

Background: Human HSV-1 glycoprotein gC-1 is heavily O-glycosylated by cellular GalNAc transferases.
Results: O-Glycosylation of the gC-1 mucin domain was characterized by LC-MS/MS and expression of GalNAc-transferases by quantitative PCR and immunohistochemistry.
Conclusion: O-Glycosylation of gC-1 occurs in a site-specific and ordered manner related to specific GalNAc transferases.
Significance: Different target cells may generate different viral glycoprofiles affecting serological responses and infectious properties.
Keywords: Glycoprotein Biosynthesis, Glycosylation, Glycosyltransferase, Herpesvirus, Mass Spectrometry (MS)
Abstract
The herpes simplex virus type 1 (HSV-1) glycoprotein gC-1, participating in viral receptor interactions and immunity interference, harbors a mucin-like domain with multiple clustered O-linked glycans. Using HSV-1-infected diploid human fibroblasts, an authentic target for HSV-1 infection, and a protein immunoaffinity procedure, we enriched fully glycosylated gC-1 and a series of its biosynthetic intermediates. This fraction was subjected to trypsin digestion and a LC-MS/MS glycoproteomics approach. In parallel, we characterized the expression patterns of the 20 isoforms of human GalNAc transferases responsible for initiation of O-linked glycosylation. The gC-1 O-glycosylation was regulated in an orderly manner initiated by synchronous addition of one GalNAc unit each to Thr-87 and Thr-91 and one GalNAc unit to either Thr-99 or Thr-101, forming a core glycopeptide for subsequent additions of in all 11 GalNAc residues to selected Ser and Thr residues of the Thr-76–Lys-107 stretch of the mucin domain. The expression patterns of GalNAc transferases in the infected cells suggested that initial additions of GalNAc were carried out by initiating GalNAc transferases, in particular GalNAc-T2, whereas subsequent GalNAc additions were carried out by followup transferases, in particular GalNAc-T10. Essentially all of the susceptible Ser or Thr residues had to acquire their GalNAc units before any elongation to longer O-linked glycans of the gC-1-associated GalNAc units was permitted. Because the GalNAc occupancy pattern is of relevance for receptor binding of gC-1, the data provide a model to delineate biosynthetic steps of O-linked glycosylation of the gC-1 mucin domain in HSV-1-infected target cells.
Introduction
Herpes simplex virus type 1 (HSV-1) encodes 12 different glycoprotein species that are exposed on the viral envelope and during the late phases of the infectious cycle also in the membranes of the infected cells (1, 2). Five of these glycoproteins, designated gB-1, gC-1, gD-1, gH-1, and gL-1, are implicated in viral attachment and subsequent uptake of virus particles into the infected cell (1–3). One of the first steps in this process is binding of virus-associated gC-1 to specific glycosaminoglycan species at the surface of the target cell (4, 5). In addition to its functions in viral attachment, gC-1 also contributes to immune evasion by functioning as a decoy receptor for factor C3b of the complement system (6), and recent data suggest that gC-1 may also express selectin ligands, e.g. sialyl Lewis X (sLex), with the capacity to participate in viral pathogenesis (7, 8).
The peptide sequence of gC-1 comprises 511 amino acid residues, and the protein is heavily glycosylated, containing 9 consensus sites for N-linked glycosylation scattered along the peptide sequence and a major mucin-like domain containing numerous clustered O-linked glycans in a peptide stretch delimited by amino acids 30 and 124 (9). The molecular size of the O-linked glycans of the gC-1 mucin domain may vary from monosaccharides to sialylated tetrasaccharides (7, 10, 11) but possibly also larger structures (8, 12). In vitro studies using HSV-1 mutants expressing gC-1 that lack its mucin domain indicated that this domain is involved not only in viral binding to its target cell but also in cell-to-cell spread of virus, which is affected by the number of O-linked glycans in this domain (13, 14). Moreover, the mucin domain of gC-1 appears to be involved in expression of virus-induced selectin ligands, a phenomenon of possible relevance for viraemic spread of HSV-1 (8, 12, 15, 16). Despite the significance of the gC-1 mucin domain it remains unclear how this region acquires its content of O-linked glycans.
Initiation of O-linked glycosylation of viral glycoproteins and thus the addition of a GalNAc residue to a serine (Ser) or threonine (Thr) residue of the mucin domain is carried out in the Golgi network by one or more of 20 host cell-encoded polypeptide GalNAc transferases, designated GalNAc-Ts (encoded by the human genes GALNT1 through T20) (17, 18). The 20 different GalNAc-Ts add GalNAc to Ser/Thr residues in mucin domains rich in Ser, Thr, and Pro, with different but partly overlapping specificities regarding the amino acid sequence in the peptide acceptors. Based on similarities in these peptide acceptor specificities, the GalNAc-Ts may be divided in several subgroups (17, 19–21). There are examples of Ser/Thr-containing proteins accepting GalNAc addition by only 1 of the 20 different GalNAc-Ts (20–23), demonstrating the existence of unique peptide specificities among the GalNAc-Ts. All but one of the GalNAc-Ts (GalNAc-T20) also possess a lectin domain that can bind to GalNAc residues of an acceptor mucin domain, thereby affecting the specificity of the enzyme (24–26).
According to the current notion, the addition of the innermost GalNAc units to their respective Ser and Thr sites in the mucin domains is carried out in two steps (26, 27). First, specific GalNAc-Ts (e.g. the ubiquitously expressed isoforms GalNAc-T1 and -T2 referred to as the “initiating” GalNAc-Ts) add GalNAc units to the naked polypeptide. This initial glycosylation paves the way for the subsequent action of “followup” GalNAc-Ts (e.g. GalNAc-T4, -T7, and -T10 (28, 29)), which depends on the presence of preexisting GalNAc units on the peptide backbone enabling the addition of GalNAc to neighboring free Ser or Thr amino acid residues via lectin domain-mediated interactions of these enzymes. The expression pattern of GalNAc-Ts varies tremendously in a tissue-dependent manner, and consequently, the site occupancy of Ser and Thr residues of any given O-GalNAc-glycosylated protein expressed in different tissues and/or cell type may differ considerably depending on the spatiotemporal expression pattern of GalNAc-Ts.
It is established that defined features of the peptide sequence of the gC-1 mucin domain determines the extent of O-linked glycosylation (30). However, despite its significance for the final structures achieved by the O-linked glycans of the gC-1 mucin domain, very little is known about the regulatory and molecular events controlling mucin type O-glycosylation of this domain. Therefore, in this study we analyzed the early molecular events controlling O-linked glycosylation of gC-1 in diploid human fibroblasts (HEL cells) infected with HSV-1. This type of cell was chosen because (a) viral infection of fibroblastoid tissue is of significance in the pathogenesis not only of HSV-1 but also a number of other herpes viruses (31) and (b) the nature of HEL cells as a non-transformed, true HSV-1-permissive, diploid cell line constitutes an excellent experimental system for studies on the effect multiple host gene products have on posttranslational modifications of viral proteins. Here we have performed a glycoproteomics approach that has enabled a detailed mapping of the order by which individual Ser and Thr residues of the gC-1 mucin domain acquire GalNAc residues.
MATERIALS AND METHODS
Cells and Viruses
Human embryonic lung (HEL)2 fibroblasts were grown in Eagle's minimal essential medium supplemented with 10% fetal calf serum (FCS), 1% l-glutamine, and 1% penicillin-streptomycin. Green monkey kidney cells were grown in Eagle's minimal essential medium supplemented with 10% FCS and 1% penicillin-streptomycin. During HSV-1 infection, completely confluent monolayers of cells were used, and FCS was omitted from the medium. The HSV-1 strain Syn17+ (32) was used throughout the study, and the virus titer was determined by plaque titration on green monkey kidney cells as previously described (14). Briefly, green monkey kidney cells were grown in 427-cm2 roller bottles, Syn17+ was added at a multiplicity of infection (m.o.i.) of 5–10 plaque forming units (pfu)/cell, and the cells were incubated at 37 °C for 24–48 h until a cytopathic effect of +++ was reached. Thereafter the bottles were freeze-thawed at −80 °C, and aliquots were stored at −80 °C. For plaque titration of the virus, 500 μl per well of a dilution series was applied on green monkey kidney cells grown in 9-cm2 wells. The virus was allowed to attach to the cells for 1 h, and then the cells were overlaid by a mixture of 1.5 ml of Hanks' balanced salt solution and 1.5 ml of 3% methylcellulose in phosphate-buffered saline (PBS). The cells were incubated in 37 °C for 48 h. Finally the overlay was removed, and the cells were fixated by the addition of 5 drops crystal violet.
Viral Replication Assay
HEL fibroblasts were grown to a density of 80,000 cells/cm2 in 9 cm2 wells. Virus stock was added to the confluent HEL fibroblasts at an m.o.i. of 10 pfu/cell and allowed to bind to the cells for 1 h. The overlay was removed, and fresh Eagle's minimal essential medium was added. After incubation in 37 °C and 5% CO2 the growth medium and the cells were harvested and stored in −20 °C until titration assay on green monkey kidney cells was performed as described above. Alternatively, the cell lysate was subjected to DNA isolation using a MagNaPure instrument (Roche Diagnostics) according to the manufacturer's instructions, and the DNA was analyzed for viral DNA using quantitative PCR (33) for quantification of herpesvirus DNA.
Isolation of Cytoplasmic RNA
HEL fibroblasts grown in 9-cm2 wells were infected at a m.o.i. of 10 as described above. At the end of infection the cells were washed in 2 ml of PBS and harvested by the addition of RLN Buffer (Qiagen protocol cytoplasmic RNA animal cells, 50 mm Tris-Cl, pH 8.0, 140 mm NaCl, 1.5 mm MgCl2, 0.5% Nonidet P-40). The cells were collected, incubated for 5 min on ice, centrifuged at 4 °C for 2 min at 300 × g, and the supernatant was kept at −20 °C before isolation with Qiagen RNeasy kit (Qiagen, Alameda, CA) according to manufacturer's instructions. DNase treatment was performed with Turbo DNAfree (Ambion, Austin, TX) according to the manufacturer's instructions. The concentration of the resulting RNA was determined by measuring the absorbance at 260/280 using an Ultrospec 2100 pro UV/visible spectrophotometer.
Quantitative Reverse Transcription Real-time PCR
To determine gene expression patterns, quantitative reverse transcription real-time PCR was performed as previously described and according to the manufacturer's instructions regarding TaqMan™ chemistry (12). In brief, 500 ng of total RNA extracted from HSV-infected HEL cells or mock-transfected HEL cells was used for reverse transcription with Superscript III (Invitrogen) in 20 μl (RNA, H2O, 1 μl of dNTP, and 1 μl of random primers, incubation at 65 °C for 5 min, incubation on ice, the addition of 4 μl of 5× of first strand synthesis buffer, 0.5 μl of RNasin, 1 μl of DTT, 1 μl of reverse transcriptase) for each reaction. Quantitative reverse transcription real-time PCR assays were run on a Step-One-Plus Real-time PCR system (Invitrogen) using chemistry and recommendations by the manufacturer and using individually optimized TaqMan primer probe sets for all 20 GALNTs (17) as well as for fucosyltransferase 6 (FUT6 (16)) and 18 S rRNA (Applied Biosystems, Carlsbad, CA). Relative concentrations of transcripts from different GalNAc transferase genes and the fucosyltransferase gene were determined using the ΔCT method (34) and normalized and linearized against 18 S RNA and the detection limit of 40 cycles. Although the different GALNT and FUT6 assays both are calculated where cycle 40 has an expression of 1, it must be noted that expression levels of different assays are not comparable. Only GalNAc transferases that were detectable are displayed in Fig. 2.
FIGURE 2.

Quantitative reverse transcriptase real time PCR analysis of RNA expressed by a selection of human GALNT genes in HEL fibroblasts. As a positive control for a bona fide HSV-1-inducible human gene, transcription of human FUT6 was analyzed. The expression levels are normalized against 18 S RNA, and the value 1 is chosen for the detection limit for each method. The different assays do not permit direct comparison of the absolute expression levels between different glycosyltransferase genes.
Immunofluorescence
HEL fibroblasts grown in confluent monolayers in 162 cm2 flasks were trypsinized and resuspended in Eagle's minimal essential medium supplemented with 1% penicillin-streptomycin, 1% l-glutamine, and 10% FCS. The cells were seeded on and allowed to adhere to Teflon-coated object slides for 24 h. The cells were thereafter infected with HSV-1 at an m.o.i. of 5–10 pfu/cell and incubated for the indicated times at 37 °C and 5% CO2 in a humid atmosphere. At the end of infection the object slides were washed in PBS, fixated in ice-cold acetone for 5 min, and stored at −80 °C until immunofluorescence staining. Before immunofluorescence the glass slides were incubated in blocking solution (PBS with 3% bovine serum albumin (BSA)) for 30 min. To visualize gC-1 a rabbit anti-gC-1 antibody (clone KF922) (12) was applied at a dilution factor of 1:100, and Golgi protein giantin was detected using a rabbit anti-giantin antibody (Abcam, Cambridge, UK) at a dilution of 1:500. Mouse monoclonal antibodies to human GalNAc-T1 (UH3, 4D8), -T2 (UH4, 4C4), -T4 (UH6, 4G2), -T5 (5F11), -T10 (6D5), and -T12 (1F9), prepared as described (35), were used at stock concentration. After incubation with the primary antibodies at 4 °C overnight the glass slides were washed in PBS and distilled water. A second incubation with a FITC-conjugated polyclonal anti mouse antibody and a TRITC-conjugated polyclonal anti rabbit antibody (DAKO, Glostrup, Denmark) applied at dilutions of 1:100 and 1:200, respectively, was performed at 37 °C for 45 min. Finally the glass slides were washed in PBS and distilled water, air-dried. and mounted with Prolong Gold Anti-fade containing 4′,6-diamidino-2-phenylindole (DAPI) (Invitrogen). The immunofluorescence was analyzed in a Zeiss LSM 510 Meta confocal microscope (Carl Zeiss, Oberkochen, Germany) using a Plan-Apochromat 63× objective in oil immersion.
Protein Immunoaffinity Purification
Immunosorbent purification was carried out essentially as previously described (7). Briefly, HEL fibroblasts were grown in 427-cm2 roller bottles to a density of 80,000 cells/cm2 and infected with HSV-1 at a m.o.i. of 5–10 pfu/cell. The virus was allowed to attach to the cells for 1 h before the inoculum was removed and fresh Eagle's supplemented with 1% penicillin-streptomycin and 1% l-glutamine was applied to the cells. The cells were incubated at 37 °C until 100% of the cells demonstrated cytopathic effect (24–48 h). The infected cells were harvested using a rubber policeman and centrifuged at 1200 × g for 10 min. The supernatant was removed and stored at −80 °C until isolation of viral particles. The cell pellet was resuspended in a small amount of supernatant and stored at −80 °C. To solubilize the proteins, the cell pellet was thawed and diluted in equal parts with 2× buffer (Tris phosphate-buffered saline (TBS) supplemented with 2% 3α,12α-dihydroxycholamic acid sodium salt), 2% Nonidet P-40, and 1 mm Pefabloc®SC (4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride (AEBSF). The cell solution was transferred to a glass homogenizer and kept on ice for 1 h while mixing regularly. The sample was transferred to 25 × 89-mm ultracentrifuge tubes (Beckman Coulter, Brea, CA), and the tubes were filled by adding TBS to the samples and placed in a Ti70.1 rotor and centrifuged at 100,000 × g for 1 h at 4 °C. The supernatant was collected and stored at −80 °C until further purification of gC-1. Immune-affinity columns were prepared by coupling of anti-gE-1 antibodies (clone B1E6(A5) (36)) and anti-gC-1 antibodies (clone B1C1(B4) (7)) to CNBr-activated Sepharose®4B beads (GE Healthcare) according to the manufacturer's instructions. The Sepharose-antibody beads were kept in 5 ml of Poly-Prep® Chromatography Columns (Bio-Rad). The columns were washed with 20 ml of buffer (0.01 m Tris-HCl, 0.5 m NaCl, and 0.1% Nonidet P-40) at pH 7.5. The protein lysate was first applied to the anti gE-1 column, and the flow-through was saved and applied to the anti gC-1 column. The anti gC-1 column was washed in 25 ml of buffer (0.01 m Tris-HCl, 0.5 m NaCl, and 0.1% Nonidet P-40) at pH 7.5 and subsequently washed in buffer (0.01 m Tris-HCl, 0.5 m NaCl) at pH 7.5. Finally gC-1 was released from the column using elution buffer (0.1 m glycine-HCl) at pH 2.4. Each elution fraction of 1 ml was captured in 100 μl of neutralization buffer (1 m Tris) at pH 8. The elution fractions were transferred to a 10K Macrosep® Advance Centrifugal Device (PALL, Ann Arbor, MI) and centrifuged for 30 min at 4 °C and 1500 × g. The concentrated protein was stored at −80 °C until immunoblot and LC-MS/MS were performed.
Immunoblot
gC-1 purified by immunoaffinity chromatography and crude cell lysate from mock-infected and HSV-1-infected HEL cells were mixed with SDS sample buffer and boiled for 5 min. The samples were loaded onto gradient polyacrylamide gel (4–12%; Invitrogen) and transferred to polyvinylidene fluoride membranes (Millipore, Billerica, MA) according to the manufacturer's instructions. The membranes were air-dried and stored at 4 °C until immunoblotting. The membranes were soaked in blocking solution (PBS with 0.1% Tween 20, 1% FCS, and 3% BSA) for 1 h. To detect gC-1 the membranes were incubated with a monoclonal antibody directed toward linear epitopes of the polypeptide (37) followed by an anti-mouse IgG peroxidase-conjugated antibody (DAKO). The membranes were finally incubated with SuperSignal West Dura Extended Duration Substrate (Thermo, Rockford, IL) and visualized using the ChemiDoc™ MP Imaging system (Bio-Rad).
Mass Spectrometric Analyses
The region corresponding to gC-1 in a 4–12% gel, as obtained from parallel immunoblotting, was subjected to in-gel trypsin digestion as described (38). The eluted peptides were reconstituted in 40 μl of 0.1% formic acid (eluent A), and 20 μl were separated by C18 chromatography on a 15-cm capillary column (Zorbax SB300, 0.075 mm inner diameter). A linear gradient ranging from 3% to 60% eluent B (84% acetonitrile in 0.1% formic acid) in eluent A for 50 min at a flow rate of 250–300 nl/min was used. The LC system (Ettan MDLC, GE Healthcare) was coupled to an LTQ-FTICR instrument (Thermo Fisher Scientific) via a nanoelectrospray source (Thermo Fisher Scientific). The MS1 survey scans were acquired in the FTICR mass analyzer using a m/z 300–2000 range. The five most intense peaks in each MS1, with a charge state ≥2 and an intensity above 500, were selected (data dependent) for collision-induced dissociation (CID) in the linear ion trap (LTQ). CID fragmentation was performed with a normalized collision energy of 35%, activation q = 0.25, activation time of 30 ms, and 3 microscans. In separate LC-MS/MS runs an inclusion list of m/z values corresponding to glycopeptide masses, obtained after the CID fragmentation analysis, was used to select precursors for electron capture dissociation (ECD) in the ICR cell. ECD was performed with a relative energy of 4, duration of 70 ms, and 3 microscans. For both, CID and ECD dynamic exclusion was enabled with a repeat count of 2 and then excluded for 3 min. The MS/MS spectra were converted to Mascot mgf format using extract msn. All sequences of the Swissprot database were searched. Oxidation of Met was set as variable, and carbamidomethyl modification of Cys was as fixed modification. The instrument variable was set to b- and y-ions (1+ and 2+), and peptide and fragment tolerance were 10 ppm and 0.6 Da, respectively. Glycopeptide spectral interpretations were all done manually. A list of glycopeptide fragment ions was obtained through the MS-product tool (ProteinProspector).
In Silico Prediction of GalNAC-T Isoform Preferences
The full-length peptide (Thr-76–Lys-107), representing the O-glycosylation-mapped portion of the gC-1 mucin domain, was analyzed in silico using the public database ISOGlyP to determine the probability of specific GalNAc-Ts glycosylating each possible residue in the peptide. This database is based on in vitro studies of individual GalNAc-Ts using millions of synthetic (glyco)peptides as substrates (39, 40). Three amino acids before and after each O-glycosylation site were analyzed, and for this reason QNK, the amino acids situated before Thr-76 was added to the peptide. Each Ser and Thr is scored with an enhancement value product (EVP), determining the likelihood of glycosylation by each GalNAc-T analyzed. ISOGlyP predictions of glycosylation of Ser units are not directly comparable to those of Thr, and because Thr was the predominant amino acid subjected to O-glycosylation in the Thr-76–Lys-107 peptide, we only included Thr glycosylations. We selected GalNAc-T1, -T2, -T5, -T10, -T11, and -T12 as possible enzymes expressed with regard to RNA expression levels and immunofluorescence staining. As the EVP values are estimations of glycosylation and not completely comparable between different GalNAc-Ts, we used different cut-off values to determine which GalNAc-Ts are most likely to glycosylate the different position, where EVP for the naked peptide ranged from 0.24 to 35.57. As the exact values are not comparable but give more relative information on the rate of glycosylation for each position, cut-off values were selected with regard to the variable nature of EVP. Higher EVP, i.e. >10, was set for the most rapid glycosylation, an EVP >4 for moderate glycosylation and an EVP >1 for the slowest glycosylation.
RESULTS
Expression of GalNAc-Ts in Normal and HSV-1-infected HEL Cells
To determine the replication kinetics of HSV-1 in HEL fibroblasts, the cells were infected at a high m.o.i. (10 pfu/cell), and intracellular as well as extracellular virus titers were quantified over time (Fig. 1). We found that infectious progeny virus was produced in increasing rates at least until 48 h post infection (p.i.), confirming that the cells were fully permissive for HSV-1 infection.
FIGURE 1.

Infectious progeny virus production in HEL cells. HEL cells infected with HSV-1 at a high multiplicity of infection (10 m.o.i.) were harvested at the indicated time points, and the intracellular and extracellular fractions were titrated by plaque assay.
Because O-glycosylation may be initiated by 20 different isoenzyme GalNAc-Ts, we first assessed the transcription of these genes in HSV-1-infected and mock-infected HEL cells (Fig. 2). Cells were harvested 9 h p.i. during the exponential phase of HSV-1 replication. As a positive control we included human FUT6, encoding a fucosyltransferase, known to be up-regulated during HSV-1 infection by >1 order of magnitude (15, 16). We found that 17 out of altogether 20 defined human GALNTs (17) were indeed transcribed in HEL cells, albeit at greatly different levels ranging from high (T1, T2, T10) to low levels (T13, T14, T16). The RNA level for each of these GalNAc-T genes was lower in HSV-1-infected cells than in mock-infected cells, and 5 of the 17 GALNTs expressed in HEL cells were not reliably detected after HSV-1 infection. As expected, the FUT6 RNA levels were about 20 times higher in the HSV-1-infected cells than in the corresponding mock-infected cells, establishing that the lowered GALNTs RNA levels were not due to unspecific viral cytopathic effects or a systematic assay bias.
To verify the GalNAc-T expression levels at the protein level, we analyzed HSV-1-infected and mock-infected HEL cells by immunofluorescence, combining a panel of monoclonal antibodies to GalNAc-T1, -T2, -T4, -T5, -T10, and -T12 (Fig. 3). In mock-infected cells the most prominent fluorescence was observed for GalNAc-T2, but distinct fluorescence was also observed for GalNAc-T1, -T4, -T5, and -T12, whereas only sporadic fluorescence was detected for GalNAc-T10. In the HSV-1-infected cells a similar staining was observed for all the GalNAc-Ts as in the mock-infected cells, although the intracellular localization of GalNAc-Ts changed as the infection progressed (Fig. 3). We used GalNAc-T2 as a probe to investigate this phenomenon more in detail as well as its relationship to expression of gC-1 (Figs. 4 and 5). Early in the infection gC-1 is expressed at variable degrees in individual cells despite a high m.o.i., and only at 9 h p.i. did all of the cells express detectable levels of gC-1. During the early phase of infection, gC-1 co-localized with GalNAc-T2 in Golgi-like compartments. Twenty-four hours p.i. gC-1 staining was both cytoplasmic and cell surface-associated, and GalNAc-T2 was dispersed throughout the cell (Fig. 4). To confirm the localization of GalNAc-T2 to the Golgi compartment, HSV-1-infected and mock-infected cells were double-stained with the pan-Golgi marker, giantin, and GalNAc-T2 antibodies (Fig. 5). At 9 h p.i. giantin staining indicated disruption of the Golgi compartment in some of the infected cells, and at 24 h p.i. the Golgi was disrupted into granular-like structures in all of the cells and still co-localized with GalNAc-T2. Thus, although RNA levels of GALNTs decreased during HSV-1 infection, GalNAc-Ts were still detected in the Golgi compartment and co-localized with gC-1 throughout the infectious cycle despite a disruption of Golgi late in the infection.
FIGURE 3.

Immunofluorescence staining of GalNAc-T1, -T2, -T4, -T5, -T10, and -T12 in HSV-1 and mock-infected HEL cells. Cells were infected with HSV-1 and fixed with acetone at different time points as indicated. Mock-infected cells were fixed at 24 h. Cells were stained for HSV-1 (red), GalNAc-T (green), and cell nucleus (blue).
FIGURE 4.

Expression of GalNAc-T2 and gC-1 in HSV-1 infected HEL cell at different time intervals using confocal microscopy. Indirect fluorescence with GalNAc-T2-reactive antibody (green), gC-1 reactive antibody (red), and nuclear staining with DAPI (blue) is shown. The far right panel represents a merge.
FIGURE 5.

Analysis of possible co-localization of GalNAc-T2 with the Golgi compartment. Shown is immunofluorescence staining using antibodies to GalNAc-T2 (green) and the giantin pan-Golgi marker (red) and nuclear staining with DAPI (blue), respectively. The far right panel represents a merge. The white arrow indicates an intact Golgi compartment. The red arrow indicates a dispersed Golgi compartment.
Isolation of gC-1 from HSV-1-infected HEL Cells
We purified gC-1 from HSV-1-infected HEL cells, harvested after all cells displayed a clear cytopathic effect (24–48 h p.i.), and the cell extract was subjected to immunoaffinity chromatography using a gC-1-specific monoclonal antibody (7). Two purified variants of gC-1 were isolated: one gC-1 preparation was purified from the cell sap of HSV-1-infected HEL cells, and one was from extracellular virus particles that were pelleted from the spent medium. Crude cell extract from mock-infected and HSV-1-infected HEL cells as well as the purified gC-1 preparation was checked by SDS-PAGE, and gC-1 bands were detected by immunoblot (37) (Fig. 6). Two gC-1-reactive bands were identified; one at 125 kDa, previously described for HSV-1-infected HEL cells as “completely glycosylated” gC-1 containing complex type N-linked glycans as well as O-linked glycans (41), and one band at about 100 kDa, previously identified as “precursor gC-1” (pgC-1) (11), containing only high mannose N-glycans and trimmed derivatives thereof.
FIGURE 6.
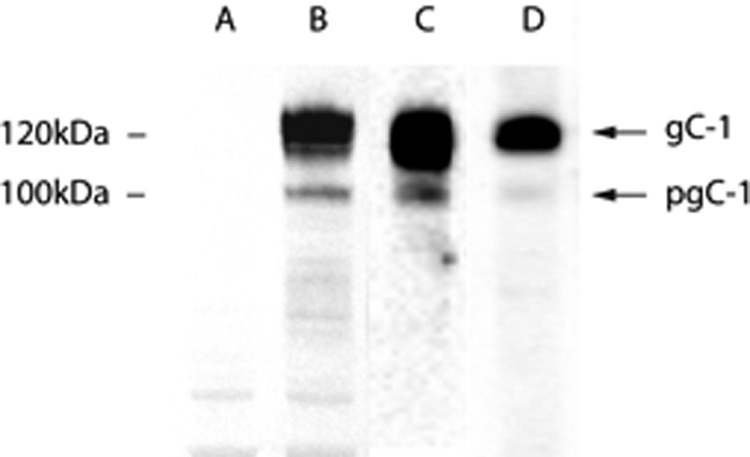
Crude cell lysate-derived gC-1 and gC-1 isolated by immunoaffinity chromatography visualized by immunoblot. Lane A, uninfected cell lysate. Lane B, HSV-1-infected HEL cell lysate. Lane C, gC isolated from HSV-1 infected HEL cells. Lane D, gC isolated from released HSV-1 viral particles. Positions of molecular weight markers and fully processed gC-1, containing complex type N-linked glycans and a variety of O-linked glycans, and precursor gC-1 (pgC), containing high mannose N-linked glycans and neglible O-linked glycans. The differences between gC-1 and pgC-1 are reviewed elsewhere (9, 14).
Mass Spectrometric Analyses of gC-1 O-Linked Glycosylation
The data from the MS analysis are presented in Fig. 7 and supplemental Figs. S1–S8. Two major tryptic glycopeptides of the gC-1 protein containing O-linked GalNAc were identified in the cellular extracts; 76TTPTEPASPPTTPKPTSTPK95 and 96SPPTSTPDPKPK107, both originating from the core mucin domain of the viral glycoprotein. The longer fragment contains nine potential O-glycosylation sites, and the shorter one contains. The tandem mass spectrometry (MS/MS) analyses aimed at pinpointing the stepwise biosynthesis of the gC-1 protein used these glycopeptides as targets for CID-MS2 and ECD experiments. Strikingly, we found that the longer peptide was modified with one-to-seven N-acetylhexosamine (HexNAc) units, in correspondence with GalNAc O-glycosylation and the shorter peptide with one-to-four HexNAc residues. Neither of the peptides showed any signs of elongated HexNAc-HexNAc structures. This indicated that (i) each HexNAc residue was attached to one individual Ser/Thr residue, (ii) the glycosylation of the Ser/Thr residues was essentially completed before the HexNAc chains were extended with hexoses and sialic acids, and (iii) there seemed to be a preference in which order the various Ser/Thr residues were glycosylated.
FIGURE 7.

MS identification of glycopeptides from the mucin domain of gC-1. Examples of mass spectra and their interpretations representing the 76TTPTEPASPPTTPKPTSTPK95 glycopeptide glycoforms are given as the following. A, summary MS1 in which the signal intensity can be compared for all glycopeptides. Glycopeptides containing a hexose in addition to HexNAc units are indicated by an asterisk. B, ECD spectrum of the diglycosylated peptide demonstrating the pinpointing of HexNAc residues using c- and z-ions. C, representation of the gC-1 amino acid sequence. The mucin domain is shaded, putative N-glycosylation sites are denoted by lollipops, and the transmembrane domain (TM) is denoted in black. The positions of three peptides of the mucin domain, relevant for this study, are depicted: peptides 76TTPTEPASPPTTPKPTSTPK95 and 96SPPTSTPDPKPK107, found to be decorated by O-linked glycans, and peptide 106PKNNTTPAK114, found to contain a complex type N-linked glycan.
For the 76TTPTEPASPPTTPKPTSTPK95 tryptic peptide the exact attachment sites of all the seven HexNAc residues could not be definitely determined from the MS/MS analysis. However, b- and y-type ions of the CID-MS2 spectra (supplemental Fig. S3) and specifically the y15 ion of the modified glycopeptides indicated that the initial 4 O-glycosylation events occur in the 81PASPPTTPKPT91 sequence. Useful ECD spectra were obtained for the di-substituted glycoform (Fig. 7B), which with the use of c11, c13, z3, and z4 ions, demonstrated clearly that Thr-87 and Thr-91 were the initial sites of O-glycosylation of the peptide. Similarly, ECD spectra of the HexNAc-tri and HexNAc-penta glycoforms indicated that the three initial HexNAc residues were found within the 83SPPTTPKPT91 sequence and that Ser-92 or Thr-93 were not glycosylated until the HexNAc-penta glycoform appeared (supplemental Fig. S3, C and E). Based on these spectra a scheme for O-glycosylation of this peptide was concluded (Fig. 8A). Extensions of the HexNAc residues with hexoses and sialic acids were observed in the precursor spectra (supplemental Fig. S2), and interestingly they seemed to follow the same general principle as described already, i.e. all HexNAc residues were substituted with Hexoses before the next sugar, sialic acid, was added.
FIGURE 8.

Time-order of the GalNAc addition to Ser and Thr residues of the glycopeptides 76TTPTEPASPPTTPKPTSTPK95 (A) and 96SPPTSTPDPKPK107 (B) based on findings of partially or fully glycosylated glycopeptides as identified in Fig. 7. GalNAc units are indicated by black squares immediately above the amino acids to which they are attached. The possible positions for ambiguous GalNAc sites are defined by brackets.
Several glycopeptides originating from the 96SPPTSTPDPKPK107 region were observed either as tryptic 96SPPTSTPDPKPK107 or as semitryptic 96SPPTSTPDPK105 glycopeptides (Table 1) and were characterized by CID and ECD (Figs. 7 and supplemental Figs. S4–S8). Taken together, the CID and ECD experiments demonstrated that similar to the 76TTPTEPASPPTTPKPTSTPK95 peptide, the initial glycosylation of the 96SPPTSTPDPKPK107 region involves the addition of three or four consecutive HexNAc residues at the four potential O-glycosites (Ser-96, Thr-99, Ser-100, Thr-101) before elongation with other monosaccharides is observed. Two isobaric forms of the diglycosylated 96SPPTSTPDPKPK107 peptide were identified, one form where Thr-99 and Ser-100 were glycosylated and another form where Ser-100 and Thr-101 were glycosylated, indicating two separate pathways at this point. However, the third HexNAc residue was attached to either Thr-101 or Thr-99 of the two separate diglycosylated peptide variants demonstrating convergence to only one triglycosylated structure. The fourth HexNAc residue was finally linked to Ser-96. Similar results were obtained also from ECD fragmentations of the semitryptic 96SPPTSTPDPK105 peptide suggesting a biosynthetic scheme as shown in Fig. 8B. The 96SPPTSTPDPKPK107 tryptic glycopeptides were also found elongated with two, three, or four core 1 like structures, one of which was also found sialylated.
TABLE 1.
Identified gC-1 N- and O-glycopeptides
The figures where the significant ions are found in the corresponding mass spectra are shown in the last column.
| Residues | Peptide sequence | Glycan composition | Measured mass [M+H]+ | Theoretical mass [M+H]+ | Δ Mass | Figure |
|---|---|---|---|---|---|---|
| ppm | ||||||
| 76–95 | TTPTEPASPPTTPKPTSTPK | 2036.0573 | 2036.0546 | 1.3 | Supplemental Fig. S1A | |
| HexNAc1 | 2239.1354 | 2239.1340 | 0.6 | Fig. 7A, supplemental Figs. S1, A and B, and S3A | ||
| HexNAc2 | 2442.2159 | 2442.2134 | 1.0 | Fig. 7, A and B, supplemental Figs. S1, A and C, and S3B | ||
| HexNAc3 | 2645.2964 | 2645.2928 | 1.4 | Fig. 7A, supplemental Figs. S1A and D, and S3C | ||
| HexNAc4 | 2848.3766 | 2848.3722 | 1.5 | Fig. 7A, supplemental Figs. S1, A and E, and S3D | ||
| HexNAc5 | 3051.4574 | 3051.4516 | 1.9 | Fig. 7A, supplemental Figs. S1, A and F, and S3E | ||
| HexNAc6 | 3254.5376 | 3254.5310 | 2.0 | Fig. 7A, supplemental Figs. S1, A and G, and S3F | ||
| HexNAc7 | 3457.6103 | 3457.6104 | 0.0 | Fig. 7A, supplemental Fig. S1A | ||
| Hex1HexNAc2 | 2604.2693 | 2604.2662 | 1.2 | Fig. 7A, supplemental Fig. S2A | ||
| Hex1HexNAc3 | 2807.3444 | 2807.3456 | −0.4 | Fig. 7A, supplemental Fig. S2A | ||
| Hex1HexNAc4 | 3010.4342 | 3010.4250 | 3.1 | Fig. 7A, supplemental Fig. S2A | ||
| Hex1HexNAc5 | 3213.5111 | 3213.5044 | 2.1 | Fig. 7A, supplemental Fig. S2A | ||
| Hex1HexNAc6 | 3416.5781 | 3416.5838 | 1.7 | Fig. 7A, supplemental Fig. S2A | ||
| Hex4HexNAc4 | 3496.5851 | 3496.5834 | 0.5 | Supplemental Fig. S2B | ||
| Hex5HexNAc5 | 3861.7220 | 3861.7156 | 1.7 | Supplemental Fig. S2B | ||
| Hex6HexNAc6 | 4226.8361 | 4226.8478 | −2.8 | Supplemental Fig. S2B | ||
| Neu5Ac1Hex6HexNAc6 | 4517.9469 | 4517.9432 | 0.8 | Supplemental Fig. S2B | ||
| 96–105 | SPPTSTPDPK | 1026.5109 | 1026.5102 | 0.7 | ||
| HexNAc1 | 1229.5903 | 1229.5896 | 0.6 | Supplemental Figs. S7, A and B, and S8D | ||
| HexNAc2 | 1432.6703 | 1432.6690 | 0.9 | Supplemental Figs. S7, A and 7C, and S8C | ||
| HexNAc3 | 1635.7493 | 1635.7484 | 0.6 | Supplemental Figs. S7, A and D, and S8B | ||
| HexNAc4 | 1838.8293 | 1838.8278 | 0.8 | Supplemental Figs. S7, A and E, and S8A | ||
| 96–107 | SPPTSTPDPKPK | 1251.6584 | 1251.6579 | 0.4 | ||
| HexNAc1 | 1454.7386 | 1454.7373 | 0.9 | Supplemental Figs. S4, A and B, and S6E | ||
| HexNAc2 | 1657.8176 | 1657.8167 | 0.5 | Supplemental Figs. S4, A and C, and S6D | ||
| HexNAc3 | 1860.8975 | 1860.8961 | 0.8 | Supplemental Figs. S4, A and D, and S6C | ||
| HexNAc4 | 2063.9771 | 2063.9755 | 0.8 | Supplemental Figs. S4, A and E, and S6B | ||
| Hex3HexNAc3 | 2347.0580 | 2347.0545 | 1.5 | Supplemental Figs. S5, A and C, and S6A | ||
| Hex1HexNAc4 | 2226.0359 | 2226.0283 | 3.4 | Supplemental Fig. S4A | ||
| Hex2HexNAc4 | 2388.0803 | 2388.0811 | −0.3 | Supplemental Fig. S5, A and B | ||
| Hex3HexNAc4 | 2550.1349 | 2550.1339 | 0.4 | Supplemental Fig. S5, A and D | ||
| Hex4HexNAc4 | 2712.1895 | 2712.1867 | 1.0 | Supplemental Fig. S5, A and E | ||
| Neu5Ac1Hex3HexNAc3 | 2638.1519 | 2638.1499 | 0.8 | Supplemental Fig. S5A | ||
| Neu5Ac1Hex3HexNAc4 | 2841.2333 | 2841.2293 | 1.4 | Supplemental Fig. S5A | ||
| Neu5Ac1Hex4HexNAc4 | 3003.2879 | 3003.2821 | 1.9 | Supplemental Fig. S5A | ||
| 106–114 | PKNNTTPAK | dHex1Hex3HexNAc4 | 2415.0680 | 2415.0655 | 1.0 | Supplemental Fig. S9A |
| dHex1Hex5HexNAc4 | 2739.1748 | 2739.1711 | 1.4 | Supplemental Figs. S9, A and B | ||
| dHex1Hex6HexNAc5 | 3104.3132 | 3104.3033 | 3.2 | Supplemental Fig. S9A | ||
| dHex1Hex7HexNAc6 | 3469.4369 | 3469.4355 | 0.4 | Supplemental Fig. S9A | ||
| dHex1Hex8HexNAc7 | 3834.5744 | 3834.5677 | 1.7 | Supplemental Fig. S9A | ||
| 108–114 | NNTTPAK | dHex1Hex3HexNAc4 | 2189.9361 | 2189.9178 | 8.4 | Supplemental Fig. S10A |
| dHex1Hex5HexNAc4 | 2514.0269 | 2514.0234 | 1.4 | Supplemental Figs. S10, A and C, and S11A | ||
| dHex1Hex6HexNAc5 | 2879.1602 | 2879.1556 | 1.6 | Supplemental Figs. S10 A and E, and S11B | ||
| dHex1Hex7HexNAc6 | 3244.2950 | 3244.2878 | 2.2 | Supplemental Fig. S10A | ||
| dHex1Hex8HexNAc7 | 3609.4256 | 3609.4200 | 1.6 | Supplemental Fig. S10A | ||
| Neu5Ac1dHex1Hex5HexNAc4 | 2805.1229 | 2805.1188 | 1.5 | Supplemental Fig. S10, B and D | ||
| Neu5Ac2dHex1Hex5HexNAc4 | 3096.2195 | 3096.2142 | 1.7 | Supplemental Fig. S10B | ||
| Neu5Ac1dHex1Hex6HexNAc5 | 3170.2598 | 3170.2510 | 2.8 | Supplemental Fig. S10, B and F | ||
| Neu5Ac2dHex1Hex6HexNAc5 | 3461.3546 | 3461.3464 | 2.4 | Supplemental Fig. S10B | ||
| Neu5Ac1dHex1Hex7HexNAc6 | 3535.3886 | 2525.3832 | 1.5 | Supplemental Fig. S10B | ||
| 146–151 | FRNSTR | dHex1Hex4HexNAc3 | 2183.9207 | 2183.9184 | 1.1 | Supplemental Fig. S12, A and C |
| dHex1Hex5HexNAc3 | 2345.9744 | 2345.9712 | 1.4 | Supplemental Figs. S12, A and E, and S13A | ||
| dHex1Hex5HexNAc4 | 2549.0531 | 2549.0506 | 1.0 | Supplemental Figs. S12. A and D, and S13C | ||
| dHex1Hex6HexNAc5 | 2914.1876 | 2914.1828 | 1.6 | Supplemental Figs. S12A and S13D | ||
| dHex1Hex7HexNAc6 | 3279.3215 | 3279.3150 | 2.0 | Supplemental Fig. S12A | ||
| dHex1Hex8HexNAc7 | 3644.4602 | 3644.4472 | 3.6 | Supplemental Fig. S12A | ||
| 361–372 | RNATGLALVLPR | Hex5HexNAc4 | 2903.3588 | 2903.3613 | −0.9 | Supplemental Fig. S14, A–C |
| dHex1Hex5HexNAc4 | 3049.4189 | 3049.4192 | −0.1 | Supplemental Fig. S14A | ||
| dHex2Hex5HexNAc4 | 3195.4790 | 3195.4771 | 0.6 | Supplemental Fig. S14A | ||
| Neu5Ac1dHex1Hex5HexNAc4 | 3340.5113 | 3340.5146 | −1.0 | Supplemental Figs. S14, A and D |
Mass Spectrometric Analyses of gC-1 N-Glycosylations
Four potential N-glycosylation sites in the 106PKNNTTPAK114, the 146FRNSTR151, and the 361RNATGLAVLPR372 peptides (Table 1) were also subjected to detailed structural analysis by CID and ECD (supplemental Figs. S9–S14). The 106PKNNTTPAK114 peptide could be modified at either Asn-108 or Asn-109 given that both residues are part of the Asn-Xaa-(Ser/Thr) consensus sequence for N-glycosylation. The actual site of glycosylation was found to be at the Asn-108 residue by ECD fragmentations (supplemental Figs. S9–S11) of the 108NNTTPAK114 glycopeptide showing the presence of c1 and z4-z6 ions for the fucosylated bi- and tri-antennary structures. The sialylated and non-sialylated N-glycoforms of this peptide were also subjected to CID-MS2 (supplemental Fig. S10) to confirm the N-glycan compositions (Table 1). The 146FRNSTR151 glycopeptide was characterized by CID-MS2 fragmentation (supplemental Fig. S12) to verify the presence of fucosylated and non-sialylated N-glycans at Asn-148, carrying either bi-/tri-/tetraantennary or hybrid-type structures. ECD spectra were used to sequence the peptide itself (supplemental Fig. S13). Finally, the 361RNATGLAVLPR372 peptide was observed with N-glycans at Asn-362 corresponding to the biantennary structure with a varying degree of fucosylation and sialylation (supplemental Fig. S14). The biantennary structure was confirmed by CID-MS2, and the peptide sequence was verified by ECD fragmentation.
The data demonstrated that the present MS approach has a capacity to concomitantly determine the structure and amino acid sequence at and around the glycosylation site also for larger N-linked glycans. Thus, gC-1 contains altogether nine formal sites for the addition of N-linked glycans (Fig. 7), and we were able to assign structures for three of those. We compared these results with predictions provided by the NetNglyc server that predicts N-glycosylation sites in human proteins using artificial neural networks that examine the sequence context of Asn-Xaa-(Ser/Thr) sequons (42). The 106PKNNTTPAK114 contains two Asn-Xaa-Thr sequons, but we found complex type N-linked glycans only associated with Asn-108 but not Asn-109, which is completely in line with the NetNglyc prediction of the Asn-109 site as insufficient for N-linked glycosylation. According to our MS analysis no O-linked glycans were associated with Thr-110 or Thr-111, probably because of steric effects as the bulky N-linked glycans were added before initiation of O-linked glycosylation. Judging from the NetNglyc server Asn-70 and Asn-74 should be susceptible to N-linked glycosylation, but this peptide stretch was not susceptible to analysis by MS/MS, probably reflecting that this domain is highly decorated by O-linked glycans and devoid of functioning trypsin sites, which results in fragments too big to be compatible with the current MS approach. Both Asn-181 and Asn-197 are predicted to be susceptible to N-linked glycosylation, but due to the lack of tryptic cleavage sites in this protein region we were unable to detect any peptide fragments covering these N-glycosylation sites.
In Silico Analysis Based on ISOGlyP for Predicting the Susceptibility of Peptides for O-Glycosylation
In an attempt to use all the data we have gathered from real-time PCR, immunofluorescence confocal microscopy, and LC-MS/MS analyses of HSV-1-infected HEL cells to provide information on which GalNAc-Ts glycosylate which sites, we performed an in silico analysis of the combination of both peptides (Thr-76–Lys-107) using the publically available database, ISOGlyP (39). ISOGlyP predicts the structural outcome of a panel of GalNAc-Ts on submitted (glyco)-peptide substrates, resulting in an EVP for each Ser and Thr and each GalNAc-T. The enzymes analyzed here were GalNAc-T1, -T2, -T5, -T10, -T11, and -T12 as their presence was considered plausible in the infected cells based on immunofluorescence or RNA expression data (Figs. 2 and 3). EVP are calculated for each GalNAc-T selected, and values >1 are considered probable glycosylation targets, whereas an EVP of 2 suggests a 2-fold rate of glycosylation. As EVP values are estimated calculations, cut-off thresholds were set to group the results in rapid (EVP > 10), moderate (EVP > 4), and slow glycosylation (EVP > 1). Applying an EVP value as high as 10 as a cut-off limit favored formation of glycopeptide 2 in Fig. 9, with initial glycosylations of Thr-87, Thr-91, Thr-99 and Thr-101, identical to the experimental results as demonstrated from the interpretation of MS data (Figs. 7 and 8). The prediction suggested involvement of GalNAc-T1, -T2, -T5, and -T11 as responsible for the addition of GalNAc to these threonine residues, but GalNAc-T2 and in particular GalNAc-T11 seemed to be more generally acting, whereas GalNAc-T1 and -T5 had high preference for only one of the threonines (Thr-101 and Thr-87, respectively). Using glycopeptide 2 as a substrate in the ISOGlyP analysis, all the enzymes expressed in HEL cells expressed EVPs <10, and therefore, an EVP of 4 was used as a cutoff value. This analysis predicted that the next threonine sites to be glycosylated would be Thr-79, Thr-86, and Thr-93, involving GalNAc-T2, -T10, and -T11, respectively, and forming glycopeptide 3. Interestingly, of the threonines analyzed Thr-76 and Thr-77 appeared to be inferior substrates to the GalNAc-Ts analyzed. GalNAc-T10 demonstrated an EVP of >1 for Thr-76 and Thr-77, respectively, whereas GalNAc-T11 demonstrated a similar value only for Thr-77, thus forming glycopeptide 4.
FIGURE 9.

In silico ISOGly-P analysis of the theoretical order of the addition of O-linked GalNAc to the Thr residues of the entire 73–107 peptide stretch. The enumeration refers to actions of GalNAc-T1, -T2, -T5, -T10, and -T11. The EVP is a score determining the likelihood of glycosylation according to the ISOGlyP public database, with increasing score indicating more rapid glycosylation. The sites of glycosylation for enzymes with EVP scores above the values indicated in the figure are shown for each peptide.
DISCUSSION
The clustered O-linked glycans of viral glycoproteins contribute to functionality in at least three important respects. First, intact O-linked glycosylation of glycoprotein mucin domains promote infectivity of human viruses using cell surface glycosaminoglycans as receptors (13). Second, the mucin domains may harbor virus-induced carbohydrate epitopes that may function as selectin ligands (8, 12). Third, O-linked glycans modulate the immunological behavior in the sense that excessive O-linked glycosylation of mucin domains shield viral envelope glycoproteins from potential neutralizing antibodies (43), whereas a moderate glycosylation of such domains augments immunogenicity by creating novel glycopeptide epitopes, accessible to circulating cognate antibodies (44, 45).
The present study has made it possible to define several of the details of the stepwise addition of GalNAc units to individual serine and threonine residues of a 32-amino acid-long peptide stretch of the mucin domain of gC-1, delimited by Thr-76 and Lys-107. Despite the reservation that the analysis was carried out on glycoforms representing two separate peptides of this domain (Thr-76 to Lys-95 and Ser-96 to Lys-107, respectively), the data permitted a model of a distinct pattern of serial additions of GalNAc units to specific positions in a highly ordered manner (Fig. 8). The first step in this model is a seemingly synchronous addition of one GalNAc unit each to Thr-87 and Thr-91 and one GalNAc unit to either Thr-99 or Thr-101, resulting in two glycoforms of this glycopeptide stretch with respect to the Thr-99/Thr-101 glycosylation status. After this stage it becomes difficult to relate the exact time order of the subsequent additions to the Thr-76–Lys-95 part of the peptide with the corresponding additions to the Ser-96–Lys-107 portion. Regarding the Ser-96–Lys-107 peptide, the next step is an addition of GalNAc to Ser-100 to both glycoforms of the monoglycosylated peptide, which is somewhat unexpected from the generally inferior GalNAc acceptor potential of serines compared with threonines (25, 39). Interestingly, only one single triglycosylated glycoform of the growing Thr-76–Lys-107 glycopeptide stretch will be created in the next step via the addition of GalNAc to either of the free Thr-101 or Thr-99 sites, respectively, of each one of the two different diglycosylated Thr-76–Lys-107 glycoforms. The data support a model of a peptide stretch containing a starting point of one or two defined GalNAc-occupied serine or threonine residues, exemplified by Thr-87 of gC-1, which may constitute a seed intermediate, a prerequisite for further addition of GalNAc units to increasingly distal serines and threonines, spreading as far as 10 amino acid residues away from the original seed Thr-87 GalNAc.
The subsequent steps, according to the model, after the addition of the initial GalNAc unit involves the addition of GalNAc to Ser-96 in parallel with gradual increase toward the N- and C-terminal directions of the glycopeptide stretch of new serine and threonine residues recruited for GalNAc addition. This process takes place in at least five steps, as resolved by the MS data (Fig. 8), but there is some uncertainty as to the exact identity of the Ser and Thr units being recruited. This is exemplified by the first step in this process, for which the MS data indicate the addition of one GalNAc to either Ser-83 or Thr-86, but it is not known whether both of the sites are equally susceptible or if only one of the sites becomes occupied. The subsequent stepwise recruitment of serine and threonine residues at the N-terminal direction takes place in a similar manner, resulting in a modified structure with a maximum of 11 of the totally 13 available serine or threonine units recruited out of which 6 of the GalNAc units can be assigned unequivocally to defined serine or threonine units. It was also indicated from the MS data that elongation of GalNAcs into core 1 structures took place essentially only after all relevant serines or threonines became decorated by GalNAc. In conclusion, the initial steps of O-glycosylation of the mucin domain of gC-1 is a dynamic process with three general characteristics: 1) the first few GalNAc residues to be added were specifically and systematically attached to only a few of all the Ser/Thr residues available for and later subjected to modification, 2) the GalNAc modifications of the Ser/Thr residues followed an ordered pattern, and 3) this process was essentially completed before the GalNAc residues were extended by other monosaccharides.
We found that all RNA levels representing the human GalNAc-Ts investigated were decreased as a consequence of HSV-1 infection. Despite this, immunofluorescence data indicated that GalNAc-Ts have a relatively long half-life with appreciable levels of several GalNAc-Ts found even after 24 h of HSV-1 infection (GalNAc-T1, -T2, -T5, -T10, -T11, and -T12). To compile all our expression data we used the ISOGlyP database (see “Materials and Methods”) to predict the capacity of these GalNAc-Ts to participate in the various stages of O-linked glycosylation of the glycopeptide stretch delimited by Gln-73 and Lys-107. Before interpreting the predictions two considerations were made. (i) Predictions regarding glycosylation of serine residues are overestimated in comparison to threonines (25, 39), and we, therefore, refrained from calculating such predictions. (ii) The ISOGly-P scores (expressed as the EVP) is a relative measurement and cannot be used for direct comparisons between isoenzymes, and we, therefore, initially only considered high EVP values in the interpretation. When applying an EVP > 10 for analysis of the effects of the GalNAc-Ts mentioned above, exactly the same four amino acid residues as were confirmed by the MS data to be initially glycosylated were predicted to be rapidly glycosylated by the ISOGlyP database (Fig. 9). Whereas GalNAcT-11 appeared to be a pan-enzyme with a high EVP for all four threonine sites, GalNAc-T1 and -T5 displayed a pronounced selectivity for only one site each. Interestingly, one of the transferases in the infected cells, GalNAc-T2, was predicted to efficiently add GalNAc to three of the four initial sites.
The GalNAc-T2, typically expressed in virus-infected cells (Fig. 2 and 3), is considered a prototype for initiating GalNAc-Ts (19–21, 26). It is, therefore, reasonable to assume that the first GalNAc residue added to either Thr-91, Thr-99, or Thr-101 is added by GalNAc-T2 in the infected cells, a conclusion supported by (a) its high abundance compared with other GalNAc-Ts in the infected cells (present work) and (b) previous in vitro studies using recombinant GalNAc-T2 and synthetic peptides representing this domain of gC-1 (30). The prototype followup GalNAc-Ts (-T4, -T10, and -T12) (24–26) appear to be expressed in HSV-1-infected HEL cells as judged from the RNA levels and corroborated by antibody detection of GalNAc-T4, -T10, and -T12 after HSV-1 infection (Figs. 2 and 3). It has been generally accepted that these enzymes belong to the distinct GalNAc-T subfamilies IIa (GalNAc-T4 and -T12) and IIb (GalNAc-T10) (17) and participate in followup glycosylation of acceptor sites that have been prior glycosylated at neighboring sites, although a recent study has shown that these followup enzymes were found to be capable of also glycosylating a subset of non-glycosylated acceptor peptide substrates (46). Of these IIa and IIb subfamily enzymes, GalNAc-T10 has been shown to be structurally distinct in the acceptor binding domain by lacking the “proline pocket” found in the other subfamily II members (47). Thus this enzyme might complete the followup glycosylation reaction in a lectin-independent manner, mechanistically distinct from the other followup reactions completed by GalNAc-T4, -T7, and -T12. In addition, GalNAc-T10 has been shown to prefer GalNAc addition if the neighboring +1 site is preoccupied by GalNAc (40). It thus seems reasonable to assume that GalNAc-T10 participates in followup glycosylation of the gC-1 mucin domain Thr-76 and Thr-86 glycosylation sites. Further detailed analysis is needed to dissect the individual role of the alternate followup GalNAc-Ts (-T4, -T7, and -T12) found to be expressed at different levels in HEL cells after infection (Figs. 2 and 3).
The diploid HEL cells used here represent a natural target for HSV-1 infection, but HSV-1 may also replicate in other cells and tissues including neural cells and circulating CD3-positive T-cells (12, 48–50). There is a considerable variation in the expression patterns of the 20 human GalNAc-Ts between these divergent tissue types (21, 26, 51). Thus, the GalNAc occupancy of the available Ser and Thr units of the gC-1 mucin region may vary considerably, dependent on the actual set of GalNAc-Ts in different target tissues for HSV-1, and that large differences may arise not only at the followup but also at the “initiation” of GalNAc-Ts.
Recently, methods were developed, designed to reveal possible differences in the serological response in panels of virus-infected patients, in the antibody response to the mucin domains of viral glycoproteins. This methodology, targeting the mucin domains of glycoproteins of two other herpes viruses, has been used for analysis of the specific antibody response in patients infected with HSV-2 and Epstein-Barr virus (44, 45). The general interpretation of these data is that the mucin domains of these two glycoproteins contain two types of glycopeptide stretches: one type of glycopeptide where the GalNAc occupancy of Ser/Thr residues is identical in viral glycoproteins for all patients and, surprisingly, one type of glycopeptide where different patients present glycopeptides to the immune response for which different combinations of Ser/Thr are decorated by GalNAc. The results presented here strongly favor a model in which combinations of constant and variable patterns in the GalNAc occupancy of Ser/Thr units affect the serological response in the virus-infected patients as well as the ability of viral cell-to-cell spread and other biological properties of herpes viruses influenced by viral glycoproteins. Obviously, the complex involvement of the 20 different GalNAc-Ts in this process, as demonstrated here, contributes to this process.
Acknowledgments
The Centre for Cellular Imaging, the Proteomics Core Facility, Sahlgrenska Academy at the University of Gothenburg, Ulla Ruetschi at the Neurochemistry laboratory, and the staff at the Laboratory of Clinical Microbiology, Sahlgrenska University Hospital are all gratefully acknowledged for assistance with fluorescence microscopy, mass spectrometric analyses, cell cultures, and HSV-1 DNA quantifications.
This work was supported by grants from the Swedish Research Council (8266 and 15283), Swedish Governmental Grants to the Sahlgrenska University Hospital, and by the Danish Research Councils and the Danish National Research Foundation (DNRF 107).

This article contains supplemental Figs. S1–S14
- HEL
- human embryonic lung
- m.o.i.
- multiplicity of infection
- TRITC
- tetramethylrhodamine isothiocyanate
- LTQ-FTICR
- linear ion trap-Fourier transform ion cyclotron resonance
- CID
- collision-induced dissociation
- ECD
- electron capture dissociation
- EVP
- enhancement value product
- p.i.
- post infection.
REFERENCES
- 1. Antoine T. E., Park P. J., Shukla D. (2013) Glycoprotein targeted therapeutics: a new era of anti-herpes simplex virus-1 therapeutics. Rev. Med. Virol. 23, 194–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Eisenberg R. J., Atanasiu D., Cairns T. M., Gallagher J. R., Krummenacher C., Cohen G. H. (2012) Herpes virus fusion and entry: a story with many characters. Viruses 4, 800–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Akhtar J., Shukla D. (2009) Viral entry mechanisms: cellular and viral mediators of herpes simplex virus entry. FEBS J. 276, 7228–7236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lycke E., Johansson M., Svennerholm B., Lindahl U. (1991) Binding of herpes simplex virus to cellular heparan sulphate, an initial step in the adsorption process. J. Gen. Virol. 72, 1131–1137 [DOI] [PubMed] [Google Scholar]
- 5. Herold B. C., WuDunn D., Soltys N., Spear P. G. (1991) Glycoprotein C of herpes simplex virus type 1 plays a principal role in the adsorption of virus to cells and in infectivity. J. Virol. 65, 1090–1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Friedman H. M., Wang L., Fishman N. O., Lambris J. D., Eisenberg R. J., Cohen G. H., Lubinski J. (1996) Immune evasion properties of herpes simplex virus type 1 glycoprotein gC. J. Virol. 70, 4253–4260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Olofsson S., Sjöblom I., Lundström M., Jeansson S., Lycke E. (1983) Glycoprotein C of herpes simplex virus type 1: characterization of O-linked oligosaccharides. J. Gen. Virol. 64, 2735–2747 [DOI] [PubMed] [Google Scholar]
- 8. Nordén R., Nyström K., Adamiak B., Halim A., Nilsson J., Larson G., Trybala E., Olofsson S. (2013) Involvement of viral glycoprotein gC-1 in expression of the selectin ligand sialyl-Lewis X induced after infection with herpes simplex virus type 1. APMIS 121, 280–289 [DOI] [PubMed] [Google Scholar]
- 9. Olofsson S. (1992) Carbohydrates in herpesvirus infections. APMIS Suppl. 27, 84–95 [PubMed] [Google Scholar]
- 10. Dall'Olio F., Malagolini N., Speziali V., Campadelli-Fiume G., Serafini-Cessi F. (1985) Sialylated oligosaccharides O-glycosidically linked to glycoprotein C from herpes simplex virus type 1. J. Virol. 56, 127–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lundström M., Olofsson S., Jeansson S., Lycke E., Datema R., Månsson J. E. (1987) Host cell-induced differences in O-glycosylation of herpes simplex virus gC-1. I. Structures of nonsialylated HPA- and PNA-binding carbohydrates. Virology 161, 385–394 [DOI] [PubMed] [Google Scholar]
- 12. Nordén R., Nyström K., Aurelius J., Brisslert M., Olofsson S. (2013) Virus-induced appearance of the selectin ligand sLeX in herpes simplex virus type 1-infected T-cells: involvement of host and viral factors. Glycobiology 23, 310–321 [DOI] [PubMed] [Google Scholar]
- 13. Ekblad M., Adamiak B., Bergefall K., Nenonen H., Roth A., Bergstrom T., Ferro V., Trybala E. (2007) Molecular basis for resistance of herpes simplex virus type 1 mutants to the sulfated oligosaccharide inhibitor PI-88. Virology 367, 244–252 [DOI] [PubMed] [Google Scholar]
- 14. Mårdberg K., Nyström K., Tarp M. A., Trybala E., Clausen H., Bergström T., Olofsson S. (2004) Basic amino acids as modulators of an O-linked glycosylation signal of the herpes simplex virus type 1 glycoprotein gC: functional roles in viral infectivity. Glycobiology 14, 571–581 [DOI] [PubMed] [Google Scholar]
- 15. Nordén R., Nyström K., Olofsson S. (2009) Activation of host antiviral RNA-sensing factors necessary for herpes simplex virus type 1-activated transcription of host cell fucosyltransferase genes FUT3, FUT5, and FUT6 and subsequent expression of sLe (x) in virus-infected cells. Glycobiology 19, 776–788 [DOI] [PubMed] [Google Scholar]
- 16. Nyström K., Nordén R., Muylaert I., Elias P., Larson G., Olofsson S. (2009) Induction of sialyl-Lex expression by herpes simplex virus type 1 is dependent on viral immediate early RNA-activated transcription of host fucosyltransferase genes. Glycobiology 19, 847–859 [DOI] [PubMed] [Google Scholar]
- 17. Bennett E. P., Mandel U., Clausen H., Gerken T. A., Fritz T. A., Tabak L. A. (2012) Control of mucin-type O-glycosylation: a classification of the polypeptide GalNAc-transferase gene family. Glycobiology 22, 736–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Raman J., Guan Y., Perrine C. L., Gerken T. A., Tabak L. A. (2012) UDP-N-acetyl-α-d-galactosamine:polypeptide N-acetylgalactosaminyltransferases: completion of the family tree. Glycobiology 22, 768–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wandall H. H., Hassan H., Mirgorodskaya E., Kristensen A. K., Roepstorff P., Bennett E. P., Nielsen P. A., Hollingsworth M. A., Burchell J., Taylor-Papadimitriou J., Clausen H. (1997) Substrate specificities of three members of the human UDP-N-acetyl-α-d-galactosamine:polypeptide N-acetylgalactosaminyltransferase family, GalNAc-T1, -T2, and -T3. J. Biol. Chem. 272, 23503–23514 [DOI] [PubMed] [Google Scholar]
- 20. Bennett E. P., Hassan H., Mandel U., Hollingsworth M. A., Akisawa N., Ikematsu Y., Merkx G., van Kessel A. G., Olofsson S., Clausen H. (1999) Cloning and characterization of a close homologue of human UDP-N-acetyl-α-d-galactosamine:polypeptide N-acetylgalactosaminyltransferase-T3, designated GalNAc-T6: evidence for genetic but not functional redundancy. J. Biol. Chem. 274, 25362–25370 [DOI] [PubMed] [Google Scholar]
- 21. Ten Hagen K. G., Fritz T. A., Tabak L. A. (2003) All in the family: the UDP-GalNAc:polypeptide N-acetylgalactosaminyltransferases. Glycobiology 13, 1R–16R [DOI] [PubMed] [Google Scholar]
- 22. Bennett E. P., Hassan H., Mandel U., Mirgorodskaya E., Roepstorff P., Burchell J., Taylor-Papadimitriou J., Hollingsworth M. A., Merkx G., van Kessel A. G., Eiberg H., Steffensen R., Clausen H. (1998) Cloning of a human UDP-N-acetyl-α-d-galactosamine:polypeptide N-acetylgalactosaminyltransferase that complements other GalNAc-transferases in complete O-glycosylation of the MUC1 tandem repeat. J. Biol. Chem. 273, 30472–30481 [DOI] [PubMed] [Google Scholar]
- 23. Schjoldager K. T., Vakhrushev S. Y., Kong Y., Steentoft C., Nudelman A. S., Pedersen N. B., Wandall H. H., Mandel U., Bennett E. P., Levery S. B., Clausen H. (2012) Probing isoform-specific functions of polypeptide GalNAc-transferases using zinc finger nuclease glycoengineered SimpleCells. Proc. Natl. Acad. Sci. U.S.A. 109, 9893–9898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hassan H., Reis C. A., Bennett E. P., Mirgorodskaya E., Roepstorff P., Hollingsworth M. A., Burchell J., Taylor-Papadimitriou J., Clausen H. (2000) The lectin domain of UDP-N-acetyl-d-galactosamine:polypeptide N-acetylgalactosaminyltransferase-T4 directs its glycopeptide specificities. J. Biol. Chem. 275, 38197–38205 [DOI] [PubMed] [Google Scholar]
- 25. Gerken T. A., Revoredo L., Thome J. J., Tabak L. A., Vester-Christensen M. B., Clausen H., Gahlay G. K., Jarvis D. L., Johnson R. W., Moniz H. A., Moremen K. (2013) The lectin domain of the polypeptide GalNAc transferase family of glycosyltransferases (ppGalNAc Ts) acts as a switch directing glycopeptide substrate glycosylation in an N- or C-terminal direction, further controlling mucin type O-glycosylation. J. Biol. Chem. 288, 19900–19914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gill D. J., Clausen H., Bard F. (2011) Location, location, location: new insights into O-GalNAc protein glycosylation. Trends Cell Biol. 21, 149–158 [DOI] [PubMed] [Google Scholar]
- 27. Pratt M. R., Hang H. C., Ten Hagen K. G., Rarick J., Gerken T. A., Tabak L. A., Bertozzi C. R. (2004) Deconvoluting the functions of polypeptide N-α-acetylgalactosaminyltransferase family members by glycopeptide substrate profiling. Chem. Biol. 11, 1009–1016 [DOI] [PubMed] [Google Scholar]
- 28. Bennett E. P., Hassan H., Hollingsworth M. A., Clausen H. (1999) A novel human UDP-N-acetyl-d-galactosamine:polypeptide N-acetylgalactosaminyltransferase, GalNAc-T7, with specificity for partial GalNAc-glycosylated acceptor substrates. FEBS Lett. 460, 226–230 [DOI] [PubMed] [Google Scholar]
- 29. Cheng L., Tachibana K., Zhang Y., Guo J. m., Kahori Tachibana K., Kameyama A., Wang H., Hiruma T., Iwasaki H., Togayachi A., Kudo T., Narimatsu H. (2002) Characterization of a novel human UDP-GalNAc transferase, pp-GalNAc-T10. FEBS Lett. 531, 115–121 [DOI] [PubMed] [Google Scholar]
- 30. Biller M., Mårdberg K., Hassan H., Clausen H., Bolmstedt A., Bergström T., Olofsson S. (2000) Early steps in O-linked glycosylation and clustered O-linked glycans of herpes simplex virus type 1 glycoprotein C: effects on glycoprotein properties. Glycobiology 10, 1259–1269 [DOI] [PubMed] [Google Scholar]
- 31. Nyström K., Grahn A., Lindh M., Brytting M., Mandel U., Larson G., Olofsson S. (2007) Virus-induced transcriptional activation of host FUT genes associated with neo-expression of Ley in cytomegalovirus-infected and sialyl-Lex in varicella-zoster virus-infected diploid human cells. Glycobiology 17, 355–366 [DOI] [PubMed] [Google Scholar]
- 32. Marsden H. S., Crombie I. K., Subak-Sharpe J. H. (1976) Control of protein synthesis in herpesvirus-infected cells: analysis of the polypeptides induced by wild type and sixteen temperature-sensitive mutants of HSV strain 17. J. Gen. Virol. 31, 347–372 [DOI] [PubMed] [Google Scholar]
- 33. Namvar L., Olofsson S., Bergström T., Lindh M. (2005) Detection and typing of Herpes Simplex virus (HSV) in mucocutaneous samples by TaqMan PCR targeting a gB segment homologous for HSV types 1 and 2. J. Clin. Microbiol. 43, 2058–2064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nyström K., Biller M., Grahn A., Lindh M., Larson G., Olofsson S. (2004) Real time PCR for monitoring regulation of host gene expression in herpes simplex virus type 1-infected human diploid cells. J. Virol. Methods 118, 83–94 [DOI] [PubMed] [Google Scholar]
- 35. Mandel U., Hassan H., Therkildsen M. H., Rygaard J., Jakobsen M. H., Juhl B. R., Dabelsteen E., Clausen H. (1999) Expression of polypeptide GalNAc-transferases in stratified epithelia and squamous cell carcinomas: immunohistological evaluation using monoclonal antibodies to three members of the GalNAc-transferase family. Glycobiology 9, 43–52 [DOI] [PubMed] [Google Scholar]
- 36. Bergström T., Sjögren-Jansson E., Jeansson S., Lycke E. (1992) Mapping neuroinvasiveness of the herpes simplex virus type 1 encephalitis-inducing strain 2762 by the use of monoclonal antibodies. Mol. Cell Probes 6, 41–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sjöblom I., Glorioso J. C., Sjögren-Jansson E., Olofsson S. (1992) Antigenic structure of the herpes simplex virus type 1 glycoprotein C: demonstration of a linear epitope situated in an environment of highly conformation-dependent epitopes. APMIS 100, 229–236 [DOI] [PubMed] [Google Scholar]
- 38. Shevchenko A., Tomas H., Havlis J., Olsen J. V., Mann M. (2006) In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1, 2856–2860 [DOI] [PubMed] [Google Scholar]
- 39. Gerken T. A., Jamison O., Perrine C. L., Collette J. C., Moinova H., Ravi L., Markowitz S. D., Shen W., Patel H., Tabak L. A. (2011) Emerging paradigms for the initiation of mucin-type protein O-glycosylation by the polypeptide GalNAc transferase family of glycosyltransferases. J. Biol. Chem. 286, 14493–14507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Perrine C. L., Ganguli A., Wu P., Bertozzi C. R., Fritz T. A., Raman J., Tabak L. A., Gerken T. A. (2009) Glycopeptide-preferring polypeptide GalNAc transferase 10 (ppGalNAc T10), involved in mucin-type O-glycosylation, has a unique GalNAc-O-Ser/Thr-binding site in its catalytic domain not found in ppGalNAc T1 or T2. J. Biol. Chem. 284, 20387–20397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lundström M., Jeansson S., Olofsson S. (1987) Host cell-induced differences in the O-glycosylation of herpes simplex virus gC-1. II. Demonstration of cell-specific galactosyltransferase essential for formation of O-linked oligosaccharides. Virology 161, 395–402 [DOI] [PubMed] [Google Scholar]
- 42. Blom N., Sicheritz-Pontén T., Gupta R., Gammeltoft S., Brunak S. (2004) Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics 4, 1633–1649 [DOI] [PubMed] [Google Scholar]
- 43. Machiels B., Lété C., Guillaume A., Mast J., Stevenson P. G., Vanderplasschen A., Gillet L. (2011) Antibody evasion by a gammaherpesvirus O-glycan shield. PLoS Pathog. 7, e1002387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cló E., Kracun S. K., Nudelman A. S., Jensen K. J., Liljeqvist J. Å., Olofsson S., Bergström T., Blixt O. (2012) Characterization of the viral O-glycopeptidome: a novel tool of relevance for vaccine design and serodiagnosis. J. Virol. 86, 6268–6278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. D'Arrigo I., Cló E., Bergström T., Olofsson S., Blixt O. (2013) Diverse IgG serum response to novel glycopeptide epitopes detected within immunodominant stretches of Epstein-Barr virus glycoprotein 350/220: diagnostic potential of O-glycopeptide microarrays. Glycoconj. J. 30, 633–640 [DOI] [PubMed] [Google Scholar]
- 46. Kong Y., Joshi H. J., Schjoldager K. T., Madsen T. D., Gerken T. A., Vester-Christensen M. B., Wandall H. H., Bennett E. P., Levery S. B., Vakhrushev S. Y., Clausen H. (2015) Probing polypeptide GalNAc-transferase isoform substrate specificities by in vitro analysis. Glycobiology 25, 55–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kubota T., Shiba T., Sugioka S., Furukawa S., Sawaki H., Kato R., Wakatsuki S., Narimatsu H. (2006) Structural basis of carbohydrate transfer activity by human UDP-GalNAc:polypeptide α-N-acetylgalactosaminyltransferase (pp-GalNAc-T10). J. Mol. Biol. 359, 708–727 [DOI] [PubMed] [Google Scholar]
- 48. Puttur F. K., Fernandez M. A., White R., Roediger B., Cunningham A. L., Weninger W., Jones C. A. (2010) Herpes simplex virus infects skin gamma delta T cells before Langerhans cells and impedes migration of infected Langerhans cells by inducing apoptosis and blocking E-cadherin downregulation. J. Immunol. 185, 477–487 [DOI] [PubMed] [Google Scholar]
- 49. Aubert M., Yoon M., Sloan D. D., Spear P. G., Jerome K. R. (2009) The virological synapse facilitates herpes simplex virus entry into T cells. J. Virol. 83, 6171–6183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kennedy P. G., Steiner I. (2013) Recent issues in herpes simplex encephalitis. J. Neurovirol. 19, 346–350 [DOI] [PubMed] [Google Scholar]
- 51. Tarp M. A., Clausen H. (2008) Mucin-type O-glycosylation and its potential use in drug and vaccine development. Biochim. Biophys. Acta 1780, 546–563 [DOI] [PubMed] [Google Scholar]