

Background: Histone deacetylase inhibitors are being evaluated to induce latent HIV reactivation, but their effect on macrophages is unknown.
Results: Histone deacetylase inhibitors induce the degradation of HIV in macrophages through a mechanism that involves autophagosome formation and maturation.
Conclusion: Histone deacetylase inhibitors inhibit HIV through an autophagy-dependent mechanism.
Significance: This is the first report of histone deacetylase inhibitors inhibiting HIV.
Keywords: Autophagy Autophagy-related Protein 7 (ATG7), Histone Deacetylase Inhibitor (HDACi), Human Immunodeficiency Virus (HIV), Macrophage
Abstract
Histone deacetylase inhibitors (HDACi) are being evaluated in a “shock-and-kill” therapeutic approach to reverse human immunodeficiency virus type-1 (HIV) latency from CD4+ T cells. Using this approach, HDACi have induced HIV RNA synthesis in latently infected cells from some patients. The hope is that the increase in viral production will lead to killing of the infected cell either by the virus itself or by the patient's immune system, a “sterilizing cure.” Although administered within the context of combination antiretroviral therapy, the infection of bystander cells remains a concern. In this study, we investigated the effect of HDACi (belinostat, givinostat, panobinostat, romidepsin, and vorinostat) on the productive infection of macrophages. We demonstrate that the HDACi tested do not alter the initial susceptibility of macrophages to HIV infection. However, we demonstrate that HDACi decrease HIV release from macrophages in a dose-dependent manner (belinostat < givinostat < vorinostat < panobinostat < romidepsin) via degradation of intracellular HIV through the canonical autophagy pathway. This mechanism involves unc-51-like autophagy-activating kinase 1 (ULK1) and the inhibition of the mammalian target of rapamycin and requires the formation of autophagosomes and their maturation into autolysosomes in the absence of increased cell death. These data provide further evidence in support of a role for autophagy in the control of HIV infection and suggest that careful consideration of off-target effects will be essential if HDACi are to be a component of a multipronged approach to eliminate latently infected cells.
Introduction
Although effective combination antiretroviral therapy (cART)3 has transformed human immunodeficiency virus type-1 (HIV) infection from an invariably fatal disease to a chronic illness, cART does not eradicate HIV infection. Even in the absence of undetectable plasma viral RNA, HIV persists in long lived resting memory CD4+ T cells, naive CD4+ T cells, astrocytes, and macrophages by establishing a “latent” infection. The molecular mechanisms of latency are complex and include the absence of nuclear forms of key host transcription factors, the absence of Tat and the associated host factors that promote efficient transcriptional elongation, epigenetic changes that inhibit HIV gene expression from the promoter or the long terminal repeat (LTR), and transcriptional interference (1). As latently infected cells are undetectable by the immune system, are unresponsive to cART, and cessation of therapy results in viral rebound within weeks, lifelong cART is required for continued viral suppression.
To date, efforts to purge the latent reservoir have focused on the activation of viral production from these cells by histone deacetylase (HDAC) inhibitors (HDACi) in the presence of cART with the rationale that viral production is cytotoxic, and cART will inhibit subsequent rounds of infection. During HIV latency, various transcription factors recruit HDACs to the HIV 5′ LTR where they induce chromatin condensation by promoting deacetylation of histone lysine residues thus rendering the LTR nuc-1 nucleosome hypoacetylated which prevents HIV transcription. HDACi counteract this mechanism by allowing remodeling of nuc-1 by histone acetyltransferases with subsequent hyperacetylation by cellular transcription factors or by HIV Tat allowing activation of virus production (2). This strategy, known as “shock and kill,” is being investigated in HIV-infected patients. Administration of the HDACi vorinostat was well tolerated in patients on cART and, in one study, induced a 4.8-fold increase in HIV RNA expression in their resting CD4+ T cells (3). However, a major clinical concern with this strategy is that, although administered in the context of cART, infection of new uninfected CD4+ T cells and macrophages by induced virus during periods of viral activation from latency may occur. Indeed, a recent study has demonstrated that although vorinostat does not affect virus-CD4+ T cell fusion, it increases the kinetics of post-entry events such as reverse transcription and integration thereby promoting productive infection of CD4+ T cells and potentially reseeding the viral reservoirs being purged (4). Another important HIV reservoir is the macrophage. These long lived cells reside within multiple tissue compartments. Moreover, the efficacy of cART within macrophages may be reduced as HIV infection increases transcription of ATP-binding cassette subfamily B (MDR/TAP) member 5 and ATP-binding cassette subfamily C (CFTR/MRP) member (ABCC) 1, ABCC4, and ABCC5, which may favor the efflux of drugs used as part of cART, thereby decreasing their pharmacological effects (5). However, no study has examined the effect of HDACi on viral infectivity of macrophages. Therefore, we investigated the effect of belinostat, givinostat, panobinostat, romidepsin, and vorinostat on the susceptibility of macrophages to HIV infection. We report that HDACi have no impact on the initial infection events, but through the induction of macroautophagy (hereafter referred to as autophagy) they induce the degradation of intracellular viral particles that lead to a reduction in viral release. Autophagy is a highly conserved degradation pathway whereby cytosolic double membrane-bound compartments termed autophagosomes engulf cytoplasmic constituents such as subcellular organelles and microbial pathogens, fuse with lysosomes, and degrade the engulfed components. Moreover, our data demonstrate that the HDACi-mediated autophagic degradation of HIV requires both the nucleation and formation of autophagosomes as well as their subsequent maturation, and this leads to a decrease in virus release.
EXPERIMENTAL PROCEDURES
Ethics Statement
Venous blood was drawn from HIV seronegative subjects using a protocol that was reviewed and approved by the Human Research Protections Program of the University of California, San Diego (Project 09-0660), in accordance with the requirements of the Code of Federal Regulations on the Protection of Human Subjects (45 CFR 46 and 21 CFR 50 and 56). Written informed consent was obtained from subjects prior to their participation.
Cells and Reagents
Monocyte-derived macrophages were obtained from peripheral blood mononuclear cells as described previously (6). All experiments were performed in RPMI 1640 medium supplemented with 10% (v/v) heat-inactivated FBS and 10 ng/ml macrophage colony-stimulating factor. TZM-bl cells were obtained through the AIDS Research and Reference Reagent Program, contributed by Drs. John C. Kappes, Xiaoyun Wu, and Tranzyme Inc. (7). Cell death was estimated using the lactate dehydrogenase (LDH) cytotoxicity detection kitPLUS (Roche Applied Science) and the single-stranded DNA (ssDNA) ELISA (Millipore), as described previously (8).
Belinostat (PXD101), givinostat (ITF2357), panobinostat (LBH589), romidepsin (FK228), and vorinostat (suberoylanilide hydroxamic acid) were purchased from Selleck Chemicals. Bafilomycin A1, SID 26681509, and sirolimus were purchased from Sigma. Bafilomycin A1 was used at 100 nmol/liter and SID 26681509 at 50 nmol/liter with pretreatment for 1 h before addition of HDACi or sirolimus. Maraviroc was purchased from Toronto Research Chemicals and was used at 10 nmol/liter. Concentrations of HDACi used were based upon previously published whole blood Cmax and Cmin data for each drug (9–13).
Virus
HIVBa-L was obtained through the AIDS Research and Reference Reagent Program, contributed by Dr. Suzanne Gartner and Dr. Robert Gallo (14, 15). Virus stocks and titers were prepared as described previously using the Alliance HIV p24 antigen ELISA (PerkinElmer Life Sciences) (16). Macrophages were infected with 105 TCID50/ml HIVBa-L per 5 × 105 cells as described previously (6). Viral binding and entry were assessed as described previously (17). Productive infection of TZM-bl cells 48 h post-HIV exposure was detected using the β-gal staining set (Roche Applied Science).
Immunoblotting
unc-51-like autophagy-activating kinase 1 (ULK1; D9D7), eukaryotic translation initiation factor 4E-binding protein 1 (EIF4EBP1; 53H11), ribosomal protein S6 kinase, 70 kDa, polypeptide 2 (RPS6KB2; 49D7), phospho-ULK1 (Ser757), phospho-EIF4EBP1 (Thr37/46; 236B4), phospho-RPS6KB2 (Thr389), autophagy-related (ATG) 7, and ATG5 antibodies were obtained from Cell Signaling. Sequestosome 1 (SQSTM1; ab56416), HIV p24 (39/5.4A), and HIV Nef (3D12) antibodies were obtained from Abcam. β-Actin (ACTB; AC-74) and microtubule-associated protein 1 light chain 3β (LC3B; NB100-2220) antibodies were obtained from Sigma and Novus Biologicals, respectively. Cell lysates were prepared using 20 mmol/liter HEPES, 150 mmol/liter NaCl, 1 mmol/liter EDTA supplemented with 1% (v/v) 4-(1,1,3,3-tetramethylbutyl)phenyl-polyethylene glycol (all Sigma) and 1% (v/v) Halt protease and phosphatase inhibitor mixture (Thermo Scientific). Cell lysates were resolved using 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol-buffered 12% polyacrylamide gel (Novex) and transferred to 0.2 μm pore-size PVDF membranes (Thermo Scientific), followed by detection with alkaline phosphatase-tagged secondary antibodies (Invitrogen) and 0.25 mm disodium 2-chloro-5-(4-methoxyspiro[1,2-dioxetane-3,2′-(5-chlorotricyclo[3.3.1.13.7]decan])-4-yl]-1-phenyl phosphate supplemented with 5% (v/v) Nitro-Block II (both from Applied Biosystems). Relative densities of the target bands compared with the reference ACTB bands were calculated using ImageJ (National Institutes of Health). Each data point was normalized to the vehicle and then log2 transformed.
shRNA Transduction
Lentiviral transduction of macrophages with MISSION lentiviral particles containing shRNAs targeting ATG5 (SHCLNV-NM_004849/TRCN0000150940 and TRCN0000151963), ATG7 (SHCLNV-NM_006395/TRCN0000007584 and TRCN0000435480), or scrambled nontarget negative control (SHC002V) was performed according to the manufacturer's protocol (Sigma). SHC002V was used as nontargeting negative control as it activates the RNA-induced silencing complex and the RNAi pathway, but it does not target any human gene allowing the examination of the effects of shRNA transduction and RNAi activation on gene expression. Macrophages were transduced with nonspecific scrambled shRNA or target shRNA and selected using 3 μg/ml puromycin (Invitrogen). Five days later, cells were analyzed for target gene silencing and used in experiments. Validation was performed by phenotypic rescue using custom shRNA-insensitive open reading frame (ORF) cDNA lentiviral vectors (LentiORF; GeneCopoeia). For rescue experiments, macrophages were transduced with target shRNA with a neomycin resistance marker and LentiORF constructs. Selection was performed using 100 μg/ml G418 (Invitrogen) combined with 3 μg/ml puromycin (for ATG7) or 200 μg/ml hygromycin B (Invitrogen) (for ATG5) as selection agents.
Quantitative Real Time PCR (qPCR)
Strong-stop HIV DNA quantification was measured using qPCR with the LightCycler 1.5 instrument and FastStart DNA Master SYBR Green I (both Roche Applied Science). Primers and run conditions were as described previously (16). Data were analyzed using the Pfaffl method (18). The ratio between HIV LTR DNA and polymerase (RNA) II (DNA-directed) polypeptide A (POLR2A) was calculated and normalized so that HIV LTR DNA in untreated cells equals 1.00. Data were then log2 transformed.
HIV integration was assessed using a nested Alu-LTR qPCR assay as described previously (19, 20) using a GeneAmp PCR system 9700 (Applied Biosystems) and a Light Cycler 480 II (Roche Applied Science). Concentrations of genomic DNA were normalized using a NanoDrop Lite (Thermo Fisher).
Flow Cytometry
Cell surface expression of chemokine (C-C motif) receptor 5 (CCR5) and CD4 was measured using allophycocyanin-tagged CCR5, peridin-chlorophyll protein-tagged CD4, and FITC-tagged CD14 monoclonal antibodies (all BD Biosciences) followed by fixation in Dulbecco's PBS supplemented with 4% (w/v) paraformaldehyde. Data were collected using a FACSCalibur flow cytometer (BD Biosciences) and analyzed using FlowJo (Tree Star).
Statistics
Data were assessed for symmetry, or skewness, using Pearson's skewness coefficient. Fold change data were log2 transformed to convert the ratio to a difference that better approximates the normal distribution on a log scale. Comparisons between groups were performed using the paired, two-tailed Student's t test. Differences were considered to be statistically significant when p < 0.05.
RESULTS
HDACi Decrease HIV Release from Human Macrophages
The recent efforts to purge the latent reservoir of HIV have focused on the activation of viral production from latently infected cells using HDACi in the presence of cART. However, the activity of these drugs in non-T cell reservoirs is unknown. Moreover, although HDACi shock-and-kill will be performed in the presence of cART, suboptimal viral inhibition could occur in the context of patient noncompliance, viral resistance, or sanctuary sites with poor drug penetration. Therefore, we determined whether HDACi influence HIV infection and replication in primary macrophages by comparing the extent to which HDACi treatment influences p24 antigen accumulation in the supernatants of productively infected macrophages. All HDACi induced a dose-dependent decrease in HIV p24 release into the culture supernatants that became significant by day 3 post-infection at the highest concentrations tested (p < 0.01). Moreover, the magnitude of the decrease escalated until cultures were discontinued on day 10 post-infection (Fig. 1A). Romidepsin induced the greatest decline in HIV p24 antigen release with 500 pmol/liter sufficient for a 98% reduction by 10 days post-infection (p = 0.002). Belinostat and givinostat were the least effective with 1 nmol/liter reducing p24 antigen release by 9% (p = 0.2) and 18% (p = 0.17), respectively, and at 10 nmol/liter by 39% (p = 0.001) and 48% (p = 0.009), respectively. This is in contrast to panobinostat that achieved a 51% (p = 0.01) and 86% decrease in HIV p24 antigen release (p = 0.003) at 1 and 10 nmol/liter, respectively. Interestingly, even a low dose of 25 nmol/liter vorinostat was sufficient to decrease extracellular HIV p24 antigen accumulation by 83% (p = 0.003). As HDACi have been reported to exert cytotoxic effects in cancer cells (21), it was important to confirm that the cells were not undergoing cell death at the concentrations being used. Therefore, we assayed for plasma membrane breakdown (as a sign of cytotoxicity) using the LDH assay. We observed that at 10 days post-HIV infection, HDACi induced no significant cytotoxic effects at the concentrations used (p > 0.06; Fig. 1B). Moreover, based on the selective thermal denaturation of apoptotic ssDNA using low heat and formamide and subsequent detection using a monoclonal antibody, a more specific indicator of apoptosis than TUNEL (22), we also observed no difference in ssDNA accumulation at the same time point (p > 0.09; Fig. 1C).
FIGURE 1.

HDACi decrease HIV p24 antigen release from macrophages. Macrophages were incubated with HDACi for 24 h before infection with HIV for 3 h. Cells were then washed and incubated with HDACi for 10 days. A, extracellular release of HIV p24 antigen into the cell supernatant at days 0, 3, 5, 7, and 10 was detected by ELISA. B, extracellular release of LDH was measured spectrophotometrically using the cytotoxicity detection kitPLUS (LDH) assay at 10 days post-infection. C, quantification of the percentage of cells with apoptotic ssDNA using the ssDNA ELISA at 10 days post-infection. Results are reported as mean ± S.E., n = 5. *, p < 0.05.
To understand how HDACi affects HIV replication, we examined sequential steps of viral replication. Treatment of cells with HDACi did not significantly affect the expression of either CD4 or CCR5, which are required for R5 HIV binding and entry into macrophages (Fig. 2A). Consistent with this finding, binding of HIV to HDACi-treated cells, as measured by ELISA of cell-associated p24 Gag protein, was similar to that of untreated cells (p > 0.3; Fig. 2B). Moreover, the quantity of intracellular p24 Gag (trypsin-resistant) in cells exposed to virus for 5 h in both untreated and HDACi-treated cells were similar (p > 0.29; Fig. 2C). As controls, sirolimus and maraviroc were used. Sirolimus is known to down-regulate CCR5 expression (23), and maraviroc is a CCR5 antagonist (24), thereby providing a control for CCR5 binding. Combined, these results suggest that, collectively, HDACi have no effect on either HIV binding or entry.
FIGURE 2.

HDACi do not affect HIV entry. A, macrophages were treated with vehicle or HDACi. After 4 h, cells were harvested and analyzed for cell surface CD4 and CCR5 expression by flow cytometry. B, macrophages treated with HDACi, sirolimus, or maraviroc for 4 h were exposed to replication-competent HIV. Binding was measured at 3 h postinfection by washing cells extensively, then lysing, and analyzing p24 by ELISA. C, entry was measured at 5 h postinfection by washing cells extensively, then trypsinizing, lysing, and subsequently analyzing intracellular p24 by ELISA. D, DNA was extracted from cells at 8 h postinfection for qPCR analysis of pre-integration strong-stop HIV DNA. POLR2A was amplified as a control. Results are expressed as the ratio between the HIV LTR DNA and POLR2A, normalized to the vehicle control, and then log2 transformed. E, macrophages lysed at 5 h post-infection were subjected to immunoblotting for both HIV Nef and ACTB. F, DNA was extracted from cells at 10 days postinfection for Alu-LTR-based nested qPCR. Results are expressed as the ratio between the Alu-LTR DNA and β2-microglobulin, normalized to the vehicle control, and then log2 transformed. G, percentage of TZM-bl cells productively infected with HIV after 4 h of pretreatment with HDACi. H, macrophages were incubated with 1 ng of HIV p24 antigen from the 10-day aliquots of cell-free supernatants post-HDACi treatment for 3 h and then cultured for 10 days with ELISA performed for HIV p24 antigen. All HDACi and sirolimus were used at 100 nmol/liter except romidepsin, which was used at 50 nmol/liter. Results are reported as mean ± S.E., n = 5. *, p < 0.05.
We then measured viral infection using qPCR for the presence and quantity of strong-stop HIV DNA (with LTR R/U5 primers) 8 h post-infection. Macrophages were treated with HDACi for 4 h prior to infection with HIV. Following 8 h of infection, HDACi had no effect on HIV reverse transcription (p > 0.1; Fig. 2D). As Nef is expressed in abundance during the early phase of HIV infection (25), we also analyzed the translation of Nef by immunoblotting. We observed a slight but nonsignificant increase in Nef production in HDACi-treated cells over vehicle controls (p > 0.18; Fig. 2E). We next assessed viral integration at 10 days post-infection and observed no significant difference in integration events post-HDACi treatment (Fig. 2F). We then assessed for productive HIV infection by assaying for Tat activity using TZM-bl cells. We pretreated TZM-bl cells with HDACi for 4 h and then exposed them to HIV. At the concentrations tested, none of the HDACi had significant inhibitory effects on HIV productive infection as measured by HIV Tat activity (p > 0.56; Fig. 2G).
In our next series of experiments, we assessed whether the decrease in cell culture supernatant HIV p24 release was due to HDACi inducing the production of replication-incompetent viral particles. 1 ng of p24 antigen from cell-free supernatants derived from cells treated with HDACi and infected for 10 days was used to infect fresh macrophages in the absence of HDACi. We observed no difference in HIV-replicative fitness post-HDACi treatment indicating that the virus being released is replication-competent (p > 0.19; Fig. 2H). Next, we examined the effect of HDACi on HIV replication at early time points post-infection by treating macrophage cultures exposed to HIV with HDACi at 4 h pre-exposure, at the time of infection and at 3, 5, and 7 days post-infection. At every time point, each HDACi induced a significant decrease in cell culture supernatant HIV p24 antigen accumulation (Fig. 3) in the absence of toxic cellular effects.
FIGURE 3.
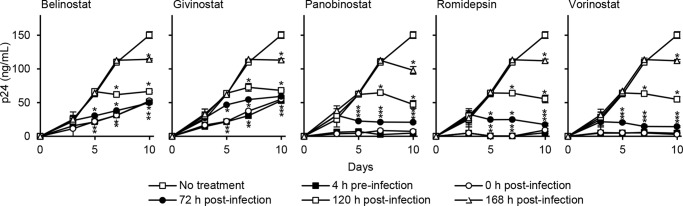
HDACi decrease HIV p24 release post-infection. Macrophages were incubated with HDACi at different time points with respect to infection with HIV. ELISAs were performed for HIV p24 antigen over 10 days. All HDACi were used at 100 nmol/liter except romidepsin, which was used at 50 nmol/liter. Results are reported as mean ± S.E., n = 6; *, p < 0.05.
HDACi Induce Autophagy in Human Primary Macrophages
HDACi are known to promote autophagic responses in a multitude of different cell types (26–34), and the pharmacological induction of autophagy is known to inhibit HIV dissemination from macrophages (6, 17, 35–37). However, the ability of HDACi to induce an autophagic response in primary human macrophages has not been investigated. During autophagy, cytosolic LC3B-I is converted to LC3B-II by a ubiquitin-like system that involves ATG7, ATG3, and the ATG12—ATG5 complex. The ATG12—ATG5 complex ligates LC3B-II to the nascent autophagosome membrane through phosphatidylethanolamine with the LC3B-II associated with the inner membrane degraded after fusion of the autophagosome with lysosomes. Therefore, the conversion of LC3B-I to LC3B-II and its turnover is an indicator of autophagy induction and flux (38). Exposure of macrophages to HDACi for 24 h led to a dose-dependent increase in LC3B-II similar to that observed with sirolimus, an inducer of autophagy through inhibition of the MTOR complex 1 (MTORC1) (Fig. 4A).
FIGURE 4.

HDACi induce autophagy in human macrophages. Macrophages were treated for 24 h with HDACi or sirolimus. A, top, representative immunoblots of LC3B isoforms and SQSTM1 using antibody to LC3B, SQSTM1, and ACTB. Bottom, densitometric analysis of immunoblots presented as means ± S.E., n = 4. *, p < 0.05. B, extracellular release of LDH was measured spectrophotometrically using the cytotoxicity detection kitPLUS (LDH) assay. The percentage of cells with apoptotic ssDNA was quantified using the ssDNA ELISA. Results are reported as mean ± S.E., n = 4; *, p < 0.05.
To verify that the increase in LC3 lipidation in HDACi-treated cells versus control cells represents increased autophagic flux rather than an accumulation of LC3B-II, the degradation of the polyubiquitin-binding protein SQSTM1 was quantified. Inhibition of autophagy leads to an increase in SQSTM1 protein levels, whereas autolysosomes degrade SQSTM1- and LC3-positive bodies during autophagic flux (39). In these experiments, HDACi treatment led to a dose-dependent decrease in SQSTM1 protein levels corresponding to the induction of autophagic flux (Fig. 4A). Although we observed an increase in autophagic markers in the absence of visible pyknosis, karyorrhexis, or plasma membrane blebbing (data not shown), it was important to confirm that the cells were not undergoing cell death at the concentrations being used, as the induction of excessive autophagy can cause cell death (40, 41). Importantly, we did not observe a significant increase in cell death due to HDACi treatment at this time point as measured by LDH release and by ssDNA accumulation (Fig. 4B).
HDACi Induce Autophagy by Inhibiting MTOR and Activating ULK1
The ULK1 kinase complex functions at the initial stages of the canonical autophagy pathway and induces autophagy by phosphorylating beclin 1 and activating phosphatidylinositol 3-kinase catalytic subunit type 3 (42–44). Under nutrient-rich conditions MTORC1, consisting of MTOR, the regulatory-associated protein of MTOR complex 1 (RPTOR), and MTOR-associated protein LST8 homolog (MLST8), phosphorylates ULK1 at Ser757 and binds it through RPTOR. Under conditions of stress such as nutrient deprivation or Toll-like receptor signaling, MTORC1 is inhibited leading to global dephosphorylation of ULK1, dissociation of ULK1 from MTORC1, and the induction of autophagosome formation. Therefore, we first examined whether ULK1 is involved in HDACi-mediated autophagy by determining whether HDACi treatment causes the inhibition of MTOR and thereby the dephosphorylation and activation of ULK1. All HDACi were found to induce significant dephosphorylation and activation of ULK1 as monitored by phospho-ULK1 (Ser757)-specific antibodies and by its faster mobility in SDS-PAGE (Fig. 5). Moreover, belinostat, givinostat, and panobinostat all induced the dose-dependent decrease in phospho-ULK1 (Ser757) (Fig. 5). We next examined whether HDACi cause global inhibition of MTORC1 by measuring the phosphorylation status of two well known MTOR substrates, RPS6KB2 and EIF4EBP1. HDACi treatment induced the dephosphorylation of RPS6KB2 and EIF4EBP1 in a similar manner to ULK1 (Fig. 5) suggesting that HDACi inhibit MTOR.
FIGURE 5.

HDACi inhibit the mammalian target of rapamycin. Macrophages were treated for 24 h with HDACi or sirolimus. Top, representative immunoblots using antibody to phospho-ULK1 (Ser757), phospho-EIF4EBP1 (Thr37/46), phospho-RPS6KB2 (Thr389), total ULK1, total EIF4EBP1, total RPS6KB2, and ACTB. Bottom, densitometric analysis of immunoblots presented as means ± S.E., n = 4. All treatments resulted in p < 0.05.
HDACi-mediated Autophagy Inhibits HIV Dissemination from Human Macrophages
To determine whether HDACi-induced autophagy contributes to the HDACi-mediated decrease of HIV p24 antigen release, we assessed the effect of ATG5 and ATG7 silencing on HIV infection post-HDACi treatment. Both ATG5 and ATG7 RNAi were effective in silencing their respective genes over the 10-day infection protocol (Figs. 6A and 7A) and were efficient at inhibiting LC3B lipidation and the degradation of SQSTM1, and thus autophagy (Figs. 6B and 7B). To validate both the selectivity of the shRNAs for ATG5 and ATG7, and the specificity of the phenotype, we performed rescue experiments employing expression constructs containing shRNA-resistant forms of the gene's open reading frame (ORF) (Figs. 6B and 7B). In the presence of shRNA-insensitive ORF transcripts, the wild type phenotype was rescued, and autophagy was restored (Figs. 6B and 7B). ATG5 RNAi abrogated the belinostat (p < 0.001) and givinostat (p < 0.001) and significantly diminished the panobinostat-, romidepsin-, and vorinostat-mediated decrease of cell culture supernatant HIV p24 antigen accumulation by day 10 (Fig. 6C). ATG7 RNAi abrogated both belinostat- and givinostat-mediated inhibition of HIV p24 accumulation by day 10 (p < 0.001). It also significantly diminished the panobinostat-, romidepsin-, and vorinostat-mediated inhibition of HIV by day 10 (p < 0.001; Fig. 7C). Importantly, in the presence of shRNA-insensitive ATG5 (Fig. 6D) or ATG7 (Fig. 7D) ORF transcripts, the HDACi-mediated decrease in extracellular supernatant HIV p24 antigen accumulation was similar to the nontargeting negative control shRNA.
FIGURE 6.

HDACi-mediated decrease of HIV p24 antigen release from macrophages requires ATG5. A, macrophages transduced with ATG5 shRNA (TRCN0000150940 or TRCN0000151963) or scrambled shRNA (shNS) were incubated with HDACi for 24 h before HIV infection under continuous HDACi treatment for 10 days. Top, representative immunoblots performed using antibodies raised to ATG5 and ACTB. Bottom, densitometric analysis of immunoblots presented as means ± S.E., n = 4. B, macrophages transduced as in A with the corresponding shRNA-insensitive open reading frame (ORF) lentiviral vectors were treated with 100 nmol/liter sirolimus for 24 h, harvested, and analyzed for autophagy proteins. A representative immunoblot of LC3B isoforms, ATG5, and SQSTM1 using antibody to LC3B, SQSTM1, ATG5, and ACTB is presented. C, macrophages transduced as in A were incubated with HDACi for 24 h before HIV infection under continuous HDACi treatment for 10 days. ELISA was performed for extracellular release of HIV p24 antigen. D, macrophages transduced with ATG5 shRNA and the corresponding rescue cDNA ORF vector were incubated with HDACi for 24 h before HIV infection under continuous HDACi treatment for 10 days. ELISA was performed for extracellular release of HIV p24 antigen. All data are reported as mean ± S.E., n = 4; *, p < 0.05.
FIGURE 7.

HDACi-mediated decrease of HIV p24 antigen release from macrophages requires ATG7. A, macrophages transduced with ATG7 shRNA (TRCN0000007584 or TRCN0000435480) or scrambled shRNA (shNS) were incubated with HDACi for 24 h before HIV infection under continuous HDACi treatment for 10 days. Top, representative immunoblots performed using antibodies raised to ATG7 and ACTB. Bottom, densitometric analysis of immunoblots presented as means ± S.E., n = 4. B, macrophages transduced as in A with the corresponding shRNA-insensitive open reading frame (ORF) lentiviral vectors were treated with 100 nmol/liter sirolimus for 24 h, harvested, and analyzed for autophagy proteins. A representative immunoblot of LC3B isoforms, ATG7, and SQSTM1 using antibody to LC3B, SQSTM1, ATG7, and ACTB is presented. C, macrophages transduced as in A were incubated with HDACi for 24 h before HIV infection under continuous HDACi treatment for 10 days. ELISA was performed for extracellular release of HIV p24 antigen. D, macrophages transduced with ATG7 shRNA and the corresponding rescue cDNA ORF vector were incubated with HDACi for 24 h before HIV infection under continuous HDACi treatment for 10 days. ELISA was performed for extracellular release of HIV p24 antigen. All data are reported as mean ± S.E., n = 4; *, p < 0.05.
We next investigated whether autophagosome acidification, a late stage event during autophagy, is required for the HDACi-mediated autophagic decrease in HIV release. During autophagy, lysosomes fuse with autophagosomes to form autolysosomes. Macrophages were treated with bafilomycin A1, an inhibitor of the vacuolar H+-ATPase and autophagosome-lysosome fusion, and subsequently infected with HIV. Bafilomycin A1 reversed the HDACi-mediated decrease of extracellular HIV accumulation (Fig. 8A), and in the case of belinostat and givinostat, it completely abrogated the decrease in HIV release suggesting that the acidic pH of autolysosomes is required for the autophagy-mediated control of HIV.
FIGURE 8.

HDACi-mediated decrease of HIV p24 antigen release from macrophages requires the completion of autophagic flux. A, macrophages were pretreated with vehicle control (top) or bafilomycin A1 (bottom) before incubation with HDACi for 24 h before infection with HIV. Cells were then washed and incubated with HDACi and bafilomycin A1 for 10 days. ELISA was performed for extracellular HIV p24 antigen release over 10 days. Results are reported as mean ± S.E., n = 5. B, macrophages were pretreated with vehicle control (top) or SID 26681509 (bottom) before incubation with HDACi for 24 h before infection with HIV. Cells were then washed and incubated with HDACi and SID 26681509 for 10 days. ELISA was performed for extracellular HIV p24 antigen release over 10 days. Results are reported as mean ± S.E., n = 5. *, p < 0.05.
After lysosomes fuse with autophagosomes to form autolysosomes, the sequestered components are degraded by lysosomal hydrolases and are released into the cytosol by lysosomal efflux permeases. We investigated whether lysosomal hydrolases are important for the HDACi-mediated decrease in HIV release through autophagy using SID 26681509, a novel thiocarbazate-specific inhibitor of the lysosome hydrolase cathepsin L. Importantly, in the absence of HDACi, SID 26681509 induced no net inhibition of HIV (Fig. 8B). Moreover, in the presence of HDACi, SID 26681509 reversed the observed HDACi-mediated decline in extracellular accumulation of HIV, and similar to bafilomycin A1, it completely abrogated both belinostat- and givinostat-mediated decrease with a less pronounced but still significant effect on vorinostat, romidepsin, and panobinostat (Fig. 8B).
Finally, we examined the effect of HDACi on intracellular HIV p24 antigen levels at a late time point post-infection by treating macrophage cultures exposed to HIV with HDACi at 7 days post-infection in the presence or absence of bafilomycin A1. As expected, HIV p24 antigen levels were increased in the presence of bafilomycin A1 alone. Moreover, at the concentrations tested, HDACi induced a significant decrease in intracellular HIV p24 antigen by day 10 post-infection (Fig. 9). Importantly, the degradation of HIV induced by HDACi was abrogated in the presence of bafilomycin A1. Collectively, these data indicate that HDACi induce the degradation of HIV through the induction of autophagy, which leads to a reduction in the number of virions that are then released.
FIGURE 9.
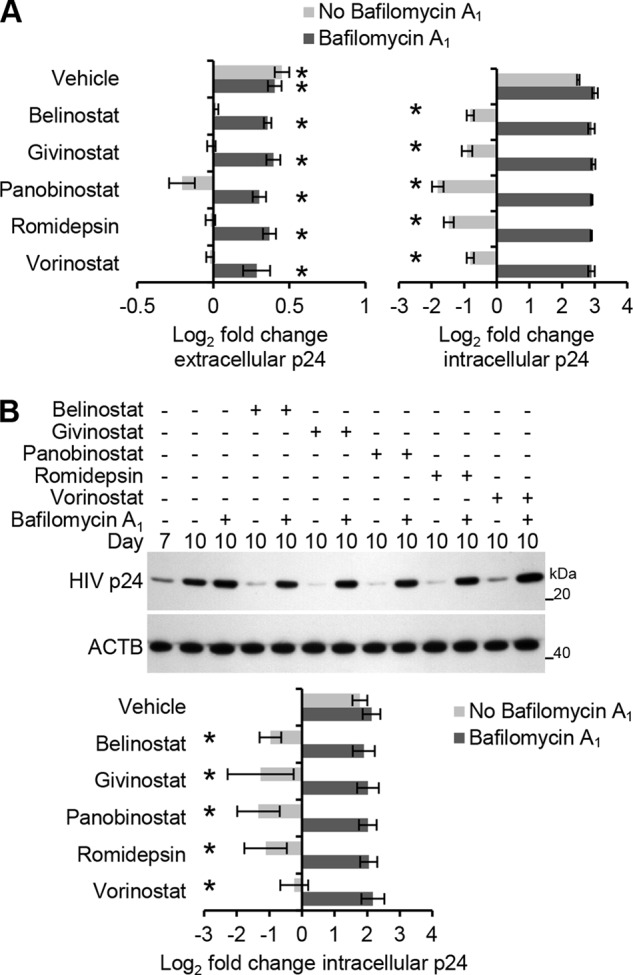
HDACi induce the lysosomal degradation of intracellular HIV. Macrophages were infected for 7 days with HIV and then treated with HDACi in the presence or absence of bafilomycin A1 for 3 days. All HDACi were used at 100 nmol/liter except romidepsin, which was used at 50 nmol/liter. A, ELISA was performed for both extracellular and intracellular HIV p24 antigen at 7 and 10 days post-infection. n = 3. B, top, representative immunoblots of HIV p24 antigen using antibody to HIV p24 antigen and ACTB. Bottom, densitometric analysis of immunoblots presented as means ± S.E., n = 4. *, p < 0.05.
DISCUSSION
Several HDACi have been evaluated for their ability to activate HIV production from latently infected cells (45, 46). To date, only vorinostat (3, 47, 48), valproic acid (48), MRK1 (49), and romidepsin (12) have demonstrated an ability to reactivate HIV from resting CD4+ T cells from HIV-infected patients on suppressive cART, the gold standard for studying latently infected CD4+ T cells. However, vorinostat was found to promote HIV infection of uninfected CD4+ T cells in a dose- and time-dependent manner that was independent of receptor and coreceptor usage raising concerns for the clinical use of vorinostat as it could reseed the viral reservoirs intended to be purged (4). To our knowledge, no study, in vitro or in vivo, had investigated the effect of HDACi on HIV infection of macrophages, an important site of HIV infection and persistence. Macrophages, along with dendritic cells, are an important reservoir due to their longevity and widespread dissemination within multiple tissue compartments. However, there is currently a lack of scientific data demonstrating that HIV can be cleared from macrophages. In this study, we investigated the effect of the HDACi belinostat, givinostat, panobinostat, romidepsin, and vorinostat on HIV infection of macrophages. Although the HDACi tested had no effect on viral binding, entry, reverse transcription, integration, or protein synthesis events, they dose-dependently inhibited HIV through the degradative canonical autophagy pathway involving ULK1 that required the formation of autophagosomes and their subsequent maturation into autolysosomes. These results reveal a previously unknown mechanism of HDACi on HIV infection that is distinct from their ability to reactivate viral transcription from latently infected CD4+ T cells. Our findings with vorinostat are in contrast to those reported by Lucera et al. (4) in uninfected CD4+ T cells outlining the importance of studying the different HDACi and their actions on different cell types. For instance, the pan-HDAC inhibitor valproic acid induces acetylation at the integrated HIV proviral promoter and reactivates latent HIV expression from resting CD4+ T cells of aviremic patients without activating or rendering uninfected CD4+ T cells more permissive for de novo infection (50). Furthermore, structurally related HDACi have been shown to have both anti- and pro-inflammatory responses in vivo; therefore, predicting their effects in the context of in vivo HIV infection may be difficult (51, 52). As an example, the anti-inflammatory pan-HDACi vorinostat can stimulate thymic production of natural regulatory T cells (Tregs), promote peripherally generated induced Tregs, and enhance the immune suppressive function of human Tregs (53, 54). In addition, the structurally related trichostatin A increases forkhead box P3 (FOXP3) acetylation thereby protecting it from proteasomal degradation (55). Thus, hydroxamic acid HDACi-induced immune suppression via Tregs may impact the course of HIV infection by permitting virus-associated excess inflammation that drives disease progression in untreated HIV infection and causes premature immunosenescence and morbidity in persons on cART (56). However, entinostat, a benzamide HDACi, transcriptionally down-regulates FOXP3 expression and blocks the suppressive function of Tregs without affecting T effector cells (57). These findings further demonstrate the complex and diverse biological roles of HDACi drugs.
In our studies, all HDACi induced the significant dephosphorylation and activation of ULK1 indicative of the inactivation of MTOR. However, it is unknown whether these HDACi inactivate MTOR via histone deacetylation and therefore transcription of certain autophagic genes or via the deacetylation inhibition of a non-histone protein, which regulates MTOR function. Importantly, unlike reports from studies involving cancer cell lines, at the biologically relevant concentrations tested, these HDACi failed to induce either nonapoptotic cell death or apoptosis of primary macrophages further demonstrating that the effects of HDACi can vary significantly depending upon the cell type studied.
This study provides support for the potential benefits of supplementing cART with HDACi in a cure strategy. Well controlled clinical trials are needed to determine whether HDACi supplementation is of value as adjunctive treatment in HIV-infected persons. However, if hydroxamic acid-based HDACi are to be employed in a shock-and-kill strategy, increasing cART may help reduce the likelihood of reseeding the CD4+ T cell viral reservoirs and may play a role in decreasing the size of the viral reservoir in treated patients. This is especially important when considering that local drug exposure and viral dynamics differ significantly in HIV sanctuary sites when compared with the systemic compartment. Combined with patient noncompliance and viral resistance, this may potentially endanger the efficacy of both cART and thereby the shock-and-kill strategy in the long term, and it may even make eradication of HIV impossible under such circumstances.
Our recent studies have demonstrated that the pharmacological induction of autophagy inhibits HIV replication in macrophages (6, 17, 35–37). Consequently, this study adds a new drug class that is not only able to decrease HIV release from macrophages but also degrades viral particles through autophagy. Further highlighting autophagy in the control of HIV, a recent study found that peripheral blood mononuclear cells from elite controllers contain significantly more autophagic vesicles and express more autophagic markers than normal progressors. Moreover, the same peripheral blood mononuclear cells from elite controllers were more responsive to sirolimus treatment leading to an enhanced autophagic response and a greater reduction in virus production (58). Dissecting the molecular mechanisms by which HIV utilizes autophagy has the potential to lead to the identification of novel drug candidates to treat HIV infection and related opportunistic infections. The induction and modulation of autophagy through pharmacological means to enhance HIV treatment are both attractive and novel as autophagy works at the host cellular level to improve intracellular killing of both replicating and nonreplicating HIV, although resistance is unlikely to develop. However, it remains to be determined whether the general beneficial effects of increased autophagy for treating infections and other disorders outweigh its possible harmful effects on normal cellular functions.
Acknowledgments
We thank Terrence Robinson and Zachary Pallack for technical assistance.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 NS084912 from NINDS, AI084573 from the NIAID. This work was also supported by the International Maternal Pediatric Adolescent AIDS Clinical Trials Network and California HIV/AIDS Research Program ID12-SD-255. This work was presented in part at the 21st Conference on Retroviruses and Opportunistic Infections, March 3–6, 2014, Boston, MA.
- cART
- combined antiretroviral therapy
- ABCC
- ATP-binding cassette, subfamily C (CFTR/MRP), member
- ACTB
- β-actin
- CCR5
- chemokine (C-C motif) receptor 5
- HDACi
- histone deacetylase inhibitor
- MTOR
- mammalian target of rapamycin
- MTORC1
- MTOR complex 1
- qPCR
- quantitative real-time PCR
- SQSTM1
- sequestosome 1
- Tregs
- regulatory T cells
- ULK1
- unc-51 like autophagy activating kinase 1
- LDH
- lactate dehydrogenase
- ssDNA
- single-stranded DNA.
REFERENCES
- 1. Siliciano R. F., Greene W. C. (2011) HIV latency. Cold Spring Harb. Perspect. Med. 1, a007096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Manson McManamy M. E., Hakre S., Verdin E. M., Margolis D. M. (2014) Therapy for latent HIV-1 infection: the role of histone deacetylase inhibitors. Antivir. Chem. Chemother. 23, 145–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Archin N. M., Liberty A. L., Kashuba A. D., Choudhary S. K., Kuruc J. D., Crooks A. M., Parker D. C., Anderson E. M., Kearney M. F., Strain M. C., Richman D. D., Hudgens M. G., Bosch R. J., Coffin J. M., Eron J. J., Hazuda D. J., Margolis D. M. (2012) Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature 487, 482–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lucera M., Tilton C. A., Mao H., Dobrowolski C., Tabler C. O, Haqqani A. A., Karn J., Tilton J. C. (2014) The histone deacetylase inhibitor vorinostat (SAHA) increases the susceptibility of uninfected CD4+ T cells to HIV by increasing the kinetics and efficiency of post-entry viral events. J. Virol. 88, 10803–10812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jorajuria S., Dereuddre-Bosquet N., Naissant-Storck K., Dormont D., Clayette P. (2004) Differential expression levels of MRP1, MRP4, and MRP5 in response to human immunodeficiency virus infection in human macrophages. Antimicrob. Agents Chemother. 48, 1889–1891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Campbell G. R., Spector S. A. (2012) Toll-like receptor 8 ligands activate a vitamin D mediated autophagic response that inhibits human immunodeficiency virus type 1. PLoS Pathog. 8, e1003017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wei X., Decker J. M., Liu H., Zhang Z., Arani R. B., Kilby J. M., Saag M. S., Wu X., Shaw G. M., Kappes J. C. (2002) Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob. Agents Chemother. 46, 1896–1905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Campbell G. R., Watkins J. D., Loret E. P., Spector S. A. (2011) Differential induction of rat neuronal excitotoxic cell death by human immunodeficiency virus type 1 clade B and C Tat proteins. AIDS Res. Hum. Retroviruses 27, 647–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Steele N. L., Plumb J. A., Vidal L., Tjørnelund J., Knoblauch P., Buhl-Jensen P., Molife R., Brown R., de Bono J. S., Evans T. R. (2011) Pharmacokinetic and pharmacodynamic properties of an oral formulation of the histone deacetylase inhibitor Belinostat (PXD101). Cancer Chemother. Pharmacol. 67, 1273–1279 [DOI] [PubMed] [Google Scholar]
- 10. Furlan A., Monzani V., Reznikov L. L., Leoni F., Fossati G., Modena D., Mascagni P., Dinarello C. A. (2011) Pharmacokinetics, safety and inducible cytokine responses during a phase 1 trial of the oral histone deacetylase inhibitor ITF2357 (givinostat). Mol. Med. 17, 353–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. DeAngelo D. J., Spencer A., Bhalla K. N., Prince H. M., Fischer T., Kindler T., Giles F. J., Scott J. W., Parker K., Liu A., Woo M., Atadja P., Mishra K. K., Ottmann O. G. (2013) Phase Ia/II, two-arm, open-label, dose-escalation study of oral panobinostat administered via two dosing schedules in patients with advanced hematologic malignancies. Leukemia 27, 1628–1636 [DOI] [PubMed] [Google Scholar]
- 12. Wei D. G., Chiang V., Fyne E., Balakrishnan M., Barnes T., Graupe M., Hesselgesser J., Irrinki A., Murry J. P., Stepan G., Stray K. M., Tsai A., Yu H., Spindler J., Kearney M., Spina C. A., McMahon D., Lalezari J., Sloan D., Mellors J., Geleziunas R., Cihlar T. (2014) Histone deacetylase inhibitor romidepsin induces HIV expression in CD4 T cells from patients on suppressive antiretroviral therapy at concentrations achieved by clinical dosing. PLoS Pathog. 10, e1004071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fujiwara Y., Yamamoto N., Yamada Y., Yamada K., Otsuki T., Kanazu S., Iwasa T., Hardwick J. S., Tamura T. (2009) Phase I and pharmacokinetic study of vorinostat (suberoylanilide hydroxamic acid) in Japanese patients with solid tumors. Cancer Sci. 100, 1728–1734 [DOI] [PubMed] [Google Scholar]
- 14. Gartner S., Markovits P., Markovitz D. M., Kaplan M. H., Gallo R. C., Popovic M. (1986) The role of mononuclear phagocytes in HTLV-III/LAV infection. Science 233, 215–219 [DOI] [PubMed] [Google Scholar]
- 15. Popovic M., Gartner S., Read-Connole E., Beaver B., Reitz M. (1988) in Retroviruses of Human AIDS and Related Animal Diseases, Colloque Des Cent Gardes (Girard M., Valette L., eds) pp. 21–27, Pasteur Vaccins, Marnes-La-Coquette, France [Google Scholar]
- 16. Campbell G. R., Spector S. A. (2008) CCL2 increases X4-tropic HIV-1 entry into resting CD4+ T cells. J. Biol. Chem. 283, 30745–30753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Campbell G. R., Spector S. A. (2011) Hormonally active vitamin D3 (1α,25-dihydroxycholecalciferol) triggers autophagy in human macrophages that inhibits HIV-1 infection. J. Biol. Chem. 286, 18890–18902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pfaffl M. W. (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29, e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Butler S. L., Hansen M. S., Bushman F. D. (2001) A quantitative assay for HIV DNA integration in vivo. Nat. Med. 7, 631–634 [DOI] [PubMed] [Google Scholar]
- 20. Lewin S. R., Murray J. M., Solomon A., Wightman F., Cameron P. U., Purcell D. J., Zaunders J. J., Grey P., Bloch M., Smith D., Cooper D. A., Kelleher A. D. (2008) Virologic determinants of success after structured treatment interruptions of antiretrovirals in acute HIV-1 infection. J. Acquir. Immune Defic. Syndr. 47, 140–147 [PubMed] [Google Scholar]
- 21. Bose P., Dai Y., Grant S. (2014) Histone deacetylase inhibitor (HDACI) mechanisms of action: Emerging insights. Pharmacol. Ther. 143, 323–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Frankfurt O. S., Krishan A. (2001) Enzyme-linked immunosorbent assay (ELISA) for the specific detection of apoptotic cells and its application to rapid drug screening. J. Immunol. Methods 253, 133–144 [DOI] [PubMed] [Google Scholar]
- 23. Donia M., McCubrey J. A., Bendtzen K., Nicoletti F. (2010) Potential use of rapamycin in HIV infection. Br. J. Clin. Pharmacol. 70, 784–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fätkenheuer G., Pozniak A. L., Johnson M. A., Plettenberg A., Staszewski S., Hoepelman A. I., Saag M. S., Goebel F. D., Rockstroh J. K., Dezube B. J., Jenkins T. M., Medhurst C., Sullivan J. F., Ridgway C., Abel S., James I. T., Youle M., van der Ryst E. (2005) Efficacy of short-term monotherapy with maraviroc, a new CCR5 antagonist, in patients infected with HIV-1. Nat. Med. 11, 1170–1172 [DOI] [PubMed] [Google Scholar]
- 25. Klotman M. E., Kim S., Buchbinder A., DeRossi A., Baltimore D., Wong-Staal F. (1991) Kinetics of expression of multiply spliced RNA in early human immunodeficiency virus type 1 infection of lymphocytes and monocytes. Proc. Natl. Acad. Sci. U.S.A. 88, 5011–5015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cao Q., Yu C., Xue R., Hsueh W., Pan P., Chen Z., Wang S., McNutt M., Gu J. (2008) Autophagy induced by suberoylanilide hydroxamic acid in Hela S3 cells involves inhibition of protein kinase B and up-regulation of Beclin 1. Int. J. Biochem. Cell Biol. 40, 272–283 [DOI] [PubMed] [Google Scholar]
- 27. New M., Olzscha H., La Thangue N. B. (2012) HDAC inhibitor-based therapies: can we interpret the code? Mol. Oncol. 6, 637–656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gammoh N., Lam D., Puente C., Ganley I., Marks P. A., Jiang X. (2012) Role of autophagy in histone deacetylase inhibitor-induced apoptotic and nonapoptotic cell death. Proc. Natl. Acad. Sci. U.S.A. 109, 6561–6565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shao Y., Gao Z., Marks P. A., Jiang X. (2004) Apoptotic and autophagic cell death induced by histone deacetylase inhibitors. Proc. Natl. Acad. Sci. U.S.A. 101, 18030–18035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Oh M., Choi I. K., Kwon H. J. (2008) Inhibition of histone deacetylase1 induces autophagy. Biochem. Biophys. Res. Commun. 369, 1179–1183 [DOI] [PubMed] [Google Scholar]
- 31. Robert T., Vanoli F., Chiolo I., Shubassi G., Bernstein K. A., Rothstein R., Botrugno O. A., Parazzoli D., Oldani A., Minucci S., Foiani M. (2011) HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature 471, 74–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shulak L., Beljanski V., Chiang C., Dutta S. M., Van Grevenynghe J., Belgnaoui S. M., Nguyên T. L., Di Lenardo T., Semmes O. J., Lin R., Hiscott J. (2014) Histone deacetylase inhibitors potentiate vesicular stomatitis virus oncolysis in prostate cancer cells by modulating NF-κB-dependent autophagy. J. Virol. 88, 2927–2940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Watanabe T., Nagase K., Chosa M., Tobinai K. (2010) Schwann cell autophagy induced by SAHA, 17-AAG, or clonazepam can reduce bortezomib-induced peripheral neuropathy. Br. J. Cancer 103, 1580–1587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wu S., Zheng S. D., Huang H. L., Yan L. C., Yin X. F., Xu H. N., Zhang K. J., Gui J. H., Chu L., Liu X. Y. (2013) Lithium down-regulates histone deacetylase 1 (HDAC1) and induces degradation of mutant huntingtin. J. Biol. Chem. 288, 35500–35510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Campbell G. R., Spector S. A. (2012) Vitamin D inhibits human immunodeficiency virus type 1 and Mycobacterium tuberculosis infection in macrophages through the induction of autophagy. PLoS Pathog. 8, e1002689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Campbell G. R., Spector S. A. (2013) Inhibition of human immunodeficiency virus type-1 through autophagy. Curr. Opin. Microbiol. 16, 349–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shoji-Kawata S., Sumpter R., Leveno M., Campbell G. R., Zou Z., Kinch L., Wilkins A. D., Sun Q., Pallauf K., MacDuff D., Huerta C., Virgin H. W., Helms J. B., Eerland R., Tooze S. A., Xavier R., Lenschow D. J., Yamamoto A., King D., Lichtarge O., Grishin N. V., Spector S. A., Kaloyanova D. V., Levine B. (2013) Identification of a candidate therapeutic autophagy-inducing peptide. Nature 494, 201–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Klionsky D. J., Abdalla F. C., Abeliovich H., Abraham R. T., Acevedo-Arozena A., Adeli K., Agholme L., Agnello M., Agostinis P., Aguirre-Ghiso J. A., Ahn H. J., Ait-Mohamed O., Ait-Si-Ali S., Akematsu T., Akira S., et al. (2012) Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 8, 445–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bjørkøy G., Lamark T., Brech A., Outzen H., Perander M., Overvatn A., Stenmark H., Johansen T. (2005) p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 171, 603–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Galluzzi L., Aaronson S. A., Abrams J., Alnemri E. S., Andrews D. W., Baehrecke E. H., Bazan N. G., Blagosklonny M. V., Blomgren K., Borner C., Bredesen D. E., Brenner C., Castedo M., Cidlowski J. A., Ciechanover A., et al. (2009) Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death Differ. 16, 1093–1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kroemer G., Levine B. (2008) Autophagic cell death: the story of a misnomer. Nat. Rev. Mol. Cell Biol. 9, 1004–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Itakura E., Mizushima N. (2010) Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy 6, 764–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Russell R. C., Tian Y., Yuan H., Park H. W., Chang Y. Y., Kim J., Kim H., Neufeld T. P., Dillin A., Guan K. L. (2013) ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 15, 741–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ganley I. G., Lam du H., Wang J., Ding X., Chen S., Jiang X. (2009) ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 284, 12297–12305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rasmussen T. A., Schmeltz Søgaard O., Brinkmann C., Wightman F., Lewin S. R., Melchjorsen J., Dinarello C., Østergaard L., Tolstrup M. (2013) Comparison of HDAC inhibitors in clinical development: effect on HIV production in latently infected cells and T-cell activation. Hum. Vaccin. Immunother. 9, 993–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wightman F., Ellenberg P., Churchill M., Lewin S. R. (2012) HDAC inhibitors in HIV. Immunol. Cell Biol. 90, 47–54 [DOI] [PubMed] [Google Scholar]
- 47. Contreras X., Schweneker M., Chen C. S., McCune J. M., Deeks S. G., Martin J., Peterlin B. M. (2009) Suberoylanilide hydroxamic acid reactivates HIV from latently infected cells. J. Biol. Chem. 284, 6782–6789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Archin N. M., Espeseth A., Parker D., Cheema M., Hazuda D., Margolis D. M. (2009) Expression of latent HIV induced by the potent HDAC inhibitor suberoylanilide hydroxamic acid. AIDS Res. Hum. Retroviruses 25, 207–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Archin N. M., Keedy K. S., Espeseth A., Dang H., Hazuda D. J., Margolis D. M. (2009) Expression of latent human immunodeficiency type 1 is induced by novel and selective histone deacetylase inhibitors. AIDS 23, 1799–1806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ylisastigui L., Archin N. M., Lehrman G., Bosch R. J., Margolis D. M. (2004) Coaxing HIV-1 from resting CD4 T cells: histone deacetylase inhibition allows latent viral expression. AIDS 18, 1101–1108 [DOI] [PubMed] [Google Scholar]
- 51. Leng C., Gries M., Ziegler J., Lokshin A., Mascagni P., Lentzsch S., Mapara M. Y. (2006) Reduction of graft-versus-host disease by histone deacetylase inhibitor suberonylanilide hydroxamic acid is associated with modulation of inflammatory cytokine milieu and involves inhibition of STAT1. Exp. Hematol. 34, 776–787 [DOI] [PubMed] [Google Scholar]
- 52. Wang D., Iclozan C., Liu C., Xia C., Anasetti C., Yu X. Z. (2012) LBH589 enhances T cell activation in vivo and accelerates graft-versus-host disease in mice. Biol. Blood Marrow Transplant. 18, 1182–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lucas J. L., Mirshahpanah P., Haas-Stapleton E., Asadullah K., Zollner T. M., Numerof R. P. (2009) Induction of Foxp3+ regulatory T cells with histone deacetylase inhibitors. Cell. Immunol. 257, 97–104 [DOI] [PubMed] [Google Scholar]
- 54. Johnson J., Pahuja A., Graham M., Hering B., Hancock W. W., Bansal-Pakala P. (2008) Effects of histone deacetylase inhibitor SAHA on effector and FOXP3+ regulatory T cells in rhesus macaques. Transplant. Proc. 40, 459–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wang L., Tao R., Hancock W. W. (2009) Using histone deacetylase inhibitors to enhance Foxp3(+) regulatory T-cell function and induce allograft tolerance. Immunol. Cell Biol. 87, 195–202 [DOI] [PubMed] [Google Scholar]
- 56. Kuller L. H., Tracy R., Belloso W., De Wit S., Drummond F., Lane H. C., Ledergerber B., Lundgren J., Neuhaus J., Nixon D., Paton N. I., Neaton J. D. (2008) Inflammatory and coagulation biomarkers and mortality in patients with HIV infection. PLoS Med. 5, e203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Shen L., Ciesielski M., Ramakrishnan S., Miles K. M., Ellis L., Sotomayor P., Shrikant P., Fenstermaker R., Pili R. (2012) Class I histone deacetylase inhibitor entinostat suppresses regulatory T cells and enhances immunotherapies in renal and prostate cancer models. PLoS One 7, e30815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Nardacci R., Amendola A., Ciccosanti F., Corazzari M., Esposito V., Vlassi C., Taibi C., Fimia G. M., Del Nonno F., Ippolito G., D'Offizi G., Piacentini M. (2014) Autophagy plays an important role in the containment of HIV-1 in nonprogressor-infected patients. Autophagy 10, 1167–1178 [DOI] [PMC free article] [PubMed] [Google Scholar]