin fibroblasts (HFF) were purchased from American Type Culture Collection (Manassas, VA). Cells were grown either in RPMI 1640 medium or DMEM (Invitrogen). All the media were supplemented with 10% FBS and antibiotics. Antibodies against c-Myc and survivin were purchased from R&D Systems (Minneapolis, MN), and antibodies against TMPRSS2, cyclin D1, CDK4, ATF3, TNF-α, and GADD45-α were from Santa Cruz Biotechnology (Santa Cruz, CA). Polyclonal antibody against Aurora kinase was purchased from Cell Signaling Technology (Danvers, MA). Polyclonal anti-5-Lox antibody was from ProteinTech (Chicago), and anti-β-actin antibody was purchased from Sigma. MK591 was obtained as a generous gift from Dr. Robert N. Young (Merck-Frosst Centre for Therapeutic Research, Quebec, Canada).
Cell Viability Assay
Cell viability was measured by Cell Titer 96® AQueous One Solution Cell Proliferation Assay (Promega Corp., Madison, WI) as described before (16–20).
Microscopy
Cells (∼3 × 105) were plated overnight in RPMI medium 1640 supplemented with 10% FBS onto 60 mm diameter tissue culture plates (Falcon) and allowed to grow for 48 h. On the day of experiment, the spent culture medium was replaced with 2 ml fresh RPMI medium and the cells were treated with inhibitors. Control cells were treated with solvent (DMSO). Photographs were taken with a Nikon digital camera attached to a LEICA microscope at ×400. Image acquisition and data processing were done with a Dell computer attached to the microscope using Q-Capture 7 software.
Gene Expression Array
LNCaP cells were plated and treated with 30 μm MK591 for 8 or 16 h at 37 °C. Then the cells were harvested, washed, and lysed in lysis buffer. RNA was isolated using Qiagen RNeasy mini kit (Qiagen, Valencia, CA). For reverse transcription, 500 ng of total RNA was used as template, and gene expression in treated and untreated samples was analyzed by Illumina Human HT-12v4 Whole Genome Gene Expression (WGGX) Array (Illumina Corp.) in duplicate.
Reverse Transcriptase and Real Time Quantitative-Polymerase Chain Reaction (RT-PCR and qPCR)
LNCaP cells were plated and treated with 30 μm MK591 at 37 °C. Then the cells were harvested and washed, and RNA was isolated from exponentially growing cells using Qiagen RNeasy mini kit from Qiagen. For reverse transcription (RT), 500 ng of total RNA was used as template, and expressions of 5-Lox and c-Myc were analyzed by PCR using human gene-specific primer sets for 5-Lox (forward, 5′-CCC GGG GCA TGG AGA GCA-3′; reverse, 5′-GCG GTC GGG CAG CGT GTC-3′) or c-Myc (forward, 5′-CAA GAG GCG AAC ACA CAA CGT C-3′; reverse, 5′-CTG TTC TCG TCG TTT CCG CAA C-3′). A primer set for β-actin (forward, 5′-CTC CTG CTT GCT GAT CCA CAT-3′; reverse, 5′-AAC CGC GAG AAG ATG ACC CAG-3′) was used as control. For the real time qPCR, 1 μg of total RNA was used for the RT reaction using high capacity cDNA-RT kit from ABI. Then the qPCRs were performed using TaqMan gene expression assay kits from ABI/Invitrogen using the ABI-7500 fast real time qPCR machine.
Western Blot
Cells (∼3 × 105) were plated in 60-mm diameter plates and allowed to grow for 48 h. The old medium was then replaced with 2 ml of fresh RPMI 1640 medium, and the cells were treated with inhibitors. After treatment, cells were harvested, washed, and lysed in lysis buffer (50 mm HEPES buffer, pH 7.4, 150 mm NaCl, 1 mm EDTA, 1 mm orthovanadate, 10 mm sodium pyrophosphate, 10 mm sodium fluoride, 1% Nonidet P-40, and a mixture of protease inhibitors). Proteins were separated by 12% SDS-PAGE and transferred to nitrocellulose membranes. Membranes were blocked with 5% nonfat milk solution and then blotted with the appropriate primary antibody followed by peroxidase-labeled secondary antibody. Bands were visualized by enhanced chemiluminescence detection kit from Pierce and analyzed with a densitometer using Kodak imaging software. Unless otherwise mentioned, protein blots were analyzed in three independent experiments.
DNA Binding Assay
LNCaP cells were treated, and nuclei were isolated using a kit from Sigma. Localization of c-Myc was detected by Western blot using nucleoporin-A as a control. DNA binding activity of c-Myc was measured using an ELISA kit from Active Motif (Carlsbad, CA) using 4 μg of nuclear extracts per assay. Free wild type (WT) and mutated E-box DNA sequences were used in parallel as positive and negative controls for assay validation.
Luciferase Assay
LNCaP cells were transfected with lentiviral E-box-luciferase constructs (>90% cells transfected), expanded, and re-plated in 96-well culture plates in triplicate. Cells were then treated with MK591, and the luciferase activity was measured by a luciferase assay kit from Promega Corp. (Madison, WI). Ibuprofen and 10058-F4 (a Myc-Max binding inhibitor) were used as negative and positive controls in parallel assays.
Invasion Assay
Invasion assay was done in Matrigel-coated Boyden transwell chambers from BD Biosciences. Transwells were soaked in 50 μl of serum-free medium for 30 min at room temperature, and then ∼40,000 cells (in RPMI 1640 medium containing 0.1% BSA) were placed into the upper chambers. These chambers were then placed in a 24-well plate (one per well) on top of 500 μl of RPMI 1640 medium containing 3% fetal serum as chemo-attractant. Inhibitors were added directly to the medium and mixed. Then the cells were incubated at 37 °C in the CO2 incubator for 16 h. Noninvaded cells along with Matrigel in the upper chambers were scraped with a cotton tip applicator, and then the membranes were fixed in methanol, stained with 0.25% crystal violet, and observed under a Leica microscope at ×200.
Soft Agar Colony Formation Assay
Colony formation assays were performed in 6-well plates by placing ∼15,000 LNCaP cells in 0.5 ml of 0.3% soft agar on top of a 2-ml base layer of 0.6% agar. Plates were allowed to settle, and then the wells were covered with 2 ml of fresh RPMI 1640 medium containing 10% FBS with or without inhibitors. Plates were incubated at 37 °C in the CO2 incubator for a maximum period of 3 weeks. Cell growth medium and inhibitors were exchanged every 4th day. At the end of the incubation, cells were stained with 0.25% crystal violet, and colonies were counted under a Leica microscope at ×150.
Immunohistochemistry
Formalin-fixed paraffin-embedded sections from treated and untreated LNCaP prostate tumors were de-paraffinized and soaked in graded concentrations of ethanol. Then the sections were treated with antigen-retrieval buffer in the microwave (power setting, high) for 1 min. After washing, sections were blocked in 10% horse serum for 1 h at room temperature and then treated either with control rabbit IgG or with primary rabbit polyclonal anti-c-Myc or anti-survivin antibody (1:100) overnight at 4 °C, followed by secondary anti-rabbit IgG labeled with HRP. After washing, slides were stained for 30 s using 3,3′-diaminobenzidine as substrate and then counter-stained with hematoxylin (Harris) for 30 s. Blocking, antibody treatment, and color development were done using Vectastain Elite impression kit (catalog no. MP-7401; Vector Laboratories, Burlingame, CA). Photographs were taken with a Nikon digital camera attached to a Leica microscope at ×600.
RESULTS
Inhibition of 5-Lox Decreases c-Myc mRNA Expression in Prostate Cancer Cells
We reported earlier that 5-Lox is continuously active in prostate cancer cells generating 5-hydroxyeicosatetraenoic acid series of metabolites and that treatment with chemical inhibitors of 5-Lox blocks production of 5-Lox metabolites and induces apoptosis in these cells (14, 16–20). Similar effects were found when these cancer cells were treated with lentiviral shRNA(s) against 5-Lox (19, 20), confirming a critical role of 5-Lox in prostate cancer cells and suggesting that 5-Lox may turn out to be an attractive therapeutic target for clinical prostate cancer. Although our findings uncovered that 5-Lox metabolites play an important role in prostate cancer cell biology, details of the underlying mechanisms by which 5-Lox metabolites promote survival and growth characteristics of prostate cancer cells have yet to be fully characterized.
To explore downstream signaling events mediated by 5-Lox activity, we performed a global gene expression array after treating prostate cancer cells with MK591, a specific second-generation chemical inhibitor of 5-Lox activity (25–29). Analysis of ∼34,000 genes in prostate cancer cells at 8 and 16 h post-MK591 treatment revealed that the expression of the c-Myc oncogene is significantly down-regulated. A set of ∼6000 significantly up- or down-regulated genes by MK591 treatment is shown in Fig. 1a. Detailed analysis of the global gene expression profile indicated that the c-Myc oncogenic signaling is presumably down-regulated in prostate cancer cells upon inhibition of 5-Lox activity (Fig. 1, b and c). Later, by PCR analysis we confirmed that expression of c-Myc mRNA is down-regulated in prostate cancer cells when treated with MK591 in a time-dependent manner (Fig. 1, d and e).
FIGURE 1.

Inhibition of 5-Lox down-regulates expression of c-Myc. a and b, LNCaP cells were plated and treated with MK591 (30 μm) at 37 °C for 8 and 16 h. Then the gene expression was analyzed by Illumina HT12-v4 whole genome gene expression array in duplicate. Data are presented as fold change in gene expression at 8 h post-treatment, which were obtained by dividing values of inhibitor-treated samples with the corresponding control solvent (0.2% DMSO)-treated samples. c shows actual values of c-Myc expression in arbitrary array units (AAU) at 8 and 16 h post-treatment presented as mean values of duplicate determination of each data point ± S.D. d, time-dependent change in the expression of c-Myc mRNA in LNCaP cells is shown after treatment with 30 μm MK591. Note: ibuprofen (a cyclooxygenase inhibitor) and PD176146 (a 15-lipoxygenase inhibitor) were used as negative controls. e, real time PCR data of c-Myc gene expression are shown in treated and control samples. Data are presented as mean values of triplicate determination of each data point ± S.E.
Inhibition of 5-Lox Dramatically Decreases the Protein Level of c-Myc in Prostate Cancer Cells
Next, we examined whether the decrease in c-Myc mRNA by MK591 is also associated with a decrease in the protein level of c-Myc. We observed that prostate cancer cells treated with MK591 show a dramatic decrease in the level of c-Myc proteins in a clear dose- and time-dependent manner, which is detectable as early as in 4 h post-treatment (Fig. 2, a and b). We also observed that MK591-induced loss of c-Myc protein is effectively prevented by 5-oxoETE, a metabolite of 5-Lox, suggesting that the MK591-induced loss of c-Myc in prostate cancer cells is mediated via inhibition of the 5-Lox activity (Fig. 2c). Additionally, we observed that knockdown of 5-Lox by shRNA dramatically decreases c-Myc protein level, confirming that in prostate cancer cells expression of c-Myc is tightly regulated by 5-Lox (Fig. 2d).
FIGURE 2.

Inhibition of 5-Lox by chemical inhibitor or shRNA decreases levels of c-Myc protein. LNCaP cells (3 × 105) were plated in 60-mm diameter plates and allowed to grow for 48 h. Then the old medium was replaced with 2 ml of fresh RPMI 1640 medium, and the cells were treated with drugs. a and b, dose- and time-dependent changes in the levels of c-Myc proteins by treatment with MK591 or ibuprofen or PD176146 are shown by Western blot. These experiments were done three times with similar results. Con, control. c shows that MK591 treatment-induced loss of c-Myc protein is significantly prevented by pretreatment with exogenous 5-oxoETE (an active metabolite of 5-Lox). d, effect of lentiviral shRNA knockdown of 5-Lox on c-Myc protein level is shown by Western blot. Representatives of two independent experiments with similar results are shown here.
Inhibition of 5-Lox Down-regulates Nuclear Accumulation, DNA Binding, and Transcriptional Activities of c-Myc
Because, the c-Myc oncoprotein is a transcription factor, we examined whether inhibition of 5-Lox affects the transcriptional activity of c-Myc using various in vitro assays. We observed a significant decrease in the nuclear accumulation of c-Myc protein upon MK591 treatment in a time-dependent manner (Fig. 3a). We also observed that the DNA binding activity of c-Myc is decreased accordingly when the cells are treated with MK591 (Fig. 3b). Moreover, we measured the transcriptional activity of c-Myc by transfecting prostate cancer cells with lentiviral luciferase constructs of the consensus c-Myc-binding sequence (E-box-Luciferase), and we observed that treatment with MK591 dramatically decreases the transcriptional activity of c-Myc in prostate cancer cells (Fig. 3, c–e). Interestingly, we found that 5-oxoETE (a metabolite of 5-Lox) prevents MK591 treatment-induced loss of c-Myc transcriptional activity, suggesting that the MK591-induced loss of c-Myc transcriptional activity in prostate cancer cells is mediated via its inhibition of the 5-Lox activity (Fig. 3f).
FIGURE 3.

Inhibition of 5-Lox down-regulates c-Myc function in LNCaP cells. a, LNCaP cells were plated and treated with MK591 (30 μm) at 37 °C for varying periods of time up to 16 h. Nuclear accumulation of c-Myc was determined by isolation of nuclei followed by detection of proteins by Western blot using nucleoporin as loading control. Ibu, ibuprofen. b, DNA binding activity of c-Myc was determined by ELISA using 4 μg of nuclear extracts per assay. Note: wild type (Wt) E-box consensus sequence neutralizes c-Myc-DNA binding activity, whereas the mutated (Mut) version of the sequence cannot. c–e, dose- and time-dependent effects of MK591 (MK) on c-Myc transcriptional activity (in terms of relative luciferase unit (RLU)) was detected by treating E-box-luciferase construct-transfected LNCaP cells with doses of inhibitors as indicated. Note: 10058-F4 (F4), an inhibitor of Myc-Max binding, was used as positive control. NT represents values obtained with the same number of nontransfected LNCaP cells under similar experimental conditions. f shows that MK591 treatment-induced loss of c-Myc-luciferase activity in LNCaP cells is significantly prevented by the 5-Lox metabolite, 5-oxoETE. These experiments were performed two to three times, and the results are shown as mean values of each data point ± S.E. (n = 3). *, p = <0.05 and **, p = <0.005; Con, control.
Inhibition of 5-Lox Alters the Expression of c-Myc Target Genes in Prostate Cancer Cells, but Not in Noncancer Cells
Because c-Myc is well known for its strong oncogenic activity that regulates the transcription of a set of genes, which in turn regulate important cell functions such as cell survival, proliferation, invasion, and metastasis, we analyzed expression of some well known c-Myc target genes after treating prostate cancer cells with MK591. The oncogenic function of c-Myc involves promotion of cell division via activation of cyclins and cyclin-dependent kinases and prevention of cell death via activation of anti-apoptotic proteins (30–35). We observed that MK591 dramatically decreases the mRNA and protein levels of several well characterized apoptosis and cell cycle-regulating c-Myc targets (such as survivin, aurora kinase, TMPRSS2, ADAMTS1, cyclin D1, Gemin4, and proliferating cell nuclear antigen) and increases the levels of apoptosis-promoting c-Myc targets (such as ATF3, GADD45, and TNF-α) in a dose-dependent manner (Fig. 4, a–c). We also observed that MK591 and the Myc-Max dimerization inhibitor (10058-F4) similarly affect c-Myc and its targets under the same experimental condition (Fig. 4d). Moreover, we observed that treatment with MK591 dramatically decreases the viability of the LNCaP human prostate cancer cells. Note: similar effects were seen in PC3 and DU145 prostate cancer cells (data not shown). Interestingly, we observed that treatment with MK591 does not alter the protein levels of c-Myc or its targets, and it does not affect the viability of normal nontransformed HFF in the same experimental conditions, showing a strong cancer-specific effect of this compound and suggesting a differential regulation of c-Myc by 5-Lox in normal versus cancer cells (Fig. 4, e–g). To understand the basis for the differential effect of MK591, we found that LNCaP cells express high levels of 5-Lox, whereas the expression of 5-Lox in HFF cells is undetectable. We also observed that a direct inhibitor of c-Myc (10058-F4) strongly affects the viability of LNCaP cells, although its effect on HFF cells was small but significant (Fig. 4, j and k).
FIGURE 4.

Alteration of transcription of c-Myc target genes in normal and cancer cells by inhibition of 5-Lox. a, LNCaP cells were treated with MK591 (30 μm) for 8 h, and changes in the expression of c-Myc target genes were analyzed by Illumina gene expression array. Results presented as mean value of each data point ± S.D. b, real time PCR data are shown in 8-h MK591-treated samples compared with controls. c, MK591 treatment-induced changes in the protein levels of Myc-targets in LNCaP cells were detected by Western blot. PD176146 (PD) (a 15-lipoxygenase inhibitor) and ibuprofen (Ibu) (a cyclooxygenase inhibitor) were used as negative controls. d, comparative effects of MK591 and 10058-F4 on the protein levels of c-Myc and targets are shown in parallel experiments. e, effects of MK591 on c-Myc and its targets are shown in the normal nontransformed HFF cells. Note: no detectable inhibition in the protein levels of either c-Myc or its targets were observed in HFF cells after MK591 treatment. f and g, comparative effects of MK591 on the viability of cancer (LNCaP) and normal (HFF) cells are shown under the same experimental conditions. LNCaP prostate cancer and normal HFF cells (2 × 104 per well in 12-well plates) were plated as described under “Experimental Procedures” and treated with MK591 for 72 h at 37 °C in the incubator. Control (C) cells were treated with vehicle only (0.2% DMSO). At the end of treatment period, cells were photographed under a microscope at ×400, and cell viability was measured by Cell Titer 96® AQueous One Solution Cell Proliferation Assay (Promega Corp.) as described before (18–20). h and i, expression of 5-Lox in HFF and LNCaP cells was detected by RT-PCR (using human 5-Lox gene-specific primer set) and Western blot respectively. Lane M, molecular weight markers. j and k, comparative effects of 1058-F4 on the viability of cancer (LNCaP) and normal (HFF) cells are shown under the same experimental conditions. **, p = <0.005.
5-Lox Inhibition-induced Degradation of c-Myc Protein in Prostate Cancer Cells Is Proteasome-mediated
The c-Myc oncoprotein undergoes rapid turnover inside the cells, and its threshold is maintained via ubiquitination followed by proteasomal degradation (33–36). Thus, next we examined the role of proteasome in 5-Lox inhibition-induced loss of c-Myc protein in prostate cancer cells. We observed that pretreatment of cells with MG132 (a proteasome inhibitor) effectively prevented MK591 treatment-induced loss of 5-Lox protein in a dose-dependent manner, whereas MG101 (an inhibitor of calpain) was ineffective, suggesting that inhibition of 5-Lox by MK591 may trigger degradation of c-Myc via activation of the proteasomal pathway (Fig. 5a). Proteasomal degradation of the c-Myc protein is preceded by its phosphorylation at Thr-58 and loss of phosphorylation at Ser-62 residues (35–37). We observed that c-Myc protein is dually phosphorylated in untreated prostate cancer cells and that treatment with MK591 increases phosphorylation of c-Myc at Thr-58 and decreases phosphorylation at Ser-62 (Fig. 5, b–d). Although the identities of the kinase(s) and phosphatase(s) responsible for these changes are not known at this time, these findings are consistent with the known pre-requisite to initiate the proteasomal degradation of c-Myc protein.
FIGURE 5.

Proteasome-mediated degradation of c-Myc proteins after MK591 treatment. a, LNCaP cells were plated as in Fig. 2b and pretreated either with a proteasome inhibitor (inh) (MG132) or a calpain inhibitor (MG101) for 30 min at the indicated concentrations, and then the cells were treated with MK591 as shown and incubated for 8 h at 37 °C. Cell lysate proteins were resolved by SDS-PAGE, and c-Myc protein levels were detected by Western blot. b, phosphorylation of c-Myc at Thr-58 and Ser-62 was detected by Western blot using corresponding phospho-specific antibodies. c and d, time-dependent changes in the amount of phosphorylation on Thr-58 and Ser-62 after MK591 treatment are shown. These experiments were done twice, and the results are shown quantitatively as mean values of each data point ± standard deviation (n = 2). C, control.
Inhibition of 5-Lox Interrupts c-Myc Oncogenic Functions in Myc-overactivated Prostate Cancer Cells
In addition to enhanced proliferation and decreased cell death, Myc-driven oncogenic characteristics in cancer cells include empowering cells to increase the metastatic abilities of cancer cells, which involves cell motility, invasion, and neo-colonization in distant sites (38, 39). Oncogenic c-Myc signaling plays a causal role especially in aggressive cancer, which can be tested in vitro by measuring the ability of cells to invade through the extracellular matrix and to form new colonies on soft agar in an anchorage-independent manner (38–43). Thus, we examined whether 5-Lox activity plays any role in regulating the invasive and colony-forming abilities of Myc-overactivated prostate cancer cells. Metastatic LNCaP human prostate cancer cells (also PC3 and DU-145) are known to harbor overactivated c-Myc, which promotes their invasive and colony-forming abilities (44–46). We observed that treatment with MK591 dramatically decreases both the in vitro invasion as well as soft agar colony formation by the LNCaP cells in a dose-dependent manner (Fig. 6, a–d). Interestingly, ibuprofen, a cyclooxygenase inhibitor, was observed to be completely ineffective in the same experimental conditions. Recently, we observed that the protein levels of both c-Myc and its targets are similarly affected by inhibition of 5-Lox in the Myc-driven transgenic mouse tumor-derived Myc-Cap prostate cancer cells, suggesting that these transgenic mouse prostate cancer cells are similar to human prostate cancer cells in regard to their response to inhibition of 5-Lox activity.3 Moreover, we also observed that treatment with MK591 remarkably reduced the invasive capacity as well as soft agar colony-forming ability of Myc-Cap cells in a clear dose-dependent manner, which suggest that the Myc-driven oncogenic characteristics of these cancer cells depend on the activity of 5-Lox. Altogether, our findings are consistent with the idea that inhibition of 5-Lox by treatment with MK591 may severely affect the metastatic capabilities of prostate cancer cells, presumably as a consequence of its inhibition of the c-Myc oncogenic functions.
FIGURE 6.

Effects of MK591 on in vitro invasion and soft agar colony formation by LNCaP cells. a, invasive capabilities of LNCaP cells were assayed using Matrigel-coated transwell chambers as described under “Experimental Procedures.” After incubation, cells were fixed and stained with crystal violet. Pictures were taken with a Leica microscope at ×200. Ibu, ibuprofen. b shows quantitative measurements of the number of invaded cells with or without drug treatment. Results represent mean values of individual data point ± S.D. (n = 3). *, p = <0.05; **, p = <0.005. c, effects of MK591 on soft agar colony formation by LNCaP cells are shown. Cells were plated on soft agar in RPMI 1640 medium as described under “Experimental Procedures.” After incubation for 3 weeks, cells were stained with crystal violet, and growing colonies were counted under microscope at ×150. Note: to distinguish growing colonies, a duplicate set of plates was used to obtain pictures of initial colonies at day 1 for a side-by-side comparison. d, results are shown quantitatively as mean values of each data point ± S.D. (n = 3). **, p = <0.005.
MK591 (an Inhibitor of 5-Lox) Decreases c-Myc Protein Levels in Prostate Tumor Cells in Vivo
We wanted to verify whether the in vitro effect of the inhibition of 5-Lox on c-Myc is valid in vivo. We implanted prostate cancer cells subcutaneously in nude mice and treated the mice with the 5-Lox inhibitor, MK591, via oral gavage for 4 weeks. Results show that treatment with MK591 significantly decreased the protein levels of c-Myc in prostate tumor cells in vivo (Fig. 7a). Moreover, protein level of survivin (a target of c-Myc) was also found to be similarly down-regulated (Fig. 7b). These findings indicate that inhibition of 5-Lox may down-regulate c-Myc even in the presence of a multitude of in vivo survival and growth-regulating factors, and they suggest that the in vivo oncogenic function of c-Myc in prostate cancer cells may be effectively inhibited by suitable 5-Lox-targeting agents.
FIGURE 7.
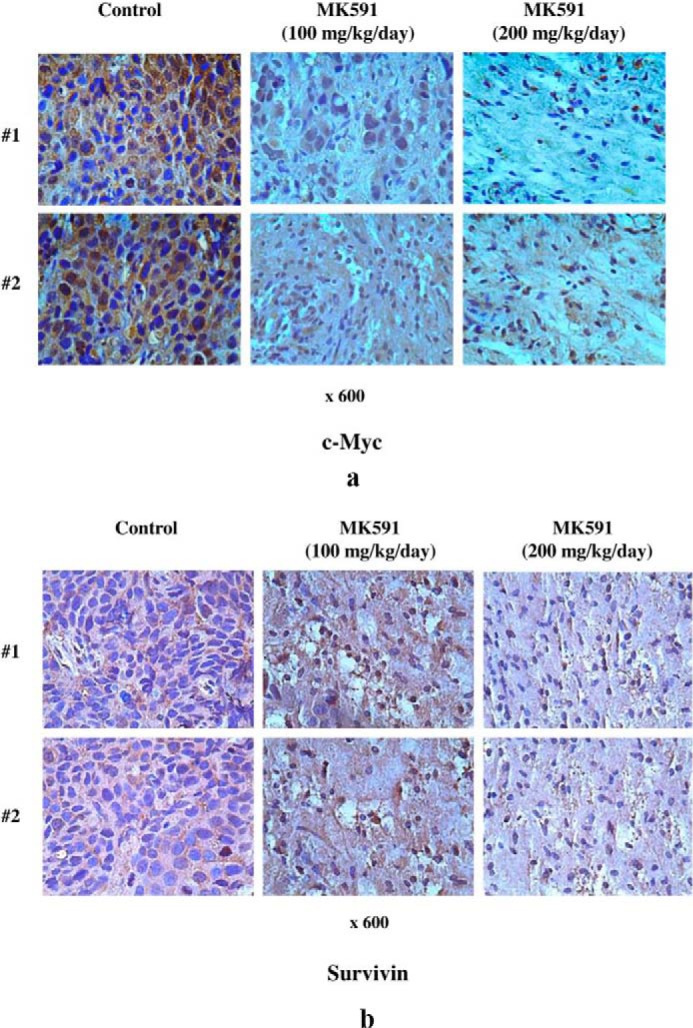
In vivo effects of MK591 on protein levels of c-Myc and survivin in prostate tumor xenografts. Prostate tumor xenografts were developed by injecting LNCaP human prostate cancer cells (2 × 106 per site) to a size of ∼100 mm3. Then tumor-bearing mice were randomly divided into three groups (n = 10). Group 1 mice were treated with solvent only (1:1:8 mixture of DMSO/Cremophor/PBS). Groups 2 and 3 were treated with 100 or 200 mg/kg/day of MK591 orally for 4 weeks. At the end of treatment period, tumors were dissected and processed for immunohistochemistry as described under “Experimental Procedures.” Expressions of c-Myc (a), and survivin (b) in tumors from treated and untreated mice were detected by corresponding rabbit polyclonal antibodies (1:100). Photographs were taken with a Leica microscope at ×600. Note: data are presented with tumor samples obtained from two representative mice in each group (#1 and #2) for side-by-side comparison.
MK591 Treatment-induced Suppression of c-Myc Protein Is Mediated via Inhibition of Stat3-mediated Transcription
To understand the mechanism underlying the loss of c-Myc protein upon MK591 treatment, we performed a “cycloheximide-chase” experiment. We found that the decrease in c-Myc protein with MK591 treatment is not faster than blockade of protein translation by cycloheximide (Fig. 8a), which triggered us to check for the involvement of transcriptional blockade in this process. We found that treatment of cells with actinomycin D rapidly decreases the c-Myc protein level, suggesting that a transcription blockade may cause c-Myc protein loss and may play a role as a mechanism for MK591 treatment-induced down-regulation of c-Myc (Fig. 8b). We addressed this by analyzing nuclear extracts and found that MK591 decreases the nuclear enrichment of Stat3 (Fig. 8c). We confirmed this by transfecting cells with Stat3-luciferase constructs that showed that MK591 exerts a strong and rapid inhibition of Stat3-mediated transcription (Fig. 8d). Moreover, we found that Stattic, an inhibitor of Stat3, mimics the inhibitory effects of MK591 on both c-Myc protein loss and its transcriptional activity (Fig. 8, e and f). Thus, MK591 may down-regulate c-Myc in prostate cancer cells, at least partially, via inhibition of Stat3-mediated transcription of c-Myc.
FIGURE 8.

Involvement of Stat3-mediated transcription in MK591 treatment-induced suppression of c-Myc protein in prostate cancer cells. a, LNCaP cells were plated as in Fig. 2b and treated either with cycloheximide (Cyclohex) or MK591 and incubated for varying periods of time as indicated at 37 °C. Cell lysate proteins were resolved by SDS-PAGE, and the c-Myc protein level was detected by Western blot. b, cells were plated and treated with actinomycin D for varying times as indicated at 37 °C, and the c-Myc protein level was detected by Western blot. c, time-dependent changes in the amounts of transcription factors in the nuclear fraction were detected by cell fractionation and Western blot. d, Stat3-luciferase-transfected LNCaP cells were plated and treated with MK591 as indicated, and the luciferase activity was measured as described under “Experimental Procedures.” Note: Stattic (an inhibitor of Stat3) was used as a positive control. NT represents the value obtained from same numbers of nontransfected LNCaP cells under the same experimental condition. This experiment was done three times, and the results are shown quantitatively as mean values of each data point ± S.E. (n = 3). e, effect of Stattic on c-Myc protein level is shown; f, the effect of Stattic on c-Myc transcriptional activity was detected by treating E-box-luciferase construct-transfected LNCaP cells with doses of inhibitors as indicated. MK591 was also used in parallel for a side-by-side comparison. Results are shown quantitatively as mean values of each data point ± S.E. (n = 3). **, p = <0.005.
DISCUSSION
Our findings show, for the first time, that the expression and function of c-Myc in prostate cancer cells are tightly regulated by 5-Lox activity, revealing a novel mechanism of action of 5-Lox metabolites in prostate cancer. Previously, we reported that the 5-Lox enzyme is continuously active in prostate cancer cells and plays an important role in the survival of prostate cancer cells (14, 16–20). However, downstream mechanisms involved in the regulation of prostate cancer cell survival and growth by 5-Lox are not fully understood. Recently, we reported that 5-Lox inhibition-induced apoptosis in prostate cancer cells occurs without the inhibition of Akt or ERK (18) but via inhibition of protein kinase C-ϵ (19, 20), suggesting the existence of an Akt- and ERK-independent survival mechanism in prostate cancer cells that is fueled by metabolism of arachidonic acid via 5-Lox. Because signaling by the Akt/PKB and ERK pathways has been well characterized to exert pro-survival effects via defined anti-apoptosis mechanisms (47–51), our findings indicate that the 5-Lox metabolites of arachidonic acid feed a survival mechanism that is independent of Akt or ERK and suggest that prostate cancer cells are equipped with additional survival mechanisms (such as PKC-ϵ) to bypass chemotherapies that are directed against Akt or ERK. However, details of the underlying mechanisms of action of 5-Lox activity in the regulation of the growth and survival characteristics of prostate cancer cells are largely unknown.
To get an insight into the signaling network regulated by 5-Lox activity, our gene array analysis revealed that several apoptosis and cell cycle-regulating genes (such as, c-Myc, TMPRSS2, survivin, aurora kinase, and cyclin D1) are down-regulated in prostate cancer cells upon inhibition of 5-Lox (Figs. 1, a and b, and 4, a–c). Because c-Myc is a well known regulator of apoptosis and promotes viability and growth of cancer cells, we wanted to explore further whether the pro-survival effects of 5-Lox involve regulation of c-Myc as a downstream mechanism. Initial observation of the inhibition of c-Myc gene expression was confirmed by RT-PCR, which showed that MK591 decreases c-Myc mRNA in a time-dependent manner (Fig. 1, d and e). Interestingly, later we observed that inhibition of 5-Lox showed a dramatic reduction in c-Myc protein level in prostate cancer cells in a dose- and time-dependent manner (Fig. 2, a and b). Moreover, we observed that 5-Lox inhibition-induced degradation of c-Myc protein is prevented by 5-oxoETE, a metabolic product of 5-Lox (Fig. 2c). Finally, we found that targeting the 5-Lox gene by shRNA down-regulated the c-Myc protein level confirming that the 5-Lox activity tightly regulates c-Myc in prostate cancer cells (Fig. 2d). These findings revealed a novel regulation of c-Myc by 5-Lox activity in prostate cancer cells. Because the c-Myc oncoprotein is a transcription factor, which coordinates expression of diverse cellular programs that together regulate a variety of cellular processes, we wanted to examine the regulation of transcriptional activity of c-Myc by 5-Lox. We observed a steep decrease in nuclear accumulation and DNA binding activities of c-Myc upon treatment with MK591 (Fig. 3, a and b). By transfecting prostate cancer cells with the luciferase construct of the c-Myc DNA-binding sequence (E-box), we observed that inhibition of 5-Lox decreases the transcriptional function of c-Myc that is prevented by 5-oxoETE, a product of 5-Lox activity, suggesting that the c-Myc function in prostate cancer cells is dependent on 5-Lox activity (Fig. 3, c–f).
Our finding of the down-regulation of c-Myc and its target genes by MK591 in prostate cancer cells, but not in the normal HFF cells, suggests that the 5-Lox activity differentially regulates the function of c-Myc in normal versus cancer cells (Fig. 4, a–e). When prostate cancer cells are treated with MK591 (a specific inhibitor of 5-Lox), a pronounced alteration in morphology was observed in a dose- and time-dependent manner, which is indicative of cells undergoing apoptosis. Interestingly, this change was not seen in the normal nontransformed HFF, which do not express 5-Lox in them, suggesting that the regulation of cell survival by 5-Lox is not a general phenomenon. Because we found that inhibition of 5-Lox kills prostate cancer cells but not normal cells (HFF) and that prostate cancer cells express 5-Lox but normal cells do not, our findings suggest that expression and function of 5-Lox are cancer-specific (Fig. 4, e–i). Degradation of c-Myc protein is known to be mediated via proteasome activity (35, 36). We found that treatment with MG132 prevents the loss of c-Myc protein, which is associated with an increase in its phosphorylation at Thr-58 and a decrease in the phosphorylation at Ser-62, suggesting that inhibition of 5-Lox triggers proteasome-mediated degradation of the c-Myc protein (Fig. 5, a–d). We also found that MK591 effectively blocked the in vitro invasion as well as the soft agar colony-forming abilities of prostate cancer cells at sub-lethal doses, suggesting that MK591 may be effective in preventing the metastatic ability of prostate cancer cells, which typically depends on characteristics regulated by c-Myc oncogenic functions (Fig. 6, a–d). Finally, our observation of the down-regulation of c-Myc in tumor xenografts by MK591 treatment suggests that inhibition of 5-Lox may effectively block c-Myc function even in the presence of various growth- and survival-regulating factors in vivo (Fig. 7, a and b). Finally, our results with cycloheximide, actinomycin D, and stattic (the inhibitor of Stat3) suggest that MK591-induced loss of c-Myc protein is not primarily due to enhanced protein degradation but rather happening as a result of its blockade of the Stat3-mediated c-Myc gene transcription (Fig. 8, a–f). Because the Myc oncogene-driven Hi-Myc mouse model closely mimics development and progression of prostate cancer in humans (52, 53), we are using this model to address the question of the regulation of c-Myc by 5-Lox activity. Preliminary observations of the expression of 5-Lox in Hi-Myc mouse prostate tumors as well as in tumor-derived cell lines and the down-regulation of c-Myc by inhibition of 5-Lox in the Myc-driven prostate cancer cells (Myc-Cap) support the concept that the regulation of expression and function of c-Myc by 5-Lox activity are not restricted to LNCaP cells only but also occur in other types of prostate cancer cells as well (data not shown).
Dysregulated expression and function of c-Myc are some of the most common abnormalities in human malignancy (1–7, 30). The importance of Myc as a cancer promoter stems from the fact that the Myc oncoprotein is a pleiotropic basic helix-loop-helix leucine zipper transcription factor that coordinates the expression of diverse cellular programs that together regulate a variety of cellular processes, including cell growth and proliferation, cell cycle progression, transcription, differentiation, apoptosis, and cell motility (30–36). Expression of c-Myc is frequently deregulated in a wide range of human cancers, including prostate cancer, and is often associated with aggressive, poorly differentiated tumors (1–7, 38–46). The c-Myc oncoprotein, with many of its target genes encoding proteins that help maintain the transformed state, can initiate or promote almost all human cancers, and discovering the role it plays in cancer may lead to advance therapy. c-Myc is also a part of a dynamic network whose members interact selectively with one another and with various transcriptional co-regulators and histone-modifying enzymes. The universal deregulation of c-Myc gene expression in tumor cells makes inhibition of c-Myc an attractive pharmacological approach for treating diverse types of cancer. However, although the mechanisms of Myc function are now well characterized, systemic Myc inhibition by direct targeting yielded no significant clinical benefit (54). Plus, this approach bears potential hazards because targeting c-Myc in general may result in damage to noncancer cells that also express low levels of c-Myc. Thus, the enthusiasm has been muted by lack of direct evidence that Myc inhibition would be therapeutically efficacious, raising concerns that it would induce serious side effects by inhibiting proliferation and maintenance of normal tissues. Recently, synthetic lethal approaches (targeting c-Myc functional dependences) are becoming popular to target c-Myc (55, 56). Under the circumstances, our current observation of the regulation of c-Myc by 5-Lox activity in cancer cells revealed a novel mechanism that may open up a new direction to monitor the oncogenic action of c-Myc and thus may help overcome practical difficulties in designing direct Myc inhibitory agents.
It is interesting to note that under normal health conditions, 5-Lox is expressed only in specific immune cells, such as neutrophils, eosinophils, and basophils, whereas its expression in nonimmune parenchymal body cells is undetectable (14, 21–24). However, overactivation of 5-Lox has been implicated in nonimmune cells associated with diseases such as asthma, arthritis, psoriasis, and some types of cancer. Expression of 5-Lox in several types of cancer cells has been observed, which suggests a strong implication of a role of fatty acids and eicosanoids in cancer cells (12, 13, 57–60). The importance of 5-Lox in the survival and growth of prostate cancer cells has been observed in various laboratories, including ours (14–20, 61–64). Interestingly, although 5-Lox plays an essential role in the survival of prostate cancer cells, experimental evidence suggests that this is not a general phenomenon. The 5-Lox knock-out mouse strains have been developed. These mice grow normally with no special abnormalities and are fertile with normal litter size, indicating that the activity of 5-Lox is not essential for normal growth and development (65, 66) and suggesting that agents that specifically block the activity of 5-Lox may turn out to be attractive tools to treat prostate cancer. MK591 is a synthetic compound (developed by Merck-Frosst Canada) that blocks the synthesis of leukotrienes by inhibiting the activity of 5-Lox via binding with its activating protein, FLAP (25–29). It does not inhibit cyclooxygenase, or epoxygenase, or 12-lipoxygenase activities. MK591 is a derivative of the parent compound, MK886, and is currently under development for the treatment of asthma because of its better bioavailability and improved target specificity. Our in vitro findings indicate that MK591 is a novel, promising compound to inhibit c-Myc functions in prostate cancer cells, but not in nontransformed HFF cells, and suggest that MK591 may turn out to be useful to prevent prostate cancer growth as well as metastasis. However, further work is needed to extensively examine the in vivo effects of MK591 to judge its suitability for therapeutic development against clinical prostate cancer.
Because arachidonic acid is a common fatty acid in high fat “Western” diets, and 5-Lox is up-regulated in prostate cancer (57, 58), identification of the signaling pathway(s) through which 5-Lox metabolites exert their pro-cancer effects will not only add significantly to our understanding about the role of 5-Lox in the biology of prostate cancer but also will open up additional targets for therapeutic intervention and better management of clinical prostate cancer in human. Our current findings indicate that prostate cancer cells obtain a clear benefit for survival and growth from the metabolism of dietary arachidonic acid via 5-Lox, and they suggest that metabolites of 5-Lox may contribute to the transformed phenotype of prostate cancer cells via signaling involving a key oncogene, c-Myc. Although in normal cells Myc is turned on transiently by growth factor signaling that instructs cell division cycle, its function in cancer cells is almost always compromised either by gene amplification or by mutation for uncontrollable cell proliferation and tumor formation. Thus, Myc (being characterized as a driver of cancer) has repeatedly been recognized as an elusive target for drug development, which in turn triggered searching for upstream or downstream signals that regulate function of Myc. Because normal cells, such as HFF, do not express 5-Lox and are not affected by MK591 treatment, our findings open up the possibility to control expression and function of c-Myc in prostate (and possibly other types of) cancer cells by small molecular inhibitors of 5-Lox, such as MK591.
Acknowledgment
We thank Dr. Ruiqun for generous help with analysis of the microarray data.
This work was supported, in whole or in part, by National Institutes of Health Grant RO1 CA152334 from NCI. This work was also supported by Department of Defense Prostate Cancer Research Program W81-XWH-05-1-0022 and the Henry Ford Health System Internal Research Grant A10203 (to J. G.).
S. Sarveswaran and J. Ghosh, unpublished observations.
- 5-Lox
- 5-lipoxygenase
- 5-oxoETE
- 5-oxoeicosatetraenoic acid
- HFF
- human foreskin fibroblast
- qPCR
- quantitative PCR.
REFERENCES
- 1. Meyer N., Penn L. Z. (2008) Reflecting on 25 years with MYC. Nat. Rev. Cancer 8, 976–990 [DOI] [PubMed] [Google Scholar]
- 2. Ponzielli R., Katz S., Barsyte-Lovejoy D., Penn L. Z. (2005) Cancer therapeutics: targeting the dark side of Myc. Eur. J. Cancer 41, 2485–2501 [DOI] [PubMed] [Google Scholar]
- 3. Wang Y. H., Liu S., Zhang G., Zhou C. Q., Zhu H. X., Zhou X. B., Quan L. P., Bai J. F., Xu N. Z. (2005) Knockdown of c-Myc expression by RNAi inhibits MCF-7 breast tumor cells growth in vitro and in vivo. Breast Cancer Res. 7, R220–R228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Soucek L., Whitfield J., Martins C. P., Finch A. J., Murphy D. J., Sodir N. M., Karnezis A. N., Swigart L. B., Nasi S., Evan G. I. (2008) Modelling Myc inhibition as a cancer therapy. Nature 455, 679–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kimura S., Maekawa T., Hirakawa K., Murakami A., Abe T. (1995) Alterations of c-myc expression by antisense oligodeoxynucleotides enhance the induction of apoptosis in HL-60 cells. Cancer Res. 55, 1379–1384 [PubMed] [Google Scholar]
- 6. Holt J. T., Redner R. L., Nienhuis A. W. (1988) An oligomer complementary to c-myc mRNA inhibits proliferation of HL-60 promyelocytic cells and induces differentiation. Mol. Cell. Biol. 8, 963–973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Iversen P. L., Arora V., Acker A. J., Mason D. H., Devi G. R. (2003) Efficacy of antisense morpholino oligomer targeted to c-myc in prostate cancer xenograft murine model and a phase I safety study in humans. Clin. Cancer Res. 9, 2510–2519 [PubMed] [Google Scholar]
- 8. Siegel R., Ward E., Brawley O., Jemal A. (2011) Cancer statistics, 2011: the impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J. Clin. 61, 212–236 [DOI] [PubMed] [Google Scholar]
- 9. Fleshner N., Bagnell P. S., Klotz L., Venkateswaran V. (2004) Dietary fat and prostate cancer. J. Urol. 171, S19–S24 [DOI] [PubMed] [Google Scholar]
- 10. Kolonel L. N., Nomura A. M., Cooney R. V. (1999) Dietary fat and prostate cancer: Current status. J. Natl. Cancer Inst. 91, 414–428 [DOI] [PubMed] [Google Scholar]
- 11. Wang Y., Corr J. G., Thaler H. T., Tao Y., Fair W. R., Heston W. D. (1995) Decreased growth of established prostate LNCaP tumors in nude mice fed a low-fat diet. J. Natl. Cancer Inst. 87, 1456–1462 [DOI] [PubMed] [Google Scholar]
- 12. Wang D., Dubois R. N. (2010) Eicosanoids and cancer. Nat. Rev. Cancer 10, 181–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nie D. (2007) Cyclooxygenases and lipoxygenases in prostate and breast cancers. Front. Biosci. 12, 1574–1585 [DOI] [PubMed] [Google Scholar]
- 14. Ghosh J., Myers C. E. (1997) Arachidonic acid stimulates prostate cancer cell growth: critical role of 5-lipoxygenase. Biochem. Biophys. Res. Commun. 235, 418–423 [DOI] [PubMed] [Google Scholar]
- 15. Anderson K. M., Seed T., Vos M., Mulshine J., Meng J., Alrefai W., Ou D., Harris J. E. (1998) 5-Lipoxygenase inhibitors reduce PC-3 cell proliferation and initiate nonnecrotic cell death. Prostate 37, 161–173 [DOI] [PubMed] [Google Scholar]
- 16. Ghosh J., Myers C. E. (1998) Inhibition of arachidonate 5-lipoxygenase triggers massive apoptosis in human prostate cancer cells. Proc. Natl. Acad. Sci. U.S.A. 95, 13182–13187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ghosh J., Myers C. E. (1999) Central role of arachidonate 5-lipoxygenase in the regulation of cell growth and apoptosis in human prostate cancer cells. Adv. Exp. Med. Biol. 469, 577–582 [DOI] [PubMed] [Google Scholar]
- 18. Sarveswaran S., Myers C. E., Ghosh J. (2010) MK591, a leukotriene biosynthesis inhibitor, induces apoptosis in prostate cancer cells: synergistic action with LY294002, an inhibitor of phosphatidylinositol 3′-kinase. Cancer Lett. 291, 167–176 [DOI] [PubMed] [Google Scholar]
- 19. Sarveswaran S., Thamilselvan V., Brodie C., Ghosh J. (2011) Inhibition of 5-lipoxygenase triggers apoptosis in prostate cancer cells via down-regulation of protein kinase C-epslilon. Biochim. Biophys. Acta 1813, 2108–2117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sarveswaran S., Ghosh J. (2013) OXER1, a G protein-coupled oxoeicosatetraenoid receptor, mediates the survival-promoting effects of arachidonate 5-lipoxygenase in prostate cancer cells. Cancer Lett. 336, 185–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ford-Hutchinson A. W., Gresser M., Young R. N. (1994) 5-Lipoxygenase. Annu. Rev. Biochem. 63, 383–417 [DOI] [PubMed] [Google Scholar]
- 22. Rådmark O., Werz O., Steinhilber D., Samuelsson B. (2007) 5-Lipoxygenase: regulation of expression and enzyme activity. Trends Biochem. Sci. 32, 332–341 [DOI] [PubMed] [Google Scholar]
- 23. Rådmark O., Samuelsson B. (2009) 5-Lipoxygenase: mechanisms of regulation. J. Lipid Res. 50, S40–S45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rådmark O., Samuelsson B. (2010) Regulation of the activity of 5-lipoxygenase, a key enzyme in leukotriene biosynthesis. Biochem. Biophys. Res. Commun. 396, 105–110 [DOI] [PubMed] [Google Scholar]
- 25. Brideau C., Chan C., Charleson S., Denis D., Evans J. F., Ford-Hutchinson A. W., Fortin R., Gillard J. W., Guay J., Guévremont D., Hutchinson J. H., Jones T. R., Leger S., Mancini J. A., McFarlane C. S., Pickett C., Piechuta H., Prasit P., Riendeau D., Rouzer C. A., Tagari P., Vickers P. J., Young R. N., Abraham W. M. (1992) Pharmacology of MK-0591 (3-[1-(4-chlorobenzyl)-3-(t-butylthio)-5-(quinolin-2-yl-methoxy)-indol-2-yl]-2,2-dimethyl propanoic acid), a potent, orally active leukotriene biosynthesis inhibitor. Can. J. Physiol. Pharmacol. 70, 799–807 [DOI] [PubMed] [Google Scholar]
- 26. Prasit P., Belley M., Blouin M., Brideau C., Chan C., Charleson S., Evans J. F., Frenette R., Gauthier J. Y., Guay J. (1993) A new class of leukotriene biosynthesis inhibitor: the development of MK-0591. J. Lipid Mediat. 6, 239–244 [PubMed] [Google Scholar]
- 27. Ferguson A. D., McKeever B. M., Xu S., Wisniewski D., Miller D. K., Yamin T. T., Spencer R. H., Chu L., Ujjainwalla F., Cunningham B. R., Evans J. F., Becker J. W. (2007) Crystal structure of inhibitor-bound human 5-lipoxygenase-activating protein. Science 317, 510–512 [DOI] [PubMed] [Google Scholar]
- 28. Evans J. F., Ferguson A. D., Mosley R. T., Hutchinson J. H. (2008) What's all the FLAP about? 5-lipoxygenase-activating protein inhibitors for inflammatory diseases. Trends Pharmacol. Sci. 29, 72–78 [DOI] [PubMed] [Google Scholar]
- 29. Sampson A. P. (2009) FLAP inhibitors for the treatment of inflammatory diseases. Curr. Opin. Investig. Drugs 10, 1163–1172 [PubMed] [Google Scholar]
- 30. Adhikary S., Eilers M. (2005) Transcriptional regulation and transformation by Myc proteins. Nat. Rev. Mol. Cell Biol. 6, 635–645 [DOI] [PubMed] [Google Scholar]
- 31. Gil J., Kerai P., Lleonart M., Bernard D., Cigudosa J. C., Peters G., Carnero A., Beach D. (2005) Immortalization of primary human prostate epithelial cells by c-Myc. Cancer Res. 65, 2179–2185 [DOI] [PubMed] [Google Scholar]
- 32. Rohan J. N., Weigel N. L. (2009) 1α,25-Dihydroxyvitamin D3 reduces c-Myc expression, inhibiting proliferation and causing G1 accumulation in C4–2 prostate cancer cells. Endocrinology 150, 2046–2054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dang C. V. (1999) c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol. Cell. Biol. 19, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dang C. V., O'Donnell K. A., Zeller K. I., Nguyen T., Osthus R. C., Li F. (2006) The c-Myc target gene network. Semin. Cancer Biol. 16, 253–264 [DOI] [PubMed] [Google Scholar]
- 35. Sears R. C. (2004) The life cycle of c-myc: from synthesis to degradation. Cell Cycle 3, 1133–1137 [PubMed] [Google Scholar]
- 36. Vervoorts J., Lüscher-Firzlaff J., Lüscher B. (2006) The ins and outs of MYC regulation by posttranslational mechanisms. J. Biol. Chem. 281, 34725–34729 [DOI] [PubMed] [Google Scholar]
- 37. Welcker M., Orian A., Jin J., Grim J. E., Grim J. A., Harper J. W., Eisenman R. N., Clurman B. E. (2004) The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc. Natl. Acad. Sci. U.S.A. 101, 9085–9090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wolfer A., Ramaswamy S. (2011) MYC and metastasis. Cancer Res. 71, 2034–2037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wolfer A., Wittner B. S., Irimia D., Flavin R. J., Lupien M., Gunawardane R. N., Meyer C. A., Lightcap E. S., Tamayo P., Mesirov J. P., Liu X. S., Shioda T., Toner M., Loda M., Brown M., Brugge J. S., Ramaswamy S. (2010) MYC regulation of a “poor-prognosis” metastatic cancer cell state. Proc. Natl. Acad. Sci. U.S.A. 107, 3698–3703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ma L., Young J., Prabhala H., Pan E., Mestdagh P., Muth D., Teruya-Feldstein J., Reinhardt F., Onder T. T., Valastyan S., Westermann F., Speleman F., Vandesompele J., Weinberg R. A. (2010) miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat. Cell Biol. 12, 247–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rapp U. R., Korn C., Ceteci F., Karreman C., Luetkenhaus K., Serafin V., Zanucco E., Castro I., Potapenko T. (2009) MYC is a metastasis gene for non-small-cell lung cancer. PLoS One 4, e6029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li X., Liu X., Xu W., Zhou P., Gao P., Jiang S., Lobie P. E., Zhu T. (2013) c-MYC-regulated miR-23a/24–2/27a cluster promotes mammary carcinoma cell invasion and hepatic metastasis by targeting Sprouty2. J. Biol. Chem. 288, 18121–18133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhang X. Y., DeSalle L. M., Patel J. H., Capobianco A. J., Yu D., Thomas-Tikhonenko A., McMahon S. B. (2005) Metastasis-associated protein 1 (MTA1) is an essential downstream effector of the c-MYC oncoprotein. Proc. Natl. Acad. Sci. U.S.A. 102, 13968–13973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nag A., Smith R. G. (1989) Amplification, rearrangement, and elevated expression of c-Myc in the human prostatic carcinoma cell line LNCaP. Prostate 15, 115–122 [DOI] [PubMed] [Google Scholar]
- 45. Jenkins R. B., Qian J., Lieber M. M., Bostwick D. G. (1997) Detection of c-myc oncogene amplification and chromosomal anomalies in metastatic prostatic carcinoma by fluorescence in situ hybridization. Cancer Res. 57, 524–531 [PubMed] [Google Scholar]
- 46. Gurel B., Iwata T., Koh C. M., Jenkins R. B., Lan F., Van Dang C., Hicks J. L., Morgan J., Cornish T. C., Sutcliffe S., Isaacs W. B., Luo J., De Marzo A. M. (2008) Nuclear MYC protein overexpression is an early alteration in human prostate carcinogenesis. Mod. Pathol. 21, 1156–1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Manning B. D., Cantley L. C. (2007) AKT/PKB signaling: navigating downstream. Cell 129, 1261–1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Engelman J. A., Luo J., Cantley L. C. (2006) The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 7, 606–619 [DOI] [PubMed] [Google Scholar]
- 49. Marte B. M., Downward J. (1997) PKB/Akt: connecting phosphoinositide3-kinase to cell survival and beyond. Trends Biochem. Sci. 22, 355–358 [DOI] [PubMed] [Google Scholar]
- 50. Altomare D. A., Testa J. R. (2005) Perturbations of the Akt signaling pathway in human cancer. Oncogene 24, 7455–7464 [DOI] [PubMed] [Google Scholar]
- 51. Hennessy B. T., Smith D. L., Ram P. T., Lu Y., Mills G. B. (2005) Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat. Rev. Drug Discov. 4, 988–1004 [DOI] [PubMed] [Google Scholar]
- 52. Ellwood-Yen K., Graeber T. G., Wongvipat J., Iruela-Arispe M. L., Zhang J., Matusik R., Thomas G. V., Sawyers C. L. (2003) Myc-driven murine prostate cancer shares molecular features with human prostate tumors. Cancer Cell 4, 223–238 [DOI] [PubMed] [Google Scholar]
- 53. Watson P. A., Ellwood-Yen K., King J. C., Wongvipat J., Lebeau M. M., Sawyers C. L. (2005) Context-dependent hormone-refractory progression revealed through characterization of a novel murine prostate cancer cell line. Cancer Res. 65, 11565–11571 [DOI] [PubMed] [Google Scholar]
- 54. Vita M., Henriksson M. (2006) The Myc oncoprotein as a therapeutic target for human cancer. Semin. Cancer Biol. 16, 318–330 [DOI] [PubMed] [Google Scholar]
- 55. Evan G. (2012) Taking a back door to target Myc. Science 335, 293–294 [DOI] [PubMed] [Google Scholar]
- 56. Kessler J. D., Kahle K. T., Sun T., Meerbrey K. L., Schlabach M. R., Schmitt E. M., Skinner S. O., Xu Q., Li M. Z., Hartman Z. C., Rao M., Yu P., Dominguez-Vidana R., Liang A. C., Solimini N. L., Bernardi R. J., Yu B., Hsu T., Golding I., Luo J., Osborne C. K., Creighton C. J., Hilsenbeck S. G., Schiff R., Shaw C. A., Elledge S. J., Westbrook T. F. (2012) A SUMOylation-dependent transcriptional subprogram is required for Myc-driven tumorigenesis. Science 335, 348–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gupta S., Srivastava M., Ahmad N., Sakamoto K., Bostwick D. G., Mukhtar H. (2001) Lipoxygenase-5 is overexpressed in prostate adenocarcinoma. Cancer 91, 737–743 [DOI] [PubMed] [Google Scholar]
- 58. Matsuyama M., Yoshimura R., Mitsuhashi M., Hase T., Tsuchida K., Takemoto Y., Kawahito Y., Sano H., Nakatani T. (2004) Expression of lipoxygenase in human prostate cancer and growth reduction by its inhibitors. Int. J. Oncol. 24, 821–827 [PubMed] [Google Scholar]
- 59. Werz O. (2002) 5-Lipoxygenase: cellular biology and molecular pharmacology. Curr. Drug Targets Inflamm. Allergy 1, 23–44 [DOI] [PubMed] [Google Scholar]
- 60. Ghosh J. (2008) Targeting 5-lipoxygenase for prevention and treatment of cancer. Curr. Enzyme Inhib. 4, 18–28 [Google Scholar]
- 61. Moretti R. M., Montagnani Marelli M., Sala A., Motta M., Limonta P. (2004) Activation of the orphan nuclear receptor RORα counteracts the proliferative effect of fatty acids on prostate cancer cells: crucial role of 5-lipoxygenase. Int. J. Cancer 112, 87–93 [DOI] [PubMed] [Google Scholar]
- 62. Yang P., Collin P., Madden T., Chan D., Sweeney-Gotsch B., McConkey D., Newman R. A. (2003) Inhibition of proliferation of PC3 cells by the branched-chain fatty acid, 12-methyltetradecanoic acid, is associated with inhibition of 5-lipoxygenase. Prostate 55, 281–291 [DOI] [PubMed] [Google Scholar]
- 63. Pham H., Vang K., Ziboh V. A. (2006) Dietary γ-linolenate attenuates tumor growth in a rodent model of prostatic adenocarcinoma via suppression of elevated generation of PGE(2) and 5S-HETE. Prostaglandins Leukot. Essent. Fatty Acids 74, 271–282 [DOI] [PubMed] [Google Scholar]
- 64. Ochoa J. J., Farquharson A. J., Grant I., Moffat L. E., Heys S. D., Wahle K. W. (2004) Conjugated linoleic acids (CLAs) decrease prostate cancer cell proliferation: different molecular mechanisms for cis-9, trans-11 and trans-10, cis-12 isomers. Carcinogenesis 25, 1185–1191 [DOI] [PubMed] [Google Scholar]
- 65. Chen X. S., Sheller J. R., Johnson E. N., Funk C. D. (1994) Role of leukotrienes revealed by targeted disruption of the 5-lipoxygenase gene. Nature 372, 179–182 [DOI] [PubMed] [Google Scholar]
- 66. Funk C. D. (1996) The molecular biology of mammalian lipoxygenases and the quest for eicosanoid functions using lipoxygenase-deficient mice. Biochim. Biophys. Acta 1304, 65–84 [DOI] [PubMed] [Google Scholar]