

Background: The role of NLRP3 inflammasomes in hyperoxic acute lung injury (HALI) remains unclear.
Results: NLRP3 deficiency exacerbated lethality and diminished Stat3 activation caused by inflammatory cells in a murine HALI model.
Conclusion: NLRP3 regulates Stat3 activation by affecting inflammatory cell infiltration independent of IL-1β.
Significance: These findings demonstrate the novel role of NLRP3 in Stat3-mediated protective effects against HALI.
Keywords: Cytokine, Inflammasome, Inflammation, Lung Injury, Signal Transduction
Abstract
Supplemental oxygen inhalation is frequently used to treat severe respiratory failure; however, prolonged exposure to hyperoxia causes hyperoxic acute lung injury (HALI), which induces acute respiratory distress syndrome and leads to high mortality rates. Recent investigations suggest the possible role of NLRP3 inflammasomes, which regulate IL-1β production and lead to inflammatory responses, in the pathophysiology of HALI; however, their role is not fully understood. In this study, we investigated the role of NLRP3 inflammasomes in mice with HALI. Under hyperoxic conditions, NLRP3−/− mice died at a higher rate compared with wild-type and IL-1β−/− mice, and there was no difference in IL-1β production in their lungs. Under hyperoxic conditions, the lungs of NLRP3−/− mice exhibited reduced inflammatory responses, such as inflammatory cell infiltration and cytokine expression, as well as increased and decreased expression of MMP-9 and Bcl-2, respectively. NLRP3−/− mice exhibited diminished expression and activation of Stat3, which regulates MMP-9 and Bcl-2, in addition to increased numbers of apoptotic alveolar epithelial cells. In vitro experiments revealed that alveolar macrophages and neutrophils promoted Stat3 activation in alveolar epithelial cells. Furthermore, NLRP3 deficiency impaired the migration of neutrophils and chemokine expression by macrophages. These findings demonstrate that NLRP3 regulates Stat3 signaling in alveolar epithelial cells by affecting macrophage and neutrophil function independent of IL-1β production and contributes to the pathophysiology of HALI.
Introduction
Supplemental oxygen inhalation is an important therapeutic strategy for respiratory failure in patients with severe pneumonia, acute lung injury, and acute respiratory distress syndrome. However, recent evidence indicates that prolonged exposure to hyperoxia causes hyperoxic acute lung injury (HALI),2 characterized by excessive inflammatory responses, endothelial and epithelial injury, and increased pulmonary permeability (1, 2). Inhibition or gene disruption of chemokines has been shown to decrease inflammatory cell infiltration and lethality in animals exposed to hyperoxia (3). In contrast, HALI has been shown to be induced in animal models that lack inflammatory cells (1). Furthermore, a recent study demonstrated that HALI and subsequent lethality may be independent of local inflammatory responses according to findings of a discrepancy between inflammation and lethality under hyperoxic conditions (4). These observations suggest that the pathophysiology of HALI is complex and multifactorial; therefore, the exact mechanism of HALI remains unclear.
Increasing evidence indicates that inflammation in the absence of pathogens, referred to as sterile inflammation, is mediated through NLRP3 (nucleotide-binding oligomerization domain-like receptor family pyrin domain containing 3; also known as NALP3 and cryopyrin) inflammasomes, which are large cytosolic multiple-protein complexes that regulate the production of the proinflammatory cytokine IL-1β (5, 6). NLRP3 inflammasomes contain NLRP3 associated with ASC (apoptosis-associated speck-like protein containing a caspase recruitment domain), which recruits caspase-1 and induces its activation. Caspase-1 is an IL-1β-converting enzyme that processes pro-IL-1β to its mature form and induces IL-1β release, causing inflammatory responses and tissue damage. We recently demonstrated the importance of NLRP3 inflammasomes in the pathogenesis of certain diseases, such as myocardial ischemia-reperfusion, atherosclerosis, vascular injury, and chronic kidney diseases (7–11). Our findings are further supported by reports demonstrating that NLRP3 inflammasomes are critical mediators of other sterile inflammatory diseases, such as gout, pseudogout, asbestosis, silicosis, Alzheimer disease, metabolic syndrome, and type 2 diabetes (6). Kolliputi et al. (12) studied HALI and reported that NLRP3 inflammasomes mediate hyperoxia-induced alveolar cell permeability, which is activated by ceramides present in macrophages (13). Furthermore, Fukumoto et al. (14) showed that NLRP3 deficiency decreases inflammatory responses and HALI in mice exposed to hyperoxia. However, the precise role of NLRP3 inflammasomes in HALI remains unknown.
In this study, we used mice deficient in NLRP3 and IL-1β and unexpectedly found that a deficiency in NLRP3 (but not IL-1β) shortened survival under hyperoxic conditions regardless of diminished inflammatory responses. Furthermore, we observed that Stat3 (signal transducer and activator of transcription 3) signaling caused hyperoxia-induced lethality in NLRP3−/− mice. These findings demonstrate a novel role for NLRP3 in the pathophysiology of HALI independent of IL-1β and provide novel insights into the mechanisms of pathogenesis of HALI.
EXPERIMENTAL PROCEDURES
Animals and Hyperoxia
C57BL/6J (WT) mice were purchased from Japan SLC, Inc. (Tokyo, Japan). NLRP3−/− and IL-1β−/− mice (C57BL/6J genetic background) were kindly provided by Dr. Vishva M. Dixit (Genentech, South San Francisco, CA) (15) and generated as described previously (16), respectively. Female 8–14-week-old mice were used. The mice were exposed to 90% oxygen (7 liters/min) in an airtight plastic chamber (40 × 35 × 30 cm) at 24–25 °C. In our preliminary experiments, we analyzed the total cell counts, neutrophils, and macrophages in bronchoalveolar lavage fluid (BALF) isolated from WT mice at 24, 48, 72, and 96 h after hyperoxia and observed a significant increase in these cells after 72 h. Therefore, we analyzed the samples at 72 h after hyperoxia. The mice had ad libitum access to food and water and were subjected to a 12-h light/dark cycle. The mice were observed every 12 h. All experiments in this study were performed in accordance with the Jichi Medical University Guide for Laboratory Animals.
BALF Analysis
BALF was obtained by cannulating the trachea with an 18-gauge catheter. After the whole lung was lavaged four times with 0.8 ml of PBS, the lavage fluid was centrifuged at 1000 rpm for 10 min at 4 °C, and cell-free supernatants were stored at −30 °C. The pellet was diluted in PBS, the cells were stained with trypan blue, and the number of cells was counted using a hemocytometer. Differential cell analysis was performed by Diff-Quik staining (Sysmex, Kobe, Japan) after cytospinning (800 rpm, 8 min, 22 °C) and by flow cytometry.
Real-time RT-PCR
RNA was extracted using ISOGEN (Nippon Gene Co., Ltd., Toyama, Japan) from lungs perfused with PBS according to the manufacturer's instructions. Real-time RT-PCR analysis was performed using a Takara TP960 PCR Thermal Cycler Dice detection system (Takara Bio Inc., Shiga, Japan) to detect mRNA. The primers (antisense and sense, respectively) were follows: Il1b, 5′-TGAAGTTGACGGACCCCAAA-3′ and 5′-TGATGTGCTGCTGTGAGATT-3′; Ccl2, 5′-GGCTCAGCCAGATGCAGTTAAC-3′ and 5′-GCCTACTCATTGGGATCATCTTG-3′; Cxcl1, 5′-GCTGGGATTCACCTCAAGAA-3′ and 5′-TCTCCGTTACTTGGGGACAC-3′; Il6, 5′-ACAACCACGGCCTTCCCTACTT-3′ and 5′-CACGATTTCCCAGAGAACATGTG-3′; Lif, 5′-ATTGTGCCCTTACTGCTGCTG-3′ and 5′-GCCAGTTGATTCTTGATCTGGT-3′; Mmp9, 5′-CCTGGAACTCACACGACATCTTC-3′ and 5′-TGGAAACTCACACGCCAGAA-3′; Bax, 5′-TTGCTGATGGCAACTTCAAC-3′ and 5′-GATCAGCTCGGGCACTTTAG-3′; Bcl2, 5′-CAGAAGATCATGCCGTCCTT-3′ and 5′-CTTTCTGCTTTTTATTTCATGAGG-3′; Pink1, 5′-GCTTGCCAATCCCTTCTATG-3′ and 5′-CTCTCGCTGGAGCAGTGAC-3′; and 18 S rRNA, 5′-GTAACCCGTTGAACCATT-3′ and 5′-CCATCCAATCGGTAGTAGCG-3′. Expression levels were quantified using a standard curve and were normalized to the content of 18 S rRNA. Each normalized value was expressed as a ratio to the value of WT mice exposed to normoxia.
Western Blot Analysis
Lysates from perfused lung tissue and cell culture were prepared using radioimmune precipitation assay buffer (20 mm Tris, 2.5 mm EDTA, 1% Triton X, 10% glycerol, 1% deoxycholic acid, 0.1% SDS, 50 mm NaF, and 10 mm Na4P2O7·10H2O) and subjected to SDS-PAGE. The proteins were electrophoretically transferred to PVDF membranes. The membranes were blocked with 2% casein or 3% BSA for 1 h at room temperature and then incubated overnight at 4 °C with primary antibodies, followed by incubation for 1 h with secondary antibodies conjugated to HRP. Immunoreactive bands were visualized using a Western BLoT chemiluminescent HRP substrate system (Takara Bio Inc.). The expression level of β-actin served as an internal control for protein loading. Primary antibodies against Stat3, phospho-Stat3 (Tyr-705), Bax, and Bcl-2 (Cell Signaling Technology, Inc., Boston, MA); NLRP3 (R&D Systems); and anti-β-actin (Sigma-Aldrich) were used. HRP-conjugated goat anti-mouse IgG (Invitrogen) and HRP-conjugated goat anti-rabbit IgG (ZyMAX grade, Zymed Laboratories Inc., South San Francisco, CA) were used as secondary antibodies. The results represent at least three independent experiments. Quantitative analysis of bands was performed using ImageJ 1.47v (National Institutes of Health, Bethesda, MD).
MMP-9 Assay
MMP-9 (matrix metalloproteinase 9) levels were measured using a mouse MMP-9 activity assay kit (QuickZyme Biosciences, Leiden, The Netherlands) according to the manufacturer's instructions.
Histology and Immunohistochemistry
Lungs were fixed by intratracheal injection of 1 ml of 10% formalin and embedded in paraffin. Lung tissue sections (5-μm thick) were stained with H&E. Neutrophils were stained using a naphthol AS-D chloroacetate esterase staining kit (Muto Pure Chemicals Co., Ltd., Tokyo, Japan), which identifies specific leukocyte esterases. The cells in 10 random fields were counted at a magnification of ×60.
Immunohistochemical analyses were performed to detect the oxidative stress marker 4-hydroxy-2-nonenal (4-HNE) and the white blood cell marker CD45. In brief, deparaffinized sections were boiled in target retrieval solution (Dako, Carpinteria, CA), blocked with normal goat serum, and incubated overnight with an antibody against CD45 (BD Biosciences). This was followed by incubation with Histofine Simple Stain Rat MAX PO (Nichirei Biosciences Inc., Tokyo, Japan). The immune complexes were detected using a 3,3′-diaminobenzidine substrate kit (Vector Labs, Burlingame, CA). For 4-HNE immunostaining, the sections were blocked with mouse IgG-blocking reagent (M.O.M. immunodetection kit, Vector Labs) and incubated overnight with an antibody against 4-HNE (clone HNEJ-2, Japan Institute for the Control of Aging, Nikken SEIL Co., Shizuoka, Japan). This was followed by incubation with biotin-conjugated secondary antibodies. The sections were treated with avidin-peroxidase (VECTASTAIN ABC kit, Vector Labs). The reaction was developed using the 3,3′-diaminobenzidine substrate kit. The sections were counterstained with hematoxylin. No signals were detected when an irrelevant IgG (Vector Labs) was used instead of the primary antibody as a negative control. The images of the stained sections were digitized and analyzed using an FSX100 microscope (Olympus, Tokyo, Japan). Five-hundred cells were randomly selected to calculate the percentage of positive cells.
Detection of Apoptosis
Apoptotic cells were identified with an in situ apoptosis detection kit (Takara Bio Inc.) using the TUNEL method and immunohistochemical staining of cleaved caspase-3. In brief, deparaffinized sections were boiled in 1 mm EDTA for 10 min in a pressure cooker. For TUNEL staining, boiled sections were blocked with 3% H2O2 and incubated at 37 °C for 90 min with terminal deoxynucleotidyltransferase enzyme. For immunohistochemical staining of cleaved caspase-3, boiled sections were blocked with 1% H2O2 and normal goat serum and incubated overnight with an antibody against cleaved caspase-3 (Cell Signaling Technology, Inc.). This was followed by incubation with Histofine Simple Stain Rabbit MAX PO (Nichirei Biosciences Inc.). The immune complexes were detected using the 3,3′-diaminobenzidine substrate kit, and the sections were counterstained with hematoxylin. The images of the stained sections were digitized and analyzed using a microscope. Five-hundred alveolar epithelial cells were randomly selected to calculate the percentage of positive cells.
Analysis of Wet/Dry Lung Weight Ratios and Protein Concentration in BALF
The extracted left lung was weighed to obtain the wet weight and kept in an oven at 75 °C for 72 h to obtain the dry weight. The protein concentration in BALF was measured using BCA protein assay reagent (Thermo Fisher Scientific Inc., Waltham, MA).
IL-1β Assay
IL-1β levels in BALF were assessed using a mouse ELISA kit (R&D Systems) according to the manufacturer's instructions.
Flow Cytometry
Cells collected from BALF were analyzed by flow cytometry. The cells were double-labeled with the following antibodies: allophycocyanin-conjugated anti-CD45 (eBioscience), FITC-conjugated anti-CD45R (eBioscience), phycoerythrin-conjugated anti-Ly6G (BD Biosciences), FITC-conjugated anti-CD11c (BD Biosciences), and phycoerythrin-conjugated anti-CD11b (eBioscience). The cells were analyzed using a FACSCalibur system with CellQuest version 3.3 (BD Biosciences). Isotype control antibodies were used as negative controls to exclude nonspecific staining.
Cell Culture
MLE 12 (murine lung epithelial cells) and MH-S (murine alveolar macrophages) cell lines were purchased from American Type Culture Collection (Manassas, VA). MLE 12 cells were grown in DMEM and 1000 mg/liter glucose (Wako Pure Chemical Industries, Ltd., Osaka, Japan) containing 10% FBS. MH-S cells were grown in RPMI 1640 medium (Sigma-Aldrich) containing 10% FBS and 0.05 mm 2-mercaptoethanol (Sigma-Aldrich). Immortalized murine bone marrow-derived macrophages (BMDMs) from WT and NLRP3 mice (provided by E. L.) (17) were cultured in DMEM and 4500 mg/liter glucose (Sigma-Aldrich) containing 10% FBS.
Primary alveolar epithelial cells and macrophages were isolated from murine lungs as described previously (18, 19). In brief, alveolar macrophages in the alveolar cavities were removed by lavaging whole lungs with PBS administered through intratracheal cannulation. After perfusion with PBS, the lungs were extracted and digested with collagenase type I (Wako Pure Chemical Industries, Ltd.) and Dispase (Dispase II, Godo Shusei Co., Ltd., Tokyo, Japan) with DNase (Worthington). To isolate alveolar epithelial cells, the cells were placed on culture dishes for 30 min to allow the alveolar macrophages to adhere securely to the bottom of the culture dishes. Non-adherent cells (alveolar epithelial cells) were removed and added to new culture dishes. The expression of type I collagen and surfactant protein C was determined in the alveolar epithelial cells. Adherent cells (alveolar macrophages) were mixed with alveolar macrophages collected from alveolar cavities. Peritoneal exudate neutrophils were isolated by intraperitoneal injection of 9% casein as described previously (20) and cultured in RPMI 1640 medium containing 10% FBS. Hyperoxia was maintained in an incubator with an atmosphere of 90% O2 and 5% CO2.
Neutrophil Migration Assay
Neutrophil migration was analyzed using a Transwell migration assay with 24-well tissue culture plates (3-μm pore polycarbonate membranes, BD Biosciences) (20). After the supernatant from MLE 12 cells or recombinant murine CXCL1 (also known as KC; PeproTech, Inc., Rocky Hill, NJ) was added to the lower chamber, neutrophils (2 × 105) were placed in the upper chamber and incubated for 1 h at 37 °C. Neutrophils in the lower chamber were collected and analyzed by flow cytometry.
Co-culture Experiments
Separated co-culture of MLE 12 cells with MH-S cells or neutrophils was performed using 6-well tissue culture plates with a Transwell culture system (0.4-μm pore polycarbonate membranes, BD Biosciences). After MLE 12 cells (1 × 106 cells) were cultured in the lower chamber for 24 h, MH-S cells (5 × 105 to 1 × 106 cells) or neutrophils (5 × 105 to 2 × 106 cells) were added to the upper chamber and incubated for 8 h, and cell lysates from the MLE 12 cells in the lower chamber were prepared. Non-separated co-culture was performed using 6-well tissue culture plates.
Adoptive Transfer of Neutrophils
Neutrophils were prepared from WT or NLRP3−/− mice as described above. NLRP3−/− mice were reconstituted with 1 × 106 WT or NLRP3−/− neutrophils by intravenous administration 5 h before exposure to hyperoxia.
Statistical Analysis
Data were analyzed using SPSS version 21 (IBM Japan Ltd., Tokyo, Japan) and expressed as the means ± S.E. An unpaired t test was used to compare two groups. For comparisons between multiple groups, the significance of differences between group means was determined by one-way analysis of variance combined with Tukey's test or the Games-Howell test. We defined p < 0.05 as statistically significant.
RESULTS
NLRP3−/− Mice Are Susceptible to Oxygen-induced Lethality
We first examined the survival times of WT, NLRP3−/−, and IL-1β−/− mice exposed to hyperoxia (90% O2), which is an established model of acute lung injury, and found that NLRP3−/− mice survived for significantly shorter times compared with WT mice (median survival time of 96 ± 6.53 h versus 120 ± 2.28 h, p < 0.01) (Fig. 1A). There was no significant difference in survival time between WT and IL-1β−/− mice (median survival time of 120 ± 0.00 h versus 108 ± 10.25 h) (Fig. 1B). In addition, there was no significant difference in IL-1β production in BALF between WT and NLRP3−/− mice under normoxic or hyperoxic conditions (Fig. 1C). Real-time RT-PCR analysis showed that although Il1b mRNA expression was significantly elevated in the lungs of hyperoxic WT mice compared with normoxic mice, there was no significant difference in elevated Il1b expression between hyperoxic WT and NLRP3−/− mice (Fig. 1D). These results indicate that NLRP3 contributes to the survival of mice exposed to lethal hyperoxia independent of IL-1β.
FIGURE 1.

NLRP3−/− mice are susceptible to oxygen-induced lethality. A, survival of WT and NLRP3−/− mice exposed to hyperoxia was analyzed using the Kaplan-Meier method (n = 18–19 for each). B, survival of WT and IL-1β−/− mice exposed to hyperoxia was analyzed using the Kaplan-Meier method (n = 9–12 for each). C, IL-1β levels were assessed in the BALF of WT and NLRP3−/− mice exposed to normoxia or hyperoxia for 72 h (n = 3–5 for each in normoxia, and n = 12–14 for each in hyperoxia). D, Il1b mRNA levels were assessed in the lung tissue of WT and NLRP3−/− mice exposed to normoxia or hyperoxia for 72 h (n = 6 for each in normoxia, and n = 9–12 for each in hyperoxia). Data are expressed as the means ± S.E. **, p < 0.01.
NLRP3−/− Mice Exhibit Similar Acute Lung Injury and Reactive Oxygen Species Generation
H&E staining showed thickening of the alveolar septae, vascular congestion, and alveolar edema present to similar extents in the lungs of hyperoxic WT and NLRP3−/− mice (Fig. 2A). Because hyperoxia generates excess amounts of reactive oxygen species in the lungs (2), we performed immunohistochemical analysis of 4-HNE, a marker of lipid peroxidation. Compared with normoxia, hyperoxia markedly increased the number of 4-HNE-positive cells in the lungs (Fig. 2, B and C); however, there was no significant difference between WT and NLRP3−/− mice with respect to the number of 4-HNE-positive cells.
FIGURE 2.

NLRP3−/− mice exhibit similar acute lung injury and generation of reactive oxygen species. Lung samples were obtained from WT and NLRP3−/− mice exposed to normoxia or hyperoxia for 72 h. A, the lung sections were stained with H&E. Representative images of H&E staining are shown (n = 3–4 for each). B, reactive oxygen species generation was assessed by immunohistochemistry with an anti-4-HNE antibody. C, quantitative analysis (n = 3–4 for each). Data are expressed as the means ± S.E. **, p < 0.01.
Lungs of NLRP3−/− Mice Exhibit Less Infiltration by Inflammatory Cells
To assess the infiltration of inflammatory cells into the lungs, we performed immunohistochemical analysis of CD45 and found that infiltration of inflammatory cells was significantly increased in the lungs of hyperoxic WT mice and that this increased infiltration was significantly suppressed in NLRP3−/− mice (Fig. 3, A and B). We determined the number and type of inflammatory cells in BALF. Consistent with the results of immunohistochemical analysis, the cell number was significantly increased by hyperoxia in the BALF of WT mice, and this increase was markedly suppressed in NLRP3−/− mice (Fig. 3C). Flow cytometric analysis revealed that the number of alveolar macrophages (CD11c+) and neutrophils (Ly6G+/CD45R−) in BALF was significantly increased in hyperoxic WT mice and significantly decreased in hyperoxic NLRP3−/− mice (Fig. 3D). Prevention of neutrophil infiltration of the lungs of hyperoxic NLRP3−/− mice was confirmed by naphthol AS-D chloroacetate esterase staining (data not shown).
FIGURE 3.

NLRP3−/− mice exhibit less inflammatory cell infiltration. Lung samples were obtained from WT and NLRP3−/− mice exposed to normoxia or hyperoxia for 72 h. A, lung sections were immunohistochemically stained with antibody against CD45. B, quantitative analysis (n = 3–4). C, the total cell count in BALF was determined. D, the number of alveolar macrophages (CD11c+) and neutrophils (Ly6G+/CD45R−) in BALF was analyzed by flow cytometry (n = 3–4 for each in normoxia, and n = 5–6 for each in hyperoxia). Data are expressed as the means ± S.E. *, p < 0.05; **, p < 0.01.
NLRP3−/− Mice Express Decreased Levels of Ccl2, Cxcl1, Il6, and Lif
To investigate whether inflammatory cytokines are involved in hyperoxia-induced lethality, we determined the levels of Ccl2, Cxcl1, Il6, and Lif in the lungs of normoxic and hyperoxic mice. Real-time RT-PCR analysis revealed that the expression of Ccl2, Cxcl1, Il6, and Lif was significantly elevated in hyperoxic WT mice compared with normoxic WT mice and that this increase was significantly suppressed in hyperoxic NLRP3−/− mice (Fig. 4).
FIGURE 4.
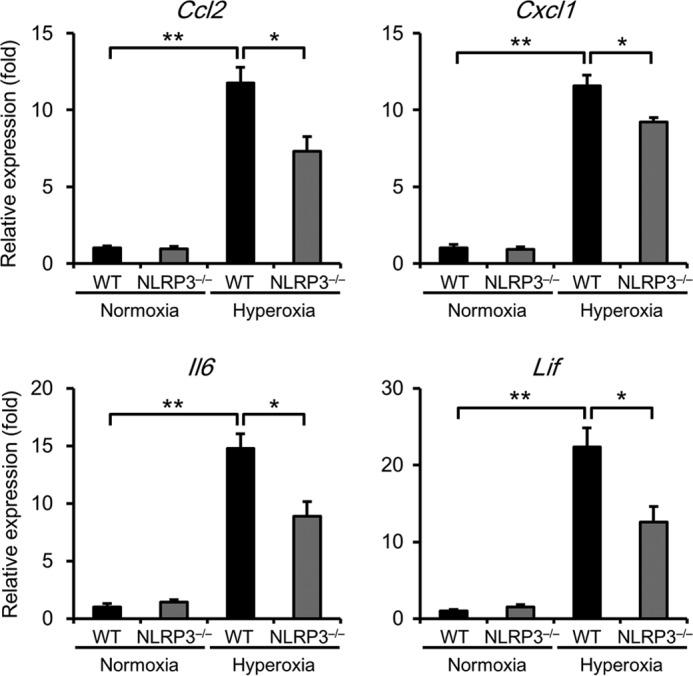
NLRP3−/− mice exhibit lower expression levels of Ccl2, Cxcl1, Il6, and Lif mRNAs. Lung samples were obtained from WT and NLRP3−/− mice exposed to normoxia or hyperoxia for 72 h. The lung mRNA levels of Ccl2, Cxcl1, Il6, and Lif were assessed by real-time RT-PCR analysis. Data are expressed as the means ± S.E. (normoxia, n = 6; hyperoxia, n = 6–12 for each). *, p < 0.05; **, p < 0.01.
NLRP3−/− Mice Exhibit Increased MMP-9 and Decreased Bcl-2 Expression
Because inflammatory responses were suppressed in the lungs of NLRP3−/− mice, we investigated whether another mechanism may explain the findings regarding hyperoxia-induced lethality. Previous studies indicate that MMP-9 and apoptosis may play a crucial role in HALI and lethality (21). Real-time RT-PCR analysis revealed that the expression of Mmp9 mRNA was markedly increased in the lungs of hyperoxic NLRP3−/− mice compared with hyperoxic WT mice (Fig. 5A). There was a consistent increase in MMP-9 protein levels in the lungs of NLRP3−/− mice compared with WT mice (Fig. 5B). Furthermore, the mRNA expression level of the anti-apoptotic molecule Bcl2 in the lungs of hyperoxic NLRP3−/− mice was lower than that in the lungs of hyperoxic WT mice (Fig. 5C). The ratio of Bax to Bcl2, a key factor in the regulation of apoptosis, was significantly increased in the lungs of hyperoxic NLRP3−/− mice. Moreover, the results of Western bot analysis showed that the protein ratio of Bax to Bcl-2 was significantly increased in the lungs of NLRP3−/− mice (Fig. 5, D and E).
FIGURE 5.

NLRP3−/− mice exhibit increased MMP-9 activity and decreased Bcl-2 expression. Lung samples were isolated from WT and NLRP3−/− mice exposed to normoxia or hyperoxia for 72 h. A, Mmp9 mRNA levels in lung tissues were assessed by real-time RT-PCR analysis (n = 4–6 for each in normoxia, and n = 7–8 for each in 72-h hyperoxia). B, MMP-9 expression was assessed in the lungs (n = 3–4 for each, 72-h hyperoxia). C, Bax and Bcl2 mRNA expression in the lungs was assessed by real-time RT-PCR analysis (n = 4–6 for each in normoxia, and n = 6–13 for each in 72-h hyperoxia). D and E, Western blot analysis of Bax and Bcl-2 in the lungs (n = 5–6 for each in 72-h hyperoxia). Data are expressed as the means ± S.E. *, p < 0.05; **, p < 0.01.
NLRP3−/− Mice Exhibit Increased Alveolar Epithelial Apoptotic Cells
Because the expression of Bcl-2 was decreased and the ratio of Bax to Bcl-2 was increased in the lungs of hyperoxic NLRP3−/− mice, we ascertained apoptosis of alveolar epithelial cells by TUNEL staining and immunohistochemical staining of cleaved caspase-3. Both TUNEL-positive cells and cleaved caspase-3-positive cells were significantly increased in alveolar epithelial cells of hyperoxic NLRP3−/− mice (Fig. 6). These results suggest that progression of apoptosis contributes to hyperoxia-induced lethality in NLRP3−/− mice.
FIGURE 6.

NLRP3−/− mice exhibit increased apoptotic alveolar epithelial cells. Lung samples were obtained from WT and NLRP3−/− mice exposed to normoxia or hyperoxia for 72 h. A, lung sections were analyzed by TUNEL staining. Arrowheads indicate TUNEL-positive cells. B, quantitative analysis (n = 3–4). C, lung sections were immunohistochemically stained with antibody against cleaved caspase-3. Arrowheads indicate cleaved caspase-3-positive cells. D, quantitative analysis (n = 3–4). Data are expressed as the means ± S.E. **, p < 0.01.
NLRP3−/− Mice Express Lower Levels of Stat3 and Phosphorylated Stat3
We next focused on Stat3 because it regulates the transcription of MMP-9 and Bcl-2 (21–25) and is in turn regulated by IL-6 and LIF (26, 27). Furthermore, Stat3 expression in alveolar epithelial cells protects against HALI and lethality in mice (21, 28). Therefore, we performed Western blot analysis to determine the levels of expression and activation (phosphorylated form) of Stat3 in the lungs. Under normoxia, the expression levels and phosphorylation of Stat3 were similar between WT and NLRP3−/− mice (Fig. 7, A and B). However, Stat3 expression and activation increased in hyperoxic WT mice, and this increase was significantly diminished in NLRP3−/− mice. These results suggest that Stat3 contributes to MMP-9 and Bcl-2 expression in hyperoxia-induced lethality in NLRP3−/− mice.
FIGURE 7.

NLRP3−/− mice exhibit lower levels of Stat3 expression and activation. Lung samples were obtained from WT and NLRP3−/− mice exposed to normoxia or hyperoxia for 72 h. A, Western blot analysis of Stat3 expression and activation in the lungs using antibodies against Stat3 and phospho-Stat3 (p-Stat3). B, quantitative analysis (n = 5–6 for each). Data are expressed as the means ± S.E. *, p < 0.05; **, p < 0.01.
Factors Produced by Alveolar Macrophages and Neutrophils Influence Stat3 Activation
To explore the mechanisms underlying reduced Stat3 levels in the lungs of NLRP3−/− mice, we performed in vitro experiments. Western blot analysis detected significant expression of NLRP3 in primary alveolar macrophages and neutrophils, but not in alveolar epithelial cells (Fig. 8, A and C). As expected, the expression of NLRP3 was undetectable in cells derived from NLRP3−/− mice. NLRP3 was expressed in MH-S cells, but not in MLE 12 cells (Fig. 8B). MLE 12 cells and primary neutrophils expressed Stat3 at significantly higher levels compared with MH-S cells (Fig. 8, B and C). Hyperoxia failed to increase the expression and activation of Stat3 in these cells, and there was no difference in Stat3 levels between WT and NLRP3−/− neutrophils.
FIGURE 8.

Macrophages and neutrophils increase Stat3 activation in alveolar epithelial cells. A–C, primary alveolar epithelial cells, macrophages, and neutrophils were isolated from WT and NLRP3−/− mice. Alveolar epithelial cells, macrophages, neutrophils, MLE 12 murine lung epithelial cells, and MH-S murine alveolar macrophage cells were exposed to normoxia or hyperoxia for the indicated times. A, Western blot analysis of NLRP3 expression in alveolar epithelial cells and macrophages (n = 3). B and C, Western blot analysis of NLRP3 expression and Stat3 expression and activation using antibodies against Stat3 and phospho-Stat3 (p-Stat3). D, MLE 12 cells were separately co-cultured with MH-S cells (5–10 × 105) or neutrophils (5–20 × 105) for 8 h. The supernatants from MH-S cells and neutrophils (diluted ×4 and ×2) were added to MLE 12 cells and incubated for 8 h. Shown are the results from Western blot analysis of Stat3 expression and activation in MLE 12 cells using antibodies against Stat3 and phospho-Stat3.
Because we hypothesized that infiltration of inflammatory cells may contribute to Stat3 activation in the lungs, we determined Stat3 levels when MLE 12 cells were co-cultured directly and indirectly with MH-S cells and neutrophils. Direct co-culture of MLE 12 with MH-S cells or neutrophils markedly increased Stat3 phosphorylation (data not shown). Alveolar epithelial cells cause HALI, and Stat3 is involved in intracellular signaling (21, 29). To determine the contribution of Stat3 in these cells, we used a Transwell co-culture system and separately co-cultured inflammatory cells with MLE 12 cells. Stat3 activation was significantly higher in MLE 12 cells when co-cultured separately from MH-S cells or neutrophils (Fig. 8D). Furthermore, the supernatant from MH-S cells or neutrophils stimulated Stat3 activation in MLE 12 cells (Fig. 8D). These results suggest that inflammatory cells, such as alveolar macrophages and neutrophils, promote Stat3 activation in alveolar epithelial cells through the production of humoral factors. In contrast, co-culture with MLE 12 cells did not induce a significant increase in Stat3 levels, indicating that the difference between WT and NLRP3−/− mice with respect to Stat3 levels in the lung tissues may involve the infiltration of inflammatory cells.
Role of Neutrophils and Macrophages in Response to Hyperoxia
Because neutrophil infiltration was markedly decreased in the lungs and BALF of NLRP3−/− mice, we prepared primary neutrophils from WT and NLRP3−/− mice and used a Transwell migration assay to determine their migration in the presence of the supernatant collected from MLE 12 cell cultures. The MLE 12 supernatant increased the number of migrating WT neutrophils, but had no detectable effect on the increased migration of NLRP3−/− neutrophils (Fig. 9A). The migration of NLRP3−/− neutrophils was lower under basal conditions. CXCL1 was used as a positive control to induce neutrophil migration.
FIGURE 9.
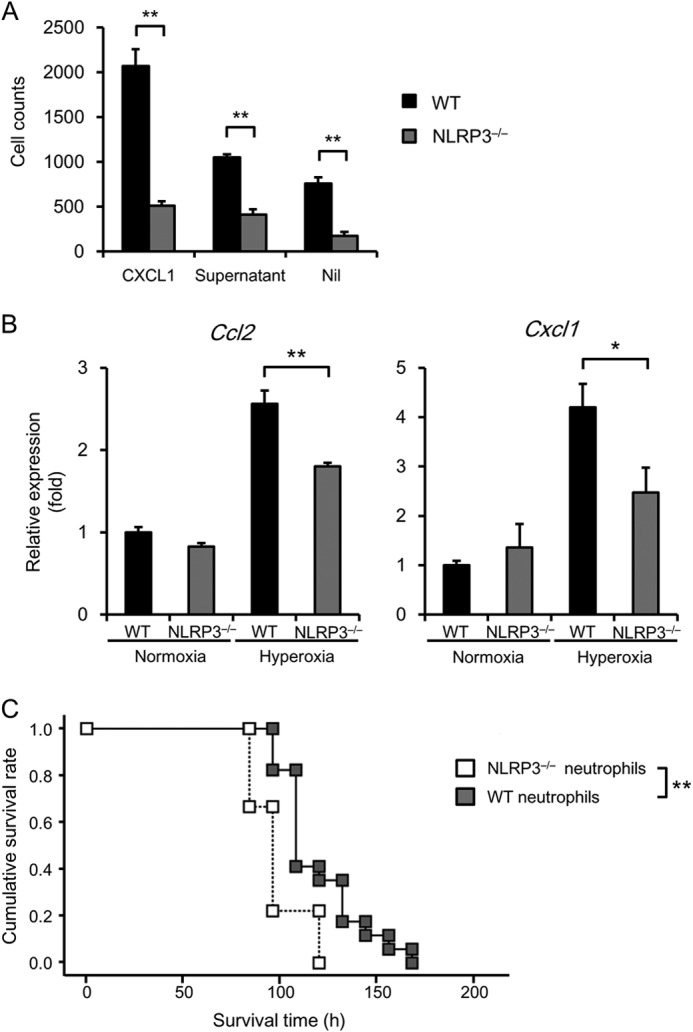
Role of neutrophils and macrophages in the response of NLRP3−/− mice to hyperoxia. A, the migration of WT and NLRP3−/− neutrophils in response to CXCL1 and the supernatants from cultures of MLE 12 cells was assessed using a Transwell migration assay (n = 3 for each). Nil, no treatment. B, total RNA was extracted from immortalized murine WT and NLRP3−/− BMDMs exposed to normoxia or hyperoxia for 48 h. Ccl2 and Cxcl1 mRNA expression was assessed by real-time RT-PCR analysis (n = 3–4 for each). C, adoptive transfer of WT or NLRP3−/− neutrophils into NLRP3−/− mice was performed 5 h before exposure to hyperoxia. The survival of NLRP3−/− mice administered WT or NLRP3−/− neutrophils was analyzed using the Kaplan-Meier method (n = 9–15 for each). Data are expressed as the means ± S.E. *, p < 0.05; **, p < 0.01.
Using immortalized murine WT and NLRP3−/− BMDMs (17), we further examined the effects of NLRP3 deficiency on the expression of chemokines CCL2 and CXCL1. Hyperoxia markedly increased Cxcl1 and Ccl2 expression by WT BMDMs, and this increased expression was significantly lower in NLRP3−/− BMDMs (Fig. 9B). Finally, to confirm the role of neutrophils in hyperoxia-induced lethality in NLRP3−/− mice, we performed adoptive transfer of neutrophils into NLRP3−/− mice. Adoptive transfer of WT neutrophils caused a significant increase in the survival of NLRP3−/− mice (median survival time of 96 ± 3.47 h versus 108 ± 3.48 h, p < 0.01) (Fig. 9C). These data indicate that NLRP3 expression by neutrophils contributes to hyperoxia-induced lethality.
DISCUSSION
The major findings of this study are as follows. 1) Under hyperoxia, NLRP3−/− mice died sooner compared with WT and IL-1β−/− mice, although there was no difference in IL-1β production in the lungs of these mice. 2) NLRP3−/− mice exhibited reduced inflammatory responses, such as inflammatory cell infiltration and cytokine expression, in hyperoxic lungs. 3) In the hyperoxic lungs of NLRP3−/− mice, the expression of MMP-9 and Bcl-2 was increased and decreased, respectively. 4) NLRP3−/− mice exhibited an increased number of apoptotic alveolar epithelial cells. 5) NLRP3−/− mice exhibited diminished expression and activation of Stat3. 6) In vitro experiments revealed that alveolar macrophages and neutrophils promoted Stat3 activation by alveolar epithelial cells. 7) NLRP3 deficiency impaired the migration of neutrophils and chemokine expression by macrophages. 8) Adoptive transfer of WT neutrophils into NLRP3−/− mice rescued hyperoxia-induced lethality. These findings indicate that NLRP3 regulates Stat3 signaling in alveolar epithelial cells by affecting macrophage and neutrophil function independent of IL-1β−/− and contributes to the pathophysiology of HALI. Our study shows a novel role of NLRP3 in HALI and provides new insights into the mechanisms underlying HALI.
HALI is characterized by inflammatory cell infiltration, increased pulmonary permeability, and endothelial and epithelial cell injury (1, 3). Although excessive inflammatory responses exacerbate lung injury, attenuation of inflammatory responses does not always improve survival from HALI. Of note, it is widely accepted that infiltration of inflammatory cells is necessary to repair lung injury and induce remodeling (30), suggesting that inflammation is not only deleterious but also beneficial in the pathogenesis of HALI. For example, Bhandari et al. (4) demonstrated that IL-13 deficiency exacerbates survival under hyperoxia through DNA injury and apoptotic cell death regardless of decreased inflammatory responses and concluded that HALI cannot be attributed solely to local tissue inflammation. Similarly, in a study by Jain et al. (31), transgenic mice overexpressing surfactant protein D experienced increased survival in HALI models, although inflammatory cell infiltration was not significantly different from that in WT mice. In our study, similar to the findings of Fukumoto et al. (14), there was less infiltration of inflammatory cells in the lungs of hyperoxic NLRP3−/− mice. In addition, we observed that there was no difference in wet/dry lung weight ratios and protein concentration in BALF between WT and NLRP3−/− mice after hyperoxia (data not shown). Contrary to our expectations, NLRP3−/− mice were significantly more sensitive to the lethal effects of hyperoxia. Furthermore, hypoxia did not affect the survival of IL-1β−/− mice, suggesting that NLRP3 may contribute to survival independent of IL-1β production. Recent investigations suggest that NLRP3 possesses inflammasome-independent functions under certain conditions (20, 32). For example, Shigeoka et al. (32) reported that ischemia-reperfusion injury is reduced in the kidneys of NLRP3−/− mice, but not of ASC−/− and caspase-1−/− mice. We recently reported similar findings for hepatic ischemia-reperfusion injury (20). Taken together, although the precise contribution of NLRP3 inflammasome activation to the development of HALI remains to be investigated in caspase-1-deficient mice, these findings suggest that NLRP3 plays a role in HALI independent of NLRP3 inflammasomes.
In this study, NLRP3 expression was detected in alveolar macrophages and neutrophils, but not in alveolar epithelial cells, indicating a significant role for these cells in hyperoxia-induced lethality in NLRP3−/− mice. For example, infiltration of alveolar macrophages and neutrophils was reduced in the lungs of NLRP3−/− mice. Supporting this, we previously showed that neutrophils derived from NLRP3−/− mice exhibit impaired chemokine-mediated signaling and functions, including activation of heterotrimeric G-proteins, [Ca2+]i elevation, Rac activation, actin assembly formation, and cell migration (20). Moreover, in the present study, in vitro experiments showed that neutrophil migration was induced in response to the supernatant collected from cultures of alveolar epithelial cells, and this increased migration was diminished in NLRP3−/− neutrophils, suggesting impaired migration even in the lungs. Furthermore, the expression of CCL2 and CXCL1 was decreased in the lungs of NLRP3−/− mice, as well as in NLRP3−/− macrophages. Considering these data together, we assume that NLRP3 regulates inflammatory cell infiltration by affecting migration activity and chemokine expression in the lungs.
Stat proteins transmit signals rapidly and directly from the cell membrane to the nucleus and mediate excessive cytokine and immune responses. Specifically, Stat3 plays a more fundamental role in cell growth and survival compared with other Stat family members (23). In this study, we assumed that Stat3 may be responsible for hyperoxia-induced lethality in NLRP3−/− mice. This assumption was supported by findings of the present and previous investigations. First, Hokuto et al. (28) showed that selective deletion of Stat3 from alveolar epithelial cells exacerbates the lethality of mice under hyperoxia. Lian et al. (21) reported that mice overexpressing activated Stat3 in alveolar epithelial cells experience improved survival under hyperoxia and that this effect is mediated by decreased MMP-9 expression. Second, Bcl-2 expression is regulated by Stat3 (22). Third, Stat3 activation is induced by IL-6 family cytokines, such as IL-6 and LIF (26, 27). In this study, we demonstrated that Stat3 expression and activation were significantly decreased in the lungs of hyperoxic NLRP3−/− mice. In addition, we observed that the number of apoptotic alveolar epithelial cells was clearly increased in hyperoxic NLRP3−/− mice. Supporting this finding, Zuurbier et al. (33) recently reported a protective effect of ischemic preconditioning in the hearts of WT and ASC−/− mice, but not in those of NLRP3−/− mice. They further showed that the impaired protective effect may be due to decreased expression of Stat3 in NLRP3−/− hearts. Moreover, our in vitro experiments revealed that alveolar macrophages and neutrophils promoted Stat3 activation via humoral factors. Because previous studies suggest the importance of Stat3 in alveolar epithelial cells in HALI models (21, 28), we postulate that Stat3 and its downstream apoptotic pathway mainly contribute to hyperoxia-induced lethality in NLRP3−/− mice.
During the preparation of this manuscript, Zhang et al. (34) reported that NLRP3−/− mice are resistant to hyperoxia. They further showed that NLRP3−/− mice exhibit higher basal and hyperoxia-induced expression of PINK1 (PTEN-induced putative kinase 1) in the lungs and concluded that PINK1 underlies the mechanism that protects against hyperoxia-induced lethality caused by NLRP3 deficiency. The results of their survival analysis are inconsistent with our findings. Although the reason for this discrepancy is unknown, we did not detect a significant increase in PINK1 expression in the lungs of WT and NLRP3−/− mice under basal or hyperoxic conditions (data not shown). In addition, the differences between our study and that of Zhang et al. are the hyperoxic protocols and mice. We exposed mice to 90% O2 to induce hyperoxia, whereas Zhang et al. exposed mice to 100% O2. The survival time during hyperoxia reported here is longer than that reported by Zhang et al. Furthermore, Lingappan et al. (35) reported that male mice are more susceptible than female mice to HALI, suggesting gender-specific differences in responses of mice with HALI. Moreover, the responses to hyperoxia and subsequent HALI are influenced by the ambient housing temperature (2). In this study, we used female mice and maintained the chamber at 24–25 °C. In contrast, Zhang et al. (34) did not describe these conditions. Therefore, the experimental protocols and conditions, including gender and temperature, may influence hyperoxia-induced lethality, and further investigations are required to determine the precise role of NLRP3 in the pathophysiology of HALI.
We propose the following mechanisms to account for our results (Fig. 10). Prolonged hyperoxia induces infiltration of macrophages and neutrophils into the lungs via chemokines, such as CCL2 and CXCL1. These infiltrated inflammatory cells augment Stat3 activation in alveolar epithelial cells through humoral factors. Activated Stat3 increases and decreases transcription of Bcl-2 and MMP-9, respectively; this in turn attenuates lung injury. In NLRP3−/− mice, infiltration of macrophages and neutrophils is decreased because of the reduced expression of CCL2 and CXCL1 and the impaired migration of neutrophils. Therefore, Stat3 activation is reduced and causes inappropriate regulation of Bcl-2 and MMP-9 transcription, which impairs the protective mechanisms of hyperoxia-induced lung injury and increases lethality. To the best of our knowledge, our study is the first to demonstrate the novel role of NLRP3 in Stat3-mediated protective effects against HALI. In addition, our findings highlight that hyperoxia-induced lethality may not be induced by excessive inflammatory responses.
FIGURE 10.

Proposed mechanism of hyperoxia-induced lethality in NLRP3−/− mice. In WT mice, hyperoxia induces infiltration of macrophages and neutrophils into the lungs via the chemokines CCL2 and CXCL1. These infiltrated inflammatory cells increase Stat3 activation through humoral factors. Activated Stat3 increases and decreases transcription of Bcl-2 and MMP-9, respectively; this may in turn attenuate lung injury. In NLRP3−/− mice, infiltration of macrophages and neutrophils decreases because of reduced expression of CCL2 and CXCL1 and impaired migration of neutrophils. Therefore, Stat3 activation is reduced and inappropriately regulates Bcl-2 and MMP-9, thereby impairing the protective mechanism of hyperoxia-induced lung injury and increasing lethality.
Acknowledgments
We thank Masako Sakurai, Yumi Ohde, and Namiko Nakada for excellent technical assistance; Dr. Vishva M. Dixit for providing NLRP3−/− mice; and Drs. Takeshi Tomita (Tokyo Women's Medical University), Manabu Ueno (Gunma University), and Hajime Kono (Teikyo University) for giving invaluable suggestions.
This work was supported by grants from the Japan Society for the Promotion of Science (JSPS) through the Funding Program for Next Generation World-Leading Researchers (NEXT Program), initiated by the Council for Science and Technology Policy (CSTP) (to M. T.); the Ministry of Education, Culture, Sports, Science, and Technology (MEXT)-supported program for the Strategic Foundation at Private Universities (to M. T.); and a grant-in-aid for research activity start-up from the Jichi Medical University (to Y. M.).
- HALI
- hyperoxic acute lung injury
- BALF
- bronchoalveolar lavage fluid
- 4-HNE
- 4-hydroxy-2-nonenal
- BMDMs
- murine bone marrow-derived macrophages.
REFERENCES
- 1. Bhandari V. (2008) Molecular mechanisms of hyperoxia-induced acute lung injury. Front. Biosci. 13, 6653–6661 [DOI] [PubMed] [Google Scholar]
- 2. Kallet R. H., Matthay M. A. (2013) Hyperoxic acute lung injury. Respir Care 58, 123–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bhandari V., Elias J. A. (2006) Cytokines in tolerance to hyperoxia-induced injury in the developing and adult lung. Free Radic Biol. Med. 41, 4–18 [DOI] [PubMed] [Google Scholar]
- 4. Bhandari V., Choo-Wing R., Homer R. J., Elias J. A. (2007) Increased hyperoxia-induced mortality and acute lung injury in IL-13 null mice. J. Immunol. 178, 4993–5000 [DOI] [PubMed] [Google Scholar]
- 5. Latz E., Xiao T. S., Stutz A. (2013) Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 13, 397–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Davis B. K., Wen H., Ting J. P. (2011) The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu. Rev. Immunol. 29, 707–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Usui F., Shirasuna K., Kimura H., Tatsumi K., Kawashima A., Karasawa T., Hida S., Sagara J., Taniguchi S., Takahashi M. (2012) Critical role of caspase-1 in vascular inflammation and development of atherosclerosis in Western diet-fed apolipoprotein E-deficient mice. Biochem. Biophys. Res. Commun. 425, 162–168 [DOI] [PubMed] [Google Scholar]
- 8. Komada T., Usui F., Shirasuna K., Kawashima A., Kimura H., Karasawa T., Nishimura S., Sagara J., Noda T., Taniguchi S., Muto S., Nagata D., Kusano E., Takahashi M. (2014) ASC in renal collecting duct epithelial cells contributes to inflammation and injury after unilateral ureteral obstruction. Am. J. Pathol. 184, 1287–1298 [DOI] [PubMed] [Google Scholar]
- 9. Yajima N., Takahashi M., Morimoto H., Shiba Y., Takahashi Y., Masumoto J., Ise H., Sagara J., Nakayama J., Taniguchi S., Ikeda U. (2008) Critical role of bone marrow apoptosis-associated speck-like protein, an inflammasome adaptor molecule, in neointimal formation after vascular injury in mice. Circulation 117, 3079–3087 [DOI] [PubMed] [Google Scholar]
- 10. Kawaguchi M., Takahashi M., Hata T., Kashima Y., Usui F., Morimoto H., Izawa A., Takahashi Y., Masumoto J., Koyama J., Hongo M., Noda T., Nakayama J., Sagara J., Taniguchi S., Ikeda U. (2011) Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation 123, 594–604 [DOI] [PubMed] [Google Scholar]
- 11. Takahashi M. (2011) Role of the inflammasome in myocardial infarction. Trends Cardiovasc. Med. 21, 37–41 [DOI] [PubMed] [Google Scholar]
- 12. Kolliputi N., Shaik R. S., Waxman A. B. (2010) The inflammasome mediates hyperoxia-induced alveolar cell permeability. J. Immunol. 184, 5819–5826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kolliputi N., Galam L., Parthasarathy P. T., Tipparaju S. M., Lockey R. F. (2012) NALP-3 inflammasome silencing attenuates ceramide-induced transepithelial permeability. J. Cell. Physiol. 227, 3310–3316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fukumoto J., Fukumoto I., Parthasarathy P. T., Cox R., Huynh B., Ramanathan G. K., Venugopal R. B., Allen-Gipson D. S., Lockey R. F., Kolliputi N. (2013) NLRP3 deletion protects from hyperoxia-induced acute lung injury. Am. J. Physiol. Cell Physiol. 305, C182–C189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lamkanfi M., Mueller J. L., Vitari A. C., Misaghi S., Fedorova A., Deshayes K., Lee W. P., Hoffman H. M., Dixit V. M. (2009) Glyburide inhibits the cryopyrin/Nalp3 inflammasome. J. Cell Biol. 187, 61–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Horai R., Asano M., Sudo K., Kanuka H., Suzuki M., Nishihara M., Takahashi M., Iwakura Y. (1998) Production of mice deficient in genes for interleukin (IL)-1α, IL-1β, IL-1α/β, and IL-1 receptor antagonist shows that IL-1β is crucial in turpentine-induced fever development and glucocorticoid secretion. J. Exp. Med. 187, 1463–1475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hornung V., Bauernfeind F., Halle A., Samstad E. O., Kono H., Rock K. L., Fitzgerald K. A., Latz E. (2008) Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 9, 847–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dobbs L. G., Gonzalez R. F. (2002) Isolation and culture of pulmonary alveolar epithelial type II cells. in Culture of Epithelial Cells (Freshney R. I., Freshney M. G., eds) 2nd Ed., pp. 277–301, John Wiley & Sons, New York [Google Scholar]
- 19. Gonzalez R. F., Dobbs L. G. (2013) Isolation and culture of alveolar epithelial type I and type II cells from rat lungs. Methods Mol. Biol. 945, 145–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Inoue Y., Shirasuna K., Kimura H., Usui F., Kawashima A., Karasawa T., Tago K., Dezaki K., Nishimura S., Sagara J., Noda T., Iwakura Y., Tsutsui H., Taniguchi S., Yanagisawa K., Yada T., Yasuda Y., Takahashi M. (2014) NLRP3 regulates neutrophil functions and contributes to hepatic ischemia-reperfusion injury independently of inflammasomes. J. Immunol. 192, 4342–4351 [DOI] [PubMed] [Google Scholar]
- 21. Lian X., Qin Y., Hossain S. A., Yang L., White A., Xu H., Shipley J. M., Li T., Senior R. M., Du H., Yan C. (2005) Overexpression of Stat3C in pulmonary epithelium protects against hyperoxic lung injury. J. Immunol. 174, 7250–7256 [DOI] [PubMed] [Google Scholar]
- 22. Yu H., Jove R. (2004) The STATs of cancer–new molecular targets come of age. Nat. Rev. Cancer 4, 97–105 [DOI] [PubMed] [Google Scholar]
- 23. Inghirami G., Chiarle R., Simmons W. J., Piva R., Schlessinger K., Levy D. E. (2005) New and old functions of STAT3: a pivotal target for individualized treatment of cancer. Cell Cycle 4, 1131–1133 [DOI] [PubMed] [Google Scholar]
- 24. You S., Li R., Park D., Xie M., Sica G. L., Cao Y., Xiao Z. Q., Deng X. (2014) Disruption of STAT3 by niclosamide reverses radioresistance of human lung cancer. Mol. Cancer Ther. 13, 606–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Alas S., Bonavida B. (2001) Rituximab inactivates signal transducer and activation of transcription 3 (STAT3) activity in B-non-Hodgkin's lymphoma through inhibition of the interleukin 10 autocrine/paracrine loop and results in down-regulation of Bcl-2 and sensitization to cytotoxic drugs. Cancer Res. 61, 5137–5144 [PubMed] [Google Scholar]
- 26. Quinton L. J., Jones M. R., Robson B. E., Simms B. T., Whitsett J. A., Mizgerd J. P. (2008) Alveolar epithelial STAT3, IL-6 family cytokines, and host defense during Escherichia coli pneumonia. Am. J. Respir. Cell Mol. Biol. 38, 699–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Quinton L. J., Mizgerd J. P., Hilliard K. L., Jones M. R., Kwon C. Y., Allen E. (2012) Leukemia inhibitory factor signaling is required for lung protection during pneumonia. J. Immunol. 188, 6300–6308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hokuto I., Ikegami M., Yoshida M., Takeda K., Akira S., Perl A. K., Hull W. M., Wert S. E., Whitsett J. A. (2004) Stat-3 is required for pulmonary homeostasis during hyperoxia. J. Clin. Invest. 113, 28–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yang L., Lian X., Cowen A., Xu H., Du H., Yan C. (2004) Synergy between signal transducer and activator of transcription 3 and retinoic acid receptor-α in regulation of the surfactant protein B gene in the lung. Mol. Endocrinol. 18, 1520–1532 [DOI] [PubMed] [Google Scholar]
- 30. Crosby L. M., Waters C. M. (2010) Epithelial repair mechanisms in the lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 298, L715–L731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jain D., Atochina-Vasserman E. N., Tomer Y., Kadire H., Beers M. F. (2008) Surfactant protein D protects against acute hyperoxic lung injury. Am. J. Respir. Crit. Care Med. 178, 805–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shigeoka A. A., Mueller J. L., Kambo A., Mathison J. C., King A. J., Hall W. F., Correia Jda S., Ulevitch R. J., Hoffman H. M., McKay D. B. (2010) An inflammasome-independent role for epithelial-expressed Nlrp3 in renal ischemia-reperfusion injury. J. Immunol. 185, 6277–6285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zuurbier C. J., Jong W. M., Eerbeek O., Koeman A., Pulskens W. P., Butter L. M., Leemans J. C., Hollmann M. W. (2012) Deletion of the innate immune NLRP3 receptor abolishes cardiac ischemic preconditioning and is associated with decreased IL-6/STAT3 signaling. PLoS ONE 7, e40643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang Y., Sauler M., Shinn A. S., Gong H., Haslip M., Shan P., Mannam P., Lee P. J. (2014) Endothelial PINK1 mediates the protective effects of NLRP3 deficiency during lethal oxidant injury. J. Immunol. 192, 5296–5304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lingappan K., Jiang W., Wang L., Couroucli X. I., Barrios R., Moorthy B. (2013) Sex-specific differences in hyperoxic lung injury in mice: implications for acute and chronic lung disease in humans. Toxicol. Appl. Pharmacol. 272, 281–290 [DOI] [PMC free article] [PubMed] [Google Scholar]