

Background: Prolyl oligopeptidase (PREP) modulates accumulation of aggregated α-synuclein both in vitro and in vivo, but the mechanism remains unknown.
Results: Using live-cell and cell-free methods, we show that PREP interacts directly with α-synuclein.
Conclusion: PREP binds to α-synuclein and enhances its dimerization.
Significance: These results establish a mechanism of how PREP inhibition reduces α-synuclein aggregation and support PREP inhibition as a novel therapy in synucleinopathies.
Keywords: α-Synuclein (a-Synuclein), Conformational Change, Parkinson Disease, Peptidase, Protein Aggregation, Protein-Protein Interaction, Inhibitor
Abstract
Prolyl oligopeptidase (PREP) accelerates the aggregation of α-synuclein (aSyn), a key protein involved in development of Parkinson disease and other synucleinopathies. PREP inhibitors reduce aSyn aggregation, but the mechanism has remained unknown. We have now used protein-fragment complementation assays (PCA) and microscale thermophoresis in parallel to show that PREP interacts directly with aSyn in both intact cells and in a cell-free system. Using split luciferase-based PCA, we first showed that PREP enhances the formation of soluble aSyn dimers in live Neuro-2A neuroblastoma cells. A PREP inhibitor, KYP-2047, reduced aSyn dimerization in PREP-expressing cells but not in cells lacking PREP expression. aSyn dimerization was also enhanced by PREP(S554A), an enzymatically inactive PREP mutant, but this was not affected by KYP-2047. PCA and microscale thermophoresis studies showed that aSyn interacts with both PREP and PREP(S554A) with low micromolar affinity. Neither the proline-rich, C-terminal domain of aSyn nor the hydrolytic activity of PREP was required for the interaction with PREP. Our results show that PREP binds directly to aSyn to enhance its dimerization and may thus serve as a nucleation point for aSyn aggregation. Native gel analysis showed that KYP-2047 shifts PREP to a compact monomeric form with reduced ability to promote aSyn nucleation. As PREP inhibition also enhances autophagic clearance of aSyn, PREP inhibitors may reduce accumulation of aSyn inclusions via a dual mechanism and are thus a novel therapeutic candidate for synucleinopathies. Our results also suggest that PREP has other cellular functions in addition to its peptidase activity.
Introduction
Accumulation of α-synuclein (aSyn)3 containing protein inclusions in the brain is the defining feature of synucleinopathies. In the most common neurodegenerative disorder with movement deficits, Parkinson disease (PD), and in dementia with Lewy bodies they are called Lewy bodies and Lewy neurites, whereas glial insoluble inclusions of aSyn are characteristic for multiple system atrophy (1–3). In addition, mutations in the gene coding for the aSyn protein, SNCA, can cause familial PD with early onset, suggesting central involvement for aSyn in PD pathophysiology (4–8). aSyn is a natively unfolded brain protein that is mainly presynaptically localized but is found also in cytosol and nucleus. Proposed aSyn functions relate to synaptic neurotransmission, e.g. trafficking, packing, and release of synaptic vesicles, but the exact physiological roles of aSyn still remain poorly understood (for review, see Ref. 9).
Due to its inherently unfolded structure, aSyn is prone to aggregation by various factors such as high concentration, posttranslational modifications, mutations, oxidative stress, low pH, and metal ions. Moreover, formation of aSyn oligomers and fibrils leads to impairments in several cellular systems including mitochondria, endoplasmic reticulum-Golgi, and other vesicular transport systems and the plasma membrane (10, 11). aSyn has three distinct domains in its protein structure: an N-terminal amphipathic region where familial PD mutations linked to aSyn aggregation are located, a hydrophobic central part (NAC) that is required for aSyn aggregation, and an acidic C terminus that has been shown to regulate fibril formation (10). aSyn aggregation is a nucleation-dependent process, and it progresses from formation of misfolded soluble monomers and oligomers to protofibrils and mature fibrils with β-sheet structure and reduced solubility (10, 12). Although deposition of insoluble aSyn in Lewy bodies and neurites is the neuropathological hallmark of synucleinopathies, it appears that soluble oligomers and protofibrils are the most toxic aSyn species. In particular, dopaminergic neurons in the substantia nigra are highly sensitive to toxic aSyn species, emphasizing the role of aSyn in PD pathology (10, 13).
Interactions of aSyn with proteins and lipids in its native cellular environment regulate its oligomerization and aggregation process. One mediator that enhances aSyn aggregation is prolyl oligopeptidase (PREP, POP, EC 3.4.21.26) (14). PREP is an 80-kDa protein with serine protease activity that is widely distributed among the species and can be found in the brain and various other tissues (15). It hydrolyzes peptides smaller than 30 amino acids, and substance P, angiotensin, thyrotropin-releasing hormone, and vasopressin are the most widely studied PREP substrates (for review, see Ref. 16). Several physiological functions for PREP have been proposed including functions in cell proliferation and differentiation, inositol-1,4,5-triphoshpate signaling, and learning and memory (for review, see Ref 17). PREP activity has been reported to increase during aging and in neurodegenerative disorders, supporting the neuropeptide hypothesis (17, 18). Various small molecule inhibitors of PREP enzyme activity have been developed, but the results of PREP inhibition on neuropeptide levels in vivo and on memory and learning are unclear (19).
The size of aSyn protein is 140 amino acids, which makes it too large to be hydrolyzed by PREP, and it has been proposed that PREP might serve as a nucleation point for aSyn aggregation (14, 20). In addition, PREP colocalizes with aSyn in the substantia nigra in human PD brains (21). PREP inhibition effectively prevents aSyn aggregation in a cell-free system, in aSyn-overexpressing cells, and in transgenic mouse models (14, 22). Recently, we have identified PREP as a negative modulator of autophagosome formation, and treatment with PREP inhibitor, KYP-2047, induced macroautophagy and autophagosome formation via a beclin 1-dependent pathway (23). This was accompanied with increased clearance of high molecular weight aSyn species in the A30P transgenic mouse brain, suggesting PREP inhibition as a novel therapeutic strategy in synucleinopathies (23).
We hypothesized that modulation of aSyn aggregation by PREP may occur via multiple mechanisms, potentially also involving a direct protein-protein interaction. Although the effect of PREP on aSyn aggregation in a cell-free system (14) indirectly supports this hypothesis, a direct interaction between aSyn and PREP has not been demonstrated. Here, we have used a combination of live-cell and cell-free methods to show that both wild-type PREP and a catalytically inactive mutant PREP with a serine-alanine mutation at residue 554 (S554A) interact with aSyn with low micromolar affinity. Both wild-type PREP and the S554A mutant enhance aSyn dimerization in cells, but only in cells expressing wild-type PREP is aSyn dimerization reduced by KYP-2047, a specific PREP inhibitor. Our data suggest that PREP modulates the early stage of aSyn aggregate formation, likely already at the dimerization stage, via a direct interaction with aSyn. These data provide further support for PREP as a nucleation point for aSyn aggregation and provide mechanistic insight in how pharmacological inhibition of PREP reduces aSyn aggregation in cells.
EXPERIMENTAL PROCEDURES
Chemicals
Chemicals used were purchased from Sigma unless otherwise specified. Ethanol was purchased from Altia (Helsinki, Finland). Recombinant porcine PREP was produced in Escherichia coli and purified as described in Venäläinen et al. (24). Inactive PREP(S554A) mutant protein was obtained from Prof. Anne-Marie Lambeir (University of Antwerp, Belgium). The PREP inhibitor, KYP-2047 (4-phenylbutanoyl-l-prolyl-2(S)-cyanopyrrolidine), was synthesized in the School of Pharmacy, University of Eastern Finland, as previously described (25). KYP-2047 was chosen as a reference compound because the biochemical and pharmacological data indicate that it is potent and selective, enters cells in culture, and crosses the blood-brain barrier effectively in rodents (22, 24, 26, 27).
DNA Constructs
The split Gaussia princeps luciferase (GLuc) expression plasmids used in this study were previously described in Nykänen et al. (28). The human aSyn cDNA used to clone aSyn-GLuc1/HA, aSyn(Δ118–140)-GLuc1/HA (with amino acid 118–140 removed), aSyn(Δ98–140)-GLuc1/HA (with amino acid 98–140 removed), and asyn-GLuc2/HA were obtained from ORFeome library (Version 3.1, Genome Biology Unit, Institute of Biotechnology, University of Helsinki, accession number BC013293) and was PCR-cloned in the Gluc vector with the HA tag using the KpnI-XhoI site (constructs graphically summarized in Figs. 2A and Fig. 4A). The PREP and PREP(S554A) plasmids used in this project were previously described in Savolainen et al. (23) and were used for cloning the PREP-GLuc2 and PREP(S554A)-GLuc2 constructs (graphically summarized in Fig. 3A). All GLuc constructs used in this study have the GLuc reporter fragment placed at the N terminus separated by a (GGGGS)2SG linker.
FIGURE 2.
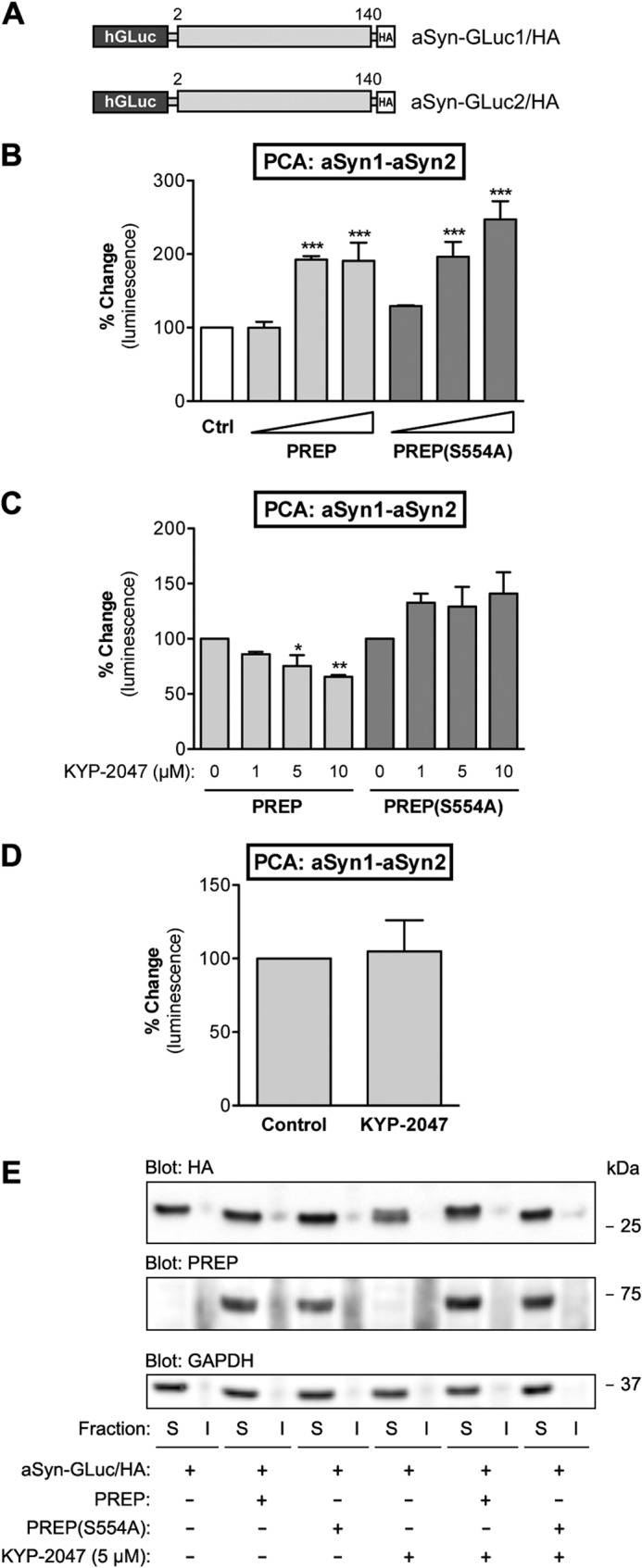
PREP co-expression promotes formation of soluble aSyn dimers in N2A cells. A, schematic presentation of GLuc fragment-tagged aSyn reporter constructs used in this study. B, the effect of co-expression of PREP or PREP(S554A) with aSyn was studied by PCA. aSyn-GLuc1 and aSyn-GLuc2 were kept constant (31.25 ng of plasmid DNA per well each), whereas increasing amounts of PREP or PREP(S554A) plasmid were transfected (12.5, 37.5, and 62.5 ng). The total amount of plasmid was adjusted to 125 ng per well with mock plasmid when needed. Both PREP and catalytically inactive PREP(S554A) significantly increased the dimerization of aSyn with increasing levels of expression (one-way ANOVA; ***, p < 0.001 PREP versus control). The average values are displayed as percent change as compared with control (cells expressing aSyn-GLuc1 and aSyn-GLuc2 only; mean ± S.E.; n = 4 independent experiments). C, the effect of KYP-2047 on aSyn dimerization in PREP-expressing cells was studied by PCA. Cells transfected with aSyn-GLuc1 and aSyn-GLuc2 (31.25 ng each) and PREP or PREP(S554A) (37.5 ng) were treated with at 1, 5, or 10 μm KYP-2047 for 4 h. Both 5 and 10 μm doses of KYP-2047 significantly decreased aSyn dimerization when incubated with active PREP (one-way ANOVA; *, p < 0.05 5 μm KYP-2047 versus control; **, p < 0.01 10 μm KYP-2047 versus control), but KYP-2047 did not show any effect on aSyn dimerization in PREP(S554A)-expressing cells. DMSO was used as vehicle control. The average values are displayed as percent change as compared with control (mean ± S.E.; n = 3 independent experiments). D, the effect of KYP-2047 on aSyn dimerization without co-expression of PREP was studied by PCA. Cells transfected with aSyn-GLuc1 and aSyn-GLuc2 (31.25 ng each) were treated with 10 μm KYP-2047 for 4 h. No change in aSyn dimerization was observed. DMSO was used as vehicle control. The average values are displayed as percent change as compared with control (mean ± S.E.; n = 4 independent experiments). E, solubility of aSyn reporter proteins with co-expression of PREP or PREP (S554A) and KYP-2047 treatment was studied by cellular fractionation and Western blot. Cells transfected with aSyn-GLuc1 and aSyn-GLuc2 (0.75 μg each), PREP, or PREP(S554A) plasmids were transfected at 1.5 μg. 5 μm KYP was added for 24 h. No shift of aSyn reporters from Triton-soluble (S) to insoluble fractions (I) was observed. Blots were stained with antibodies to PREP, HA tag (aSyn-GLuc1/2), and GAPDH as a loading control.
FIGURE 4.
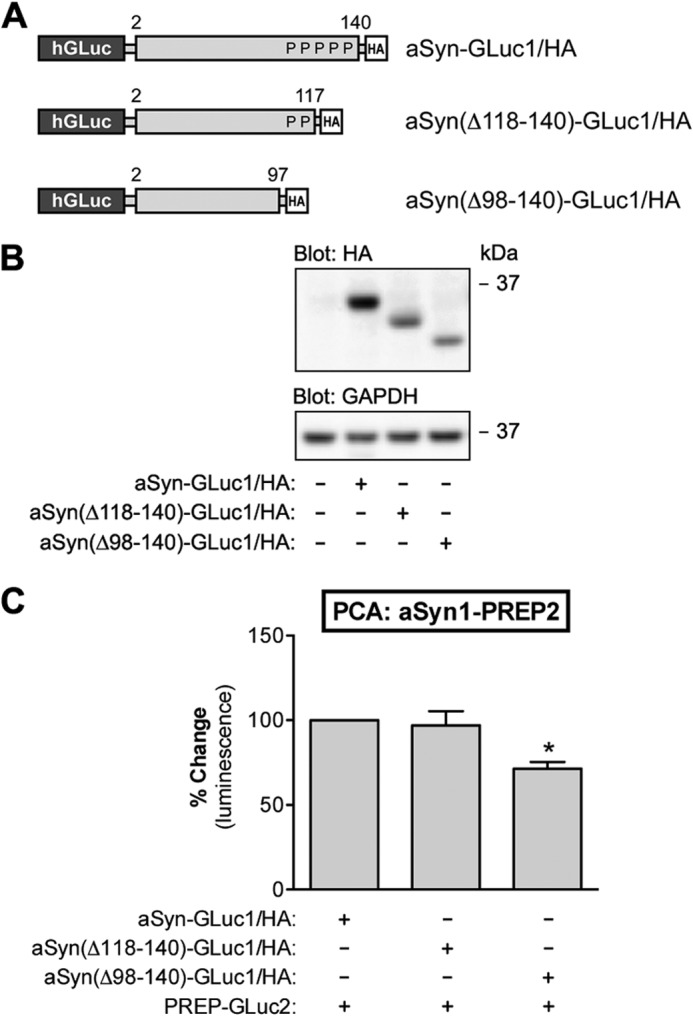
aSyn-PREP interaction does not require the proline-rich C-terminal domain of aSyn. A, schematic presentation of GLuc fragment-tagged aSyn constructs with different C-terminal truncations used in this study. P indicates a proline residue in the C-terminal tail of aSyn. B, the expression aSyn-GLuc1, aSyn(Δ118–140)-GLuc1, and aSyn(ΔC98–140)-GLuc1 was analyzed by Western blot. Each plasmid was transfected at 1.5 μg per well. All the constructs were appropriately expressed in N2A cells as determined by the anti-HA staining of the Western blot. GAPDH served as a loading control. C, the effect of C-terminal truncation of aSyn on aSyn-PREP interaction was studied by PCA. Co-expression of PREP-GLuc2 with aSyn(ΔC118–140)-GLuc1 showed similar PCA signal as the full-length aSyn-GLuc1, but co-expression of PREP-GLuc2 with aSyn(ΔC98–140)-GLuc1 showed a decrease in PCA signal. However, this may be due to the lower expression level of aSyn(Δ98–140) as compared with the full-length aSyn-GLuc1 reporter (B). The average values are displayed as percent change as compared with the control (mean ± S.E.; n = 3 independent experiments). * indicates significant difference with p < 0.05 (one-way ANOVA).
FIGURE 3.

aSyn interacts with PREP and PREP(S554A) in live cells. A, schematic presentation of GLuc fragment-tagged reporter constructs of aSyn and PREP used in this study. B, PREP protein expression after different construct transfections was studied by Western blot. All the constructs showed PREP protein expression, although PREP-GLuc2 was expressed at a lower level as compared with untagged PREP. Blot was stained with antibodies to PREP and GAPDH as a loading control. C, the enzymatic activity of PREP and PREP-GLuc2 was studied by an enzyme assay using a fluorescent substrate peptide. PREP activity in N2A cell lysates was significantly increased by PREP and PREP-GLuc2 transfections (one-way ANOVA; ***, p < 0.001 PREP versus control; *, p < 0.05 PREP-GLuc2 versus control), whereas cells transfected with PREP(S554A) or PREP(S554A)-GLuc2 did not show any enzymatic activity. D, the interaction of aSyn-GLuc1 with PREP-GLuc2 and PREP(S554A)-GLuc2 was studied by PCA. To confirm interaction specificity, the following reporter plasmids were co-expressed in pairs (62.5 ng each): PREP-GLuc2 with GLuc1 (“empty” plasmid, no insert), PREP(S554A)-GLuc2 with GLuc1, aSyn-GLuc1 with GLuc2, aSyn-GLuc1 with GSAP(16k)-GLuc2, APPC98-GLuc1 with GSAP(16k)-GLuc2. To demonstrate the aSyn-PREP interaction, aSyn-GLuc1 was co-expressed with PREP-GLuc2 or PREP(S554A)-GLuc2 (62.5 ng each). Both PREP-GLuc2 and PREP(S554A)-GLuc2 showed a significant PCA signal with aSyn-GLuc1 (one-way ANOVA; ***, p < 0.001), and interestingly, PREP(S554A)-GLuc2 had even stronger interaction with aSyn-GLuc1 than active PREP (one-way ANOVA; ***, p < 0.001). The average values are displayed as direct luminescence readout. Signals below 700 relative luminescence units (RLU) were regarded as background noise (mean ± S.E.; n = 3 independent experiments). E, the effect of KYP-2047 on aSyn-PREP and aSyn-PREP(S554A) interaction was studied by PCA. The same transfections were used as in Fig. 3B, but in addition the cells were treated with 5 μm KYP-2047 for 4 h. KYP-2047 showed a significant increase in the PCA signal of PREP-GLuc2 and aSyn-GLuc2 (one-way ANOVA; *, p < 0.05), but not with inactive PREP(S554A). DMSO was used as vehicle control. The average values are displayed as percent change as compared with the individual controls for aSyn-PREP and aSyn-PREP(S554A), respectively (mean ± S.E.; n = 3 independent experiments).
Cell Culture and Transfection
Mouse Neuro-2A (N2A) neuroblastoma cells were used throughout the whole study. N2A cells were cultured in full Dulbecco's modified Eagle's medium (DMEM) with an additional 10% (v/v) FBS (Invitrogen), 1% (v/v) l-glutamine-penicillin-streptomycin solution (Lonza) at 37 °C and 5% CO2, water-saturated air. Transfection of N2A was done using JetPei (Polyplus) according to the manufacturer's instruction.
Protein-Fragment Complementation Assay (PCA)
PCA was performed as previously described in Nykänen et al. (28). N2A cells were plated on poly-l-lysine-coated 96-well plates (PerkinElmer Life Sciences, white wall) at a density of 10,000 cells per well. 24 h post-plating, reporter plasmids were transfected (125 ng of total plasmid DNA per well). PCA signal was read 24 h post-transfection. Cells were changed to phenol red-free DMEM (Invitrogen) without serum 30 min before the measurement. A GLuc PCA signal was detected by injecting 25 μl of native coelenterazine (Nanolight Technology) per well (final concentration of 20 μm), and the emitted luminescence was read by Varioskan Flash multiplate reader (Thermo Scientific). For each experimental condition, 4 replicate wells were used, and 3–4 replicates of independent experiments were performed. A 100 mm stock solution of KYP-2047 was prepared in DMSO, further diluted to PBS, and added at various concentrations (1–10 μm) 4 h before the measurement. The corresponding amount of DMSO was used as vehicle control.
Western Blotting
The procedure was described in Nykänen et al. (28). Cells were plated on 6-well plates and transfected at 60–80% confluency with 3 μg of DNA per well. 48 h post-transfection, cells were washed twice with ice-cold PBS and collected in extraction buffer (10 mm Tris-HCl, pH 6.8, 150 mm NaCl, 1 mm EDTA, 1% Triton X-100, 0.25% Nonidet P-40, 1 μm NaF, Protease Inhibitor mixture tablets (Roche Molecular Biochemicals)) and further incubated on ice for 30 min. Cell debris was removed by centrifuging total lysates at 16,000 × g. A BCA protein assay kit (Thermo) was used to determine protein concentration, and equal amounts of protein samples (25–40 μg) were loaded per lane in 4–12% gradient Bis-Tris gels (Novex, Invitrogen) under reducing conditions. PVDF membranes (GE Healthcare) were used for blotting. The following primary antibodies were used for detection: anti-HA (Sigma), anti-PREP (R&D Systems), anti-PREP serum (collected on date 70–72 from a rabbit immunized against DPDSEQTKAFVEAQNK (PREP 33–48) peptide; specificity of collected serum against PREP was tested commercially using ELISA (Thermo)), and anti-GAPDH (Millipore). Horseradish-conjugated secondary antibodies and ECL Western blotting detection reagent (Thermo) were used for chemiluminescence signal generation. Quantitative analysis of the blot was done using QuantityOne software (Bio-Rad).
Cellular Fractionation
Total cell lysate was prepared as described above under “Western Blotting.” After 30-min of incubation on ice, each sample was adjusted to equal volumes with the extraction buffer in polycarbon centrifugation tubes (Beckman Coulter). Samples were centrifuged with a tabletop ultracentrifuge (OptimaTM ultracentrifuge, Beckman Coulter) and TLA-100 rotor at 100,000 × g for 30 min. After centrifugation, each soluble fraction was collected into a separate tube, with protein concentration measured and loaded on a gel for Western blot analysis. The insoluble pellet in each tube was washed once with milliQ-H2O and mixed with 50 μl of Laemmli buffer (75 mm Tris-HCl with pH 6.8, 3% SDS, 15% glycerol, 3.75 mm EDTA, pH 7.4). The insoluble fraction samples were further sonicated using a rod sonicator (Labsonic® M.B. Braun Biotech International) for 2 × 1 s at 0.5 cycle and an amplitude of 60% for solubilizing the pellets. Then the insoluble protein fraction was solubilized in gel-loading buffer containing 0.25% mercaptoethanol and loaded on a gel for Western blot analysis.
Native Polyacrylamide Gel Electrophoresis (PAGE)
Native-PAGE was carried out as described earlier in Szeltner et al. (29) by leaving out SDS from the PAGE-gel, sample buffer (Bio-Rad), and Tris-glycine running buffer. 6 μg of purified recombinant PREP or PREP(S554A) proteins were incubated with a 30-fold molar excess of KYP-2047 or vehicle (0.15% DMSO in PBS) for 30 min at room temperature. Samples were run on 10% native gel containing a stacking gel. The gel was then fixed for 30 min in destaining buffer (45% methanol, 10% acetic acid), stained with 0.1% Coomassie Brilliant Blue R-250 for 2 h, and destained 3–4 times for 30 min.
PREP Activity Assay
PREP activity assay was used to confirm the enzymatic activity of various PREP constructs, and it was performed as described in Myöhänen et al. (22, 30). Briefly, the cells transfected with various PREP constructs were homogenized with lysis buffer (50 mm KH2PO4, 1.5 mm MgCl2, 10 mm NaCl, 1 mm EDTA, pH 7.4), and the homogenates were centrifuged at 16,000 × g for 10 min at +4 °C. The homogenate was preincubated with assay buffer for 30 min at 37 °C. Then the substrate (4 mm Suc-Gly-Pro-7-amino-4-methylcoumarin) was added to initiate the reaction, and the incubation was continued for 60 min at 37 °C. The reaction was stopped with 1 m sodium acetate buffer, pH 4.2. The formation of 7-amino-4-methylcoumarin was measured using a Wallac 1420 Victor fluorescence plate reader (PerkinElmer Life Sciences). The excitation and emission wavelengths were 360 and 460 nm, respectively. The protein concentration of the cell homogenate was determined using BCA protein assay kit (Thermo Scientific).
Microscale Thermophoresis (MST)
MST was performed to study protein-protein interaction using purified recombinant proteins. Recombinant porcine PREP and PREP(S554A) proteins were labeled with the Monolith NT.115 protein labeling kit (NanoTemper Technologies GmbH, München, Germany) using red fluorescent dye NT-647 N-hydroxysuccinimide (amine-reactive) according to the manufacturer's instructions. Labeling reagents were removed by buffer-exchange column chromatography, and PREP and PREP(S554A) were eluted in PBS with 0.05% Tween 20. Binding assays were performed with a Monolith NT.115 Microscale Thermophoresis device using standard treated capillaries (both device and capillaries were from NanoTemper Technologies). To improve the accuracy of the Kd determination while giving a fluorescence signal above 200 units, the concentration of labeled protein was kept to a minimum (range 1–100 nmol) by diluting in assay buffer (final concentration of 0.05% Tween 20, 0.5 mg/ml bovine serum albumin (BSA), and 2.5 mm DTT in PBS) and modifying the LED power between 20 and 60%. Equal amounts of labeled protein were titrated by purified recombinant human aSyn (Sigma, #S7820) in a 1:1 series dilution starting from 35 μm or with KYP-2047 in 10:1 and 1:1 series dilution starting from 10 μm. Curve-fitting was done by NTanalysis software (NanoTemper Technologies) in the Thermophoresis + T-jump mode. Assimilated Curves generated from three replicate binding assays were analyzed in GraphPad Prism software to calculate the Kd values using non-linear regression and one site-specific binding with the Hill slope.
Surface Plasmon Resonance (SPR)
SPR sensor hydrogel was prepared according to the instructions of the instrument manufacturer (BioNavis Ltd., Tampere, Finland). The immobilization of PREP protein was performed at 20 °C, a 20 μl/min flow rate, and with 5 mm MES, pH 5.0, as the running buffer. PREP protein injection solution was 33 μg/ml in 5 mm pH 4.75 acetate buffer prepared fresh from freezer aliquots immediately before injection. The immobilization cycle was as follows. The sensor surface was cleaned with 2 m NaCl + 10 mm NaOH cleaning solution. The hydrogel binding of PREP was tested with a preconcentration injection. The surface was then cleaned again with the same cleaning solution. Then the surface was activated with l-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC)/N-hydroxysuccinimide (NHS) solution (of concentration 20/5 mg/ml), and PREP was injected in one of the flow channels; other flow channels remained as the reference channel. The surface was then deactivated with 1 m ethanolamine, pH 8.5, solution. The immobilization resulted with 140–400 ng/mm2 protein immobilized on the surface. The specificity of PREP sensor was tested with macroglobulin (1, 10, and 40 μg/ml at 30 μl/min) and BSA injections. No unspecific binding was detected.
A recombinant human aSyn (Sigma, #S7820) sample was run at 20 μl/min. The binding of α-synuclein was tested in logarithmic dilution 0.5, 5, and 50 μg/ml, which were diluted from a 500 μg/ml stock. The binding was analyzed with TraceDrawer 1.3 for BioNavis (Ridgeview Instruments AB, Uppsala, Sweden) for binding constants. The unspecific binding of aSyn and PREP was tested with a macroglobulin sensor that was prepared in same manner as the PREP sensor, except that 50 μg/ml macroglobulin solution was used. This resulted in 100 ng/mm2 protein immobilized on the sensor. No unspecific binding of aSyn or PREP was detected (data not shown).
Statistical Analyses
Statistical analyses were carried out using Student's t test (two groups) or one-way ANOVA with Bonferroni's post-tests (three or more groups) in GraphPad Prism software. Significance was set at p < 0.05.
RESULTS
PREP Overexpression Increases aSyn Reporter Dimerization in N2A Cells
PREP inhibition was previously shown to reduce chronic oxidative stress-induced accumulation of aSyn aggregates in SH-SY5Y neuroblastoma cells (22). To study the effects of PREP in the earliest stages of the aSyn aggregation process, we first developed an assay for studying aSyn oligomer formation in live cells. This system is based on PCA where complementary fragments of G. princeps luciferase (GLuc) are attached to interacting proteins of interest and a luminescence signal is generated upon protein interaction in live cells (31). We first expressed the aSyn-GLuc1 and aSyn-GLuc2 PCA reporter constructs in cells together with wild-type PREP and PREP(S554A). In N2A neuroblastoma cells, which show no detectable endogenous PREP expression, there was a subtle increase in aSyn levels upon co-expression of PREP or PREP(S554A), a catalytically inactive mutant of PREP, with the aSyn-GLuc reporters (Fig. 1, A and B) at 48 h post-transfection. We did not observe signs of aSyn cleavage in PREP-expressing cells.
FIGURE 1.
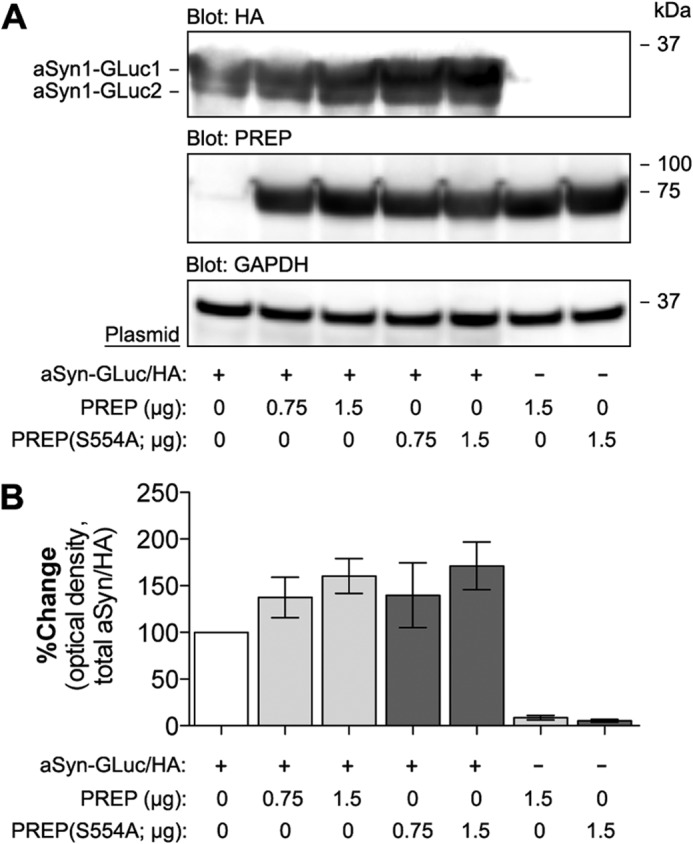
Co-expression of PREP increases levels of aSyn in N2A cells. A, the expression of PREP and PREP(S554A) together with GLuc-fragment- and HA-tagged aSyn was studied by Western blot. aSyn-GLuc1 and aSyn-GLuc2 were transfected at 0.75 μg of plasmid DNA per well (each plasmid), whereas PREP and PREP(S554A) were transfected either at 0.75 μg or 1.5 μg. The total amount of plasmid DNA was set at 3 μg per well and was adjusted by mock plasmid when needed. Cells transfected with aSyn-GLuc1 and aSyn-GLuc2 plasmids only were used as the control. Blots were stained with antibodies to PREP, HA-tag (aSyn-GLuc1/2), and GAPDH as a loading control. B, optical density-based quantification of aSyn levels in four replicate experiments (as in Fig. 1A). Both PREP and PREP(S554A) showed a trend toward increased aSyn levels, but the difference did not reach significance. The average values are displayed as the percent change as compared with control-transfected cells (mean ± S.E.; n = 4 independent experiments).
Next, we measured the PCA signal in N2A cells coexpressing the aSyn-GLuc reporters and PREP or PREP(S554A). As shown in Fig. 2B, increasing levels of PREP or PREP(S554A) in cells strongly promoted complementation with the maximal effects reaching 193 and 247% induction for PREP and PREP(S554A), respectively. The addition of PREP inhibitor KYP-2047 to the cells reduced PREP-induced aSyn complementation only in cells expressing the wild-type PREP but not in PREP(S554A) cells (Fig. 2C). KYP-2047 had no effect on aSyn reporter dimerization in control-transfected cells, suggesting that the effect of KYP-2047 is PREP-specific and requires expression of catalytically active PREP (Fig. 2D).
To confirm that the cell-based assay system and selected time points reflect the early events in the aSyn-oligomerization process, not the formation of aSyn inclusions, we extracted cells with 1% Triton X-100 for cellular fractionation and analyzed Triton-soluble and insoluble fractions on Western blot. As shown in Fig. 2E, expression of PREP or PREP(S554A) or the treatment with KYP-2047 had no effect on aSyn solubility in N2A cells at 48 h post-transfection. This further supports that in N2A cells at these early time points, with no additional stress stimuli, the aSyn-GLuc reporters remain almost entirely in the soluble fraction.
Interaction of PREP and aSyn in Live Cells
To address the potential interaction of PREP with aSyn, we generated another PCA reporter, PREP-GLuc2, carrying an N-terminal GLuc2 tag (Fig. 3A). The expression and enzymatic activity of PREP-GLuc2 was assessed to confirm the functionality of the construct. Although the PREP-GLuc2 reporter is expressed at a lower level in N2A cells as compared with the untagged wild-type PREP (Fig. 3B), the PREP-GLuc2 reporter maintains hydrolytic activity toward a fluorescently labeled substrate peptide (Fig. 3C). As shown in Fig. 3D, expression of aSyn-GLuc1 or PREP-GLuc PCA reporters alone in cells generates only background luminescence signal. However, co-expression of PREP-GLuc2 or PREP(S554A)-GLuc2 reporters with aSyn-GLuc1 reporter results in generation of significant PCA signal suggesting that these proteins are found in close enough proximity (likely <10Å) in cells to allow refolding of the GLuc reporter fragments into an active luciferase reporter protein. Interestingly, PREP(S554A) shows >30% higher PCA signal with aSyn as compared with the wild-type PREP-GLuc2-expressing cells. We also verified the specificity of the aSyn-GLuc1 reporter-derived PCA signal with GLuc2-tagged GSAP(16K), a cytoplasmic protein that interacts with γ-secretase and the cytosolic domain of the amyloid precursor protein (APP) (32). GSAP(16k)-GLuc2 produced a significant PCA signal with the APP-C98-GLuc1 control but not with aSyn-GLuc1 (Fig. 3D).
Next, we tested the effect of KYP-2047 on the PREP-aSyn interaction. KYP-2047 binds to both PREP and PREP(S554A) (our MST data, not shown). Interestingly, KYP-2047 strongly increased the PREP-aSyn interaction (+207%) but had very little effect on PREP(S554A)-aSyn interaction (+109%) as compared with vehicle-treated cells expressing the same reporters (Fig. 3E).
Because the PCA data suggest that PREP and aSyn interact in cells, we sought to further characterize the interaction. PREP has hydrolytic specificity toward the C-terminal side of proline residues (33). The acidic 45-residue C-terminal domain of aSyn contains all five proline residues of the wild-type aSyn protein. We generated C-terminal truncation mutants of the aSyn-GLuc1 reporter for further interaction studies with PREP. The aSyn(Δ118–140) mutant lacks three and the aSyn(Δ98–140) mutant lacks all five proline residues (Fig. 4A). Both truncation mutants are expressed in N2A cells, although at a slightly lower level as compared with the full-length aSyn-GLuc1 reporter protein (Fig. 4B). When the C-terminal truncation mutants of aSyn-GLuc1 were co-expressed with the PREP-GLuc2 reporter, all the reporters generated comparable levels of PCA signals (Fig. 4C). Although the aSyn(Δ98–140)-GLuc1-PREP-GLuc2 reporter pair showed a 29% lower signal as compared with the full-length aSyn-GLuc1-PREP interaction, this could be explained by the somewhat lower level of aSyn(Δ98–140)-GLuc1 reporter protein expression (Fig. 4B). Altogether, the PCA data suggest that PREP and aSyn can interact in intact cells and that neither the C-terminal proline residues of aSyn nor the enzyme activity of PREP is necessary for the interaction.
Direct Interaction of PREP and aSyn in a Cell-free System
To confirm a direct interaction of PREP and aSyn using purified proteins in a cell-free system, we turned to MST, a novel method for quantitative analysis of protein interactions in free solution (34, 35). Recombinant PREP and PREP(S554A) proteins were labeled with the red fluorescent dye NT-647. The addition of recombinant unlabeled aSyn protein to the MST capillaries containing wild-type PREP shows the interaction between the two proteins, with a Kd of 2.96 μm (Fig. 5A). In a similar MST experiment, recombinant PREP(S554A) also interacts with aSyn, with a Kd value of 1.41 μm (Fig. 5B). Fluorescence time traces detected by the MST reader suggest low or no aggregation of the proteins during the experiments (insets in the Fig. 5, A and B). These data confirm that there is a direct interaction between PREP and aSyn, with a binding affinity in the low micromolar range. Interestingly, similar to the PCA experiments, the cell-free MST experiments also suggest that the PREP(S554A) mutant has a slightly higher affinity for aSyn than the wild-type PREP.
FIGURE 5.
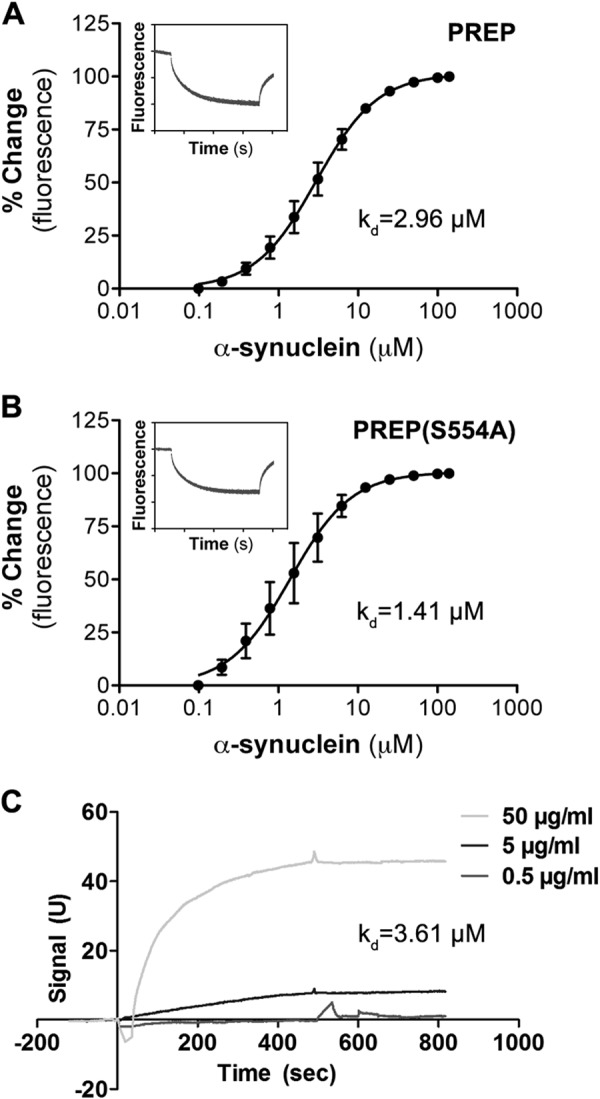
Microscale thermophoresis and surface plasmon resonance confirm a direct interaction between PREP and aSyn. The interactions between aSyn and PREP (A) and between aSyn and PREP(S554A) (B) were studied by MST. Equal amounts of fluorescently labeled (NT-647) recombinant PREP or PREP(S554A) were titrated by recombinant unlabeled aSyn at various concentrations. Similar to PCA results, aSyn binds to PREP and PREP(S554A) with a Kd of ∼2.96 and ∼1.41 μm, respectively. Fluorescence time traces taken from single experiment suggest low or no aggregation had occurred in both sets of experiments (insets in both graphs). The curve and Kd values were calculated by averaging Kd curves assimilated using NTanalysis software from three independent experiments (mean ± S.E.). C, a time versus signal plot of SPR measurement is shown, normalized by Bmax of the first order analysis. Three doses of aSyn were used in the injections (0.5, 5, and 50 μg/ml). Kd determined by SPR was 3.61 μm, and it was the mean of three independent experiments.
The interaction between PREP and aSyn that was seen in MST was also confirmed with SPR. In the SPR experiments the PREP sensor showed a clear signal with aSyn, and an affinity plot was created from three concentrations (Fig. 5C). The Kd value of the interaction was 3.61 μm (average of three assays), which was similar to the Kd value obtained from MST experiments. aSyn did not show any interaction with a macroglobulin sensor, and the PREP sensor did not react with macroglobulin or BSA, suggesting that the binding was specific. We also studied the interaction between PREP(S554A) and aSyn in SPR, but despite repeated trials PREP(S554A)-aSyn interaction was not detected by this method.
KYP-2047 Modifies Conformational Forms of Wild-type but Not Inactive PREP Mutant
It has been previously shown that KYP-2047 binding modifies conformation of PREP (29). Our data suggested that KYP-2047 binds both to PREP and PREP(S554A) but showed a decrease in aSyn reporter dimerization in PCA only when incubated with active PREP. We hypothesized that the wild-type and mutant PREP proteins may have differences in their conformational forms or KYP-2047 could modify them differently.
When wild-type PREP was applied on a native gel, three bands representing different conformational states were seen (Fig. 6). Incubation with KYP-2047 before gel application shifted wild-type PREP in a single faster migrating band, possibly representing monomeric PREP in a compact conformation (Fig. 6). Three bands were visible also for PREP(S554A), but the main form appears to migrate slightly faster as compared with wild-type PREP. Importantly, incubation with KYP-2047 had no effect on the three conformations of PREP(S554A) (Fig. 6). These data show that PREP can adopt multiple conformational states that are sensitive to inhibitor binding. The differential sensitivity of wild-type PREP and the S554A mutant to inhibitor-induced conformation shifting offers a potential explanation of why these PREP forms have different effects on aSyn dimerization in the presence of KYP-2047.
FIGURE 6.
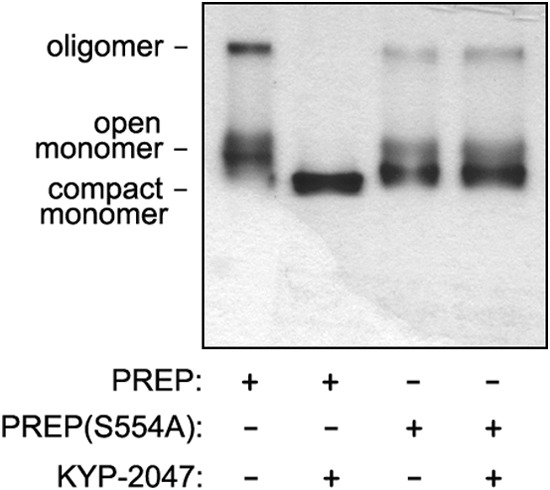
KYP-2047 alters the conformational forms of active PREP but not PREP(S554A). Recombinant PREP and PREP(S554A) proteins on native-PAGE revealed three different conformations that likely represent different open/close monomeric and oligomeric forms of the protein. However, inhibition of PREP with KYP-2047 shifts the slower migrating conformations into a single band, likely representing closed monomeric conformation of PREP. PREP(S554A) had similarly three conformations, but they were not affected by KYP-2047.
DISCUSSION
It was previously shown that PREP accelerates aSyn aggregation and that a PREP inhibitor blocks this in vitro and promotes clearance of aSyn aggregates in aSyn-overexpressing cells and in vivo models (14, 22). However, although PREP increased the aggregation of aSyn in a cell-free model, the interaction between PREP and aSyn has not been documented earlier. In the current paper we have now shown that PREP directly interacts with aSyn and increases its dimerization and that this effect can be countered with a specific PREP inhibitor, KYP-2047.
aSyn is known to interact with a variety of proteins and small molecules, such as tyrosine hydroxylase and tau, metal ions, and lipids (36–40). The N-terminal and central NAC domains of aSyn interact with lipids in cellular membranes and with some proteins (41–44), whereas the C-terminal domain was shown to bind metal ions (38). aSyn interactions with proteins, such as synphilin-1 (45) and tubulin (46), small molecules, and macromolecules, such as dopamine (47), polysaccharides (48), and metal ions (11) can enhance aggregation of aSyn. Because the aggregation process is nucleation-dependent (12, 49, 50), it is possible that some of these interaction partners function as a seeding point for aSyn oligomerization and aggregation.
In this study we have shown that PREP increases aSyn dimerization in intact cells in its native cellular environment and that it directly interacts with aSyn in vitro. This suggests that PREP acts as a seeding point for aSyn dimerization, but unlike several other aSyn-interacting proteins with aggregation-promoting properties, PREP-induced dimerization and aggregation can be blocked with specific PREP inhibitors (14, 22, 23, 51). PREP expression also increased the levels of soluble aSyn in cells that may further promote the aggregation of aSyn, as a higher concentration is a trigger for the aggregation process (12). In a previous study it was shown that PREP colocalizes with aSyn during the aggregation process in aSyn-overexpressing cells, and it was hypothesized that PREP inhibition could disturb the interaction thus leading to reduced aSyn aggregation (22).
Based on our data, it seems that PREP inhibition promotes its interaction with aSyn while decreasing aSyn dimerization in cells. Previously, it was demonstrated that PREP adopts three distinct conformations, a compact monomeric form, an open monomeric form, and an oligomeric form, and that KYP-2047 has a prominent effect on the conformational state of PREP (29, 52, 53). Our data confirm that in the presence of KYP-2047, the majority of PREP oligomers adopt the compact monomeric form. In living cells, KYP-2047 does not abolish the physical interaction between aSyn and PREP, as we hypothesized earlier (22), but changes its characteristics so that the stimulating effect on aSyn oligomerization is decreased. These observations are in line with earlier studies that indicated that PREP acts on the nucleation process of aSyn aggregation and fibril formation and that the conformational state of PREP is important for its functions inside the cell (14, 29, 54). However, whether the increased PCA signal between PREP and aSyn in KYP-2047-treated cells reflects higher affinity of inhibitor-bound monomeric PREP toward aSyn remains to be determined in future studies.
Expression of PREP(S554A) mutant in cells increased aSyn reporter dimerization similarly to active PREP, but the addition of KYP-2047 had no effect on either aSyn dimerization or aSyn-PREP(S554A) interaction. Contrary to wild-type PREP, KYP-2047 had no effect on the conformational states of PREP(S554A). This offers a plausible explanation why the PREP(S554A) mutant is insensitive to KYP-2047 regarding enhancement of aSyn dimerization. Although we failed in our attempts to isolate oligomeric and monomeric pools of PREP for testing their effects on aSyn separately, the current data suggests that aSyn nucleation is mediated by the open monomeric or oligomeric PREP, independently of its enzymatic activity, and that KYP-2047 interferes with aSyn nucleation by dissociating PREP oligomers and stabilizing the closed monomeric form.
It is not known what site in the PREP structure could act as a binding site for other proteins. One possibility could be the access loop structures of PREP as suggested earlier in the literature (29, 55). These structures contain three loops (A, B, and C), of which loop A seems to be the most important for substrate gating, and it changes its conformation to a substrate-inaccessible position in the presence of an inhibitor (55). This may in part explain the conformational change of PREP by KYP-2047 that we observed in native gel. Furthermore, based on our current results, it appears that the S554A mutation stabilizes the open conformation and probably also the interdomain loops of PREP, although it lacks the enzymatic activity.
We also attempted to define the PREP-binding site in aSyn by using C-terminal truncation mutants of aSyn. The C terminus of aSyn is proline-rich, and PREP has hydrolytic specificity toward the C-terminal side of proline residue (33), suggesting a potential interaction point. In addition, conformational changes in the aSyn C-terminal prolines are known to enhance aSyn aggregation (56). Using the C-terminal truncation mutants of aSyn, we showed that the proline-rich domain of aSyn is not required for PREP interaction. These data suggest that the PREP interaction is determined mostly by the N-terminal and NAC domains of aSyn.
It appears that PREP has a dual effect on the aSyn aggregation process; it enhances the formation of soluble aSyn oligomers in a process that involves a direct PREP-aSyn interaction, and it also negatively affects autophagy, a catabolic pathway used for clearance of insoluble aSyn oligomers and fibrils (23). Importantly, a small molecule PREP inhibitor affects both of these PREP actions that modulate accumulation of aSyn aggregates. Our current data show that PREP inhibition reduces dimerization of aSyn via a direct protein-protein interaction. Moreover, PREP inhibition increases the clearance of aSyn aggregates by increasing autophagosome formation via beclin1 (23). Taken together, these results strongly support PREP inhibition as a potential new therapy for synucleinopathies.
Acknowledgments
The plasmids encoding the split humanized GLuc fragments were kind gifts from Prof. Stephen Michnick (Université de Montréal, Canada). We thank Prof. Anne-Marie Lambeir (University of Antwerpen, Belgium) for PREP proteins, for inspiring and stimulating this research, and for proofreading the manuscript. We also thank Dr. Tapani Viitala and Dr. Niko Granqvist for excellent assistance on SPR experiments.
This work was supported by grants from the Academy of Finland (218081 and 263762 (to H. J. H.) and 267788 and 2737991 (to T. T. M.)), the Brain and Mind doctoral program (to X. Y.), the Doctoral Program in Drug Research (to M. S.), the University of Helsinki (to H. J. H. and T. T. M.), the Jane and Aatos Erkko Foundation (to T. T. M.), the Sigrid Juselius Foundation (to T. T. M.), and the Emil Aaltonen Foundation (to M. S.). H. J. H. is an employee and shareholder of Herantis Pharma plc.
- aSyn
- α-synuclein
- GLuc
- humanized G. princeps luciferase
- MST
- microscale thermophoresis
- PCA
- protein-fragment complementation assay
- PD
- Parkinson disease
- PREP
- prolyl oligopeptidease (prolyl endopeptidase)
- SPR
- surface plasmon resonance
- N2A
- Neuro-2A
- Bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- ANOVA
- analysis of variance
- APP
- amyloid precursor protein.
REFERENCES
- 1. Spillantini M. G., Schmidt M. L., Lee V. M., Trojanowski J. Q., Jakes R., Goedert M. (1997) α-Synuclein in Lewy bodies. Nature 388, 839–840 [DOI] [PubMed] [Google Scholar]
- 2. Bennett M. C. (2005) The role of α-synuclein in neurodegenerative diseases. Pharmacol. Ther. 105, 311–331 [DOI] [PubMed] [Google Scholar]
- 3. Arima K., Uéda K., Sunohara N., Arakawa K., Hirai S., Nakamura M., Tonozuka-Uehara H., Kawai M. (1998) NACP/α-synuclein immunoreactivity in fibrillary components of neuronal and oligodendroglial cytoplasmic inclusions in the pontine nuclei in multiple system atrophy. Acta Neuropathol. 96, 439–444 [DOI] [PubMed] [Google Scholar]
- 4. Zarranz J. J., Alegre J., Gómez-Esteban J. C., Lezcano E., Ros R., Ampuero I., Vidal L., Hoenicka J., Rodriguez O., Atarés B., Llorens V., Gomez Tortosa E., del Ser T., Muñoz D. G., de Yebenes J. G. (2004) The new mutation, E46K, of α-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 55, 164–173 [DOI] [PubMed] [Google Scholar]
- 5. Polymeropoulos M. H., Lavedan C., Leroy E., Ide S. E., Dehejia A., Dutra A., Pike B., Root H., Rubenstein J., Boyer R., Stenroos E. S., Chandrasekharappa S., Athanassiadou A., Papapetropoulos T., Johnson W. G., Lazzarini A. M., Duvoisin R. C., Di Iorio G., Golbe L. I., Nussbaum R. L. (1997) Mutation in the α-synuclein gene identified in families with Parkinson's disease. Science 276, 2045–2047 [DOI] [PubMed] [Google Scholar]
- 6. Conway K. A., Harper J. D., Lansbury P. T. (1998) Accelerated in vitro fibril formation by a mutant α-synuclein linked to early-onset Parkinson disease. Nat. Med. 4, 1318–1320 [DOI] [PubMed] [Google Scholar]
- 7. Krüger R., Kuhn W., Müller T., Woitalla D., Graeber M., Kösel S., Przuntek H., Epplen J. T., Schöls L., Riess O. (1998) Ala30Pro mutation in the gene encoding α-synuclein in Parkinson's disease. Nat. Genet. 18, 106–108 [DOI] [PubMed] [Google Scholar]
- 8. Satake W., Nakabayashi Y., Mizuta I., Hirota Y., Ito C., Kubo M., Kawaguchi T., Tsunoda T., Watanabe M., Takeda A., Tomiyama H., Nakashima K., Hasegawa K., Obata F., Yoshikawa T., Kawakami H., Sakoda S., Yamamoto M., Hattori N., Murata M., Nakamura Y., Toda T. (2009) Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson's disease. Nat. Genet. 41, 1303–1307 [DOI] [PubMed] [Google Scholar]
- 9. Surguchov A. (2008) Molecular and cellular biology of synucleins. Int. Rev. Cell. Mol. Biol. 270, 225–317 [DOI] [PubMed] [Google Scholar]
- 10. Bisaglia M., Mammi S., Bubacco L. (2009) Structural insights on physiological functions and pathological effects of α-synuclein. FASEB J. 23, 329–340 [DOI] [PubMed] [Google Scholar]
- 11. Uversky V. N., Li J., Fink A. L. (2001) Metal-triggered structural transformations, aggregation, and fibrillation of human α-synuclein. A possible molecular NK between Parkinson's disease and heavy metal exposure. J. Biol. Chem. 276, 44284–44296 [DOI] [PubMed] [Google Scholar]
- 12. Wood S. J., Wypych J., Steavenson S., Louis J. C., Citron M., Biere A. L. (1999) α-Synuclein fibrillogenesis is nucleation-dependent. Implications for the pathogenesis of Parkinson's disease. J. Biol. Chem. 274, 19509–19512 [DOI] [PubMed] [Google Scholar]
- 13. Mosharov E. V., Larsen K. E., Kanter E., Phillips K. A., Wilson K., Schmitz Y., Krantz D. E., Kobayashi K., Edwards R. H., Sulzer D. (2009) Interplay between cytosolic dopamine, calcium, and α-synuclein causes selective death of substantia nigra neurons. Neuron 62, 218–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Brandt I., Gérard M., Sergeant K., Devreese B., Baekelandt V., Augustyns K., Scharpé S., Engelborghs Y., Lambeir A. M. (2008) Prolyl oligopeptidase stimulates the aggregation of α-synuclein. Peptides 29, 1472–1478 [DOI] [PubMed] [Google Scholar]
- 15. Myöhänen T. T., García-Horsman J. A., Tenorio-Laranga J., Männistö P. T. (2009) Issues about the physiological functions of prolyl oligopeptidase based on its discordant spatial association with substrates and inconsistencies among mRNA, protein levels, and enzymatic activity. J. Histochem. Cytochem. 57, 831–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. García-Horsman J. A., Männistö P. T., Venäläinen J. I. (2007) On the role of prolyl oligopeptidase in health and disease. Neuropeptides 41, 1–24 [DOI] [PubMed] [Google Scholar]
- 17. Brandt I., Scharpé S., Lambeir A. M. (2007) Suggested functions for prolyl oligopeptidase: a puzzling paradox. Clin. Chim. Acta 377, 50–61 [DOI] [PubMed] [Google Scholar]
- 18. Mantle D., Falkous G., Ishiura S., Blanchard P. J., Perry E. K. (1996) Comparison of proline endopeptidase activity in brain tissue from normal cases and cases with Alzheimer's disease, Lewy body dementia, Parkinson's disease, and Huntington's disease. Clin. Chim. Acta 249, 129–139 [DOI] [PubMed] [Google Scholar]
- 19. Männisto P. T., Venäläinen J., Jalkanen A., García-Horsman J. A. (2007) Prolyl oligopeptidase: a potential target for the treatment of cognitive disorders. Drug News Perspect. 20, 293–305 [DOI] [PubMed] [Google Scholar]
- 20. Lambeir A. M. (2011) Interaction of prolyl oligopeptidase with α-synuclein. CNS Neurol. Disord. Drug Targets 10, 349–354 [DOI] [PubMed] [Google Scholar]
- 21. Hannula M. J., Myöhänen T. T., Tenorio-Laranga J., Männistö P. T., Garcia-Horsman J. A. (2013) Prolyl oligopeptidase colocalizes with α-synuclein, β-amyloid, τ protein, and astroglia in the post-mortem brain samples with Parkinson's and Alzheimer's diseases. Neuroscience 242, 140–150 [DOI] [PubMed] [Google Scholar]
- 22. Myöhänen T. T., Hannula M. J., Van Elzen R., Gerard M., Van Der Veken P., García-Horsman J. A., Baekelandt V., Männistö P. T., Lambeir A. M. (2012) A prolyl oligopeptidase inhibitor, KYP-2047, reduces α-synuclein protein levels and aggregates in cellular and animal models of Parkinson's disease. Br. J. Pharmacol. 166, 1097–1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Savolainen M. H., Richie C. T., Harvey B. K., Männistö P. T., Maguire-Zeiss K. A., Myöhänen T. T. (2014) The beneficial effect of a prolyl oligopeptidase inhibitor, KYP-2047, on α-synuclein clearance and autophagy in A30P transgenic mouse. Neurobiol. Dis. 68, 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Venäläinen J. I., Garcia-Horsman J. A., Forsberg M. M., Jalkanen A., Wallén E. A., Jarho E. M., Christiaans J. A., Gynther J., Männistö P. T. (2006) Binding kinetics and duration of in vivo action of novel prolyl oligopeptidase inhibitors. Biochem. Pharmacol. 71, 683–692 [DOI] [PubMed] [Google Scholar]
- 25. Jarho E. M., Venäläinen J. I., Huuskonen J., Christiaans J. A., Garcia-Horsman J. A., Forsberg M. M., Järvinen T., Gynther J., Männistö P. T., Wallén E. A. (2004) A cyclopent-2-enecarbonyl group mimics proline at the P2 position of prolyl oligopeptidase inhibitors. J. Med. Chem. 47, 5605–5607 [DOI] [PubMed] [Google Scholar]
- 26. Jalkanen A. J., Hakkarainen J. J., Lehtonen M., Venäläinen T., Kääriäinen T. M., Jarho E., Suhonen M., Forsberg M. M. (2011) Brain pharmacokinetics of two prolyl oligopeptidase inhibitors, JTP-4819 and KYP-2047, in the rat. Basic Clin. Pharmacol. Toxicol. 109, 443–451 [DOI] [PubMed] [Google Scholar]
- 27. Jalkanen A. J., Piepponen T. P., Hakkarainen J. J., De Meester I., Lambeir A. M., Forsberg M. M. (2012) The effect of prolyl oligopeptidase inhibition on extracellular acetylcholine and dopamine levels in the rat striatum. Neurochem. Int. 60, 301–309 [DOI] [PubMed] [Google Scholar]
- 28. Nykänen N. P., Kysenius K., Sakha P., Tammela P., Huttunen H. J. (2012) γ-Aminobutyric acid type A (GABAA) receptor activation modulates τ phosphorylation. J. Biol. Chem. 287, 6743–6752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Szeltner Z., Juhász T., Szamosi I., Rea D., Fülöp V., Módos K., Juliano L., Polgár L. (2013) The loops facing the active site of prolyl oligopeptidase are crucial components in substrate gating and specificity. Biochim. Biophys. Acta 1834, 98–111 [DOI] [PubMed] [Google Scholar]
- 30. Myöhänen T. T., Venäläinen J. I., García-Horsman J. A., Piltonen M., Männistö P. T. (2008) Distribution of prolyl oligopeptidase in the mouse whole-body sections and peripheral tissues. Histochem. Cell Biol. 130, 993–1003 [DOI] [PubMed] [Google Scholar]
- 31. Remy I., Michnick S. W. (2006) A highly sensitive protein-protein interaction assay based on Gaussia luciferase. Nat. Methods 3, 977–979 [DOI] [PubMed] [Google Scholar]
- 32. He G., Luo W., Li P., Remmers C., Netzer W. J., Hendrick J., Bettayeb K., Flajolet M., Gorelick F., Wennogle L. P., Greengard P. (2010) γ-Secretase activating protein is a therapeutic target for Alzheimer's disease. Nature 467, 95–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Walter R., Shlank H., Glass J. D., Schwartz I. L., Kerenyi T. D. (1971) Leucylglycinamide released from oxytocin by human uterine enzyme. Science 173, 827–829 [DOI] [PubMed] [Google Scholar]
- 34. Wienken C. J., Baaske P., Rothbauer U., Braun D., Duhr S. (2010) Protein-binding assays in biological liquids using microscale thermophoresis. Nat. Commun. 1, 100. [DOI] [PubMed] [Google Scholar]
- 35. Seidel S. A., Dijkman P. M., Lea W. A., van den Bogaart G., Jerabek-Willemsen M., Lazic A., Joseph J. S., Srinivasan P., Baaske P., Simeonov A., Katritch I., Melo F. A., Ladbury J. E., Schreiber G., Watts A., Braun D., Duhr S. (2013) Microscale thermophoresis quantifies biomolecular interactions under previously challenging conditions. Methods 59, 301–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Perez R. G., Waymire J. C., Lin E., Liu J. J., Guo F., Zigmond M. J. (2002) A role for α-synuclein in the regulation of dopamine biosynthesis. J. Neurosci. 22, 3090–3099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jensen P. H., Hager H., Nielsen M. S., Hojrup P., Gliemann J., Jakes R. (1999) α-Synuclein binds to τ and stimulates the protein kinase A-catalyzed τ phosphorylation of serine residues 262 and 356. J. Biol. Chem. 274, 25481–25489 [DOI] [PubMed] [Google Scholar]
- 38. Bisaglia M., Tessari I., Mammi S., Bubacco L. (2009) Interaction between α-synuclein and metal ions, still looking for a role in the pathogenesis of Parkinson's disease. Neuromolecular Med. 11, 239–251 [DOI] [PubMed] [Google Scholar]
- 39. Lai Y., Kim S., Varkey J., Lou X., Song J. K., Diao J., Langen R., Shin Y. K. (2014) Nonaggregated α-synuclein influences SNARE-dependent vesicle docking via membrane binding. Biochemistry 53, 3889–3896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jo E., McLaurin J., Yip C. M., St George-Hyslop P., Fraser P. E. (2000) α-Synuclein membrane interactions and lipid specificity. J. Biol. Chem. 275, 34328–34334 [DOI] [PubMed] [Google Scholar]
- 41. Jensen P. H., Hojrup P., Hager H., Nielsen M. S., Jacobsen L., Olesen O. F., Gliemann J., Jakes R. (1997) Binding of Aβ to α- and β-synucleins: identification of segments in α-synuclein/NAC precursor that bind Aβ and NAC. Biochem. J. 323, 539–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pfefferkorn C. M., Jiang Z., Lee J. C. (2012) Biophysics of α-synuclein membrane interactions. Biochim. Biophys. Acta 1818, 162–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lorenzen N., Lemminger L., Pedersen J. N., Nielsen S. B., Otzen D. E. (2014) The N terminus of α-synuclein is essential for both monomeric and oligomeric interactions with membranes. FEBS Lett. 588, 497–502 [DOI] [PubMed] [Google Scholar]
- 44. Lee D., Lee S. Y., Lee E. N., Chang C. S., Paik S. R. (2002) α-Synuclein exhibits competitive interaction between calmodulin and synthetic membranes. J. Neurochem. 82, 1007–1017 [DOI] [PubMed] [Google Scholar]
- 45. Engelender S., Kaminsky Z., Guo X., Sharp A. H., Amaravi R. K., Kleiderlein J. J., Margolis R. L., Troncoso J. C., Lanahan A. A., Worley P. F., Dawson V. L., Dawson T. M., Ross C. A. (1999) Synphilin-1 associates with α-synuclein and promotes the formation of cytosolic inclusions. Nat. Genet. 22, 110–114 [DOI] [PubMed] [Google Scholar]
- 46. Alim M. A., Hossain M. S., Arima K., Takeda K., Izumiyama Y., Nakamura M., Kaji H., Shinoda T., Hisanaga S., Ueda K. (2002) Tubulin seeds α-synuclein fibril formation. J. Biol. Chem. 277, 2112–2117 [DOI] [PubMed] [Google Scholar]
- 47. Cappai R., Leck S. L., Tew D. J., Williamson N. A., Smith D. P., Galatis D., Sharples R. A., Curtain C. C., Ali F. E., Cherny R. A., Culvenor J. G., Bottomley S. P., Masters C. L., Barnham K. J., Hill A. F. (2005) Dopamine promotes α-synuclein aggregation into SDS-resistant soluble oligomers via a distinct folding pathway. FASEB J. 19, 1377–1379 [DOI] [PubMed] [Google Scholar]
- 48. Uversky V. N., M Cooper E., Bower K. S., Li J., Fink A. L. (2002) Accelerated α-synuclein fibrillation in crowded milieu. FEBS Lett. 515, 99–103 [DOI] [PubMed] [Google Scholar]
- 49. Uversky V. N., Li J., Fink A. L. (2001) Evidence for a partially folded intermediate in α-synuclein fibril formation. J. Biol. Chem. 276, 10737–10744 [DOI] [PubMed] [Google Scholar]
- 50. Uversky V. N. (2007) Neuropathology, biochemistry, and biophysics of α-synuclein aggregation. J. Neurochem. 103, 17–37 [DOI] [PubMed] [Google Scholar]
- 51. Dokleja L., Hannula M. J., Myöhänen T. T. (2014) Inhibition of prolyl oligopeptidase increases the survival of α-synuclein overexpressing cells after rotenone exposure by reducing α-synuclein oligomers. Neurosci. Lett. 583, 37–42 [DOI] [PubMed] [Google Scholar]
- 52. Szeltner Z., Morawski M., Juhász T., Szamosi I., Liliom K., Csizmók V., Tölgyesi F., Polgár L. (2010) GAP43 shows partial co-localisation but no strong physical interaction with prolyl oligopeptidase. Biochim. Biophys. Acta 1804, 2162–2176 [DOI] [PubMed] [Google Scholar]
- 53. Tarragó T., Martín-Benito J., Sabidó E., Claasen B., Madurga S., Gairí M., Valpuesta J. M., Giralt E. (2009) A new side opening on prolyl oligopeptidase revealed by electron microscopy. FEBS Lett. 583, 3344–3348 [DOI] [PubMed] [Google Scholar]
- 54. Di Daniel E., Glover C. P., Grot E., Chan M. K., Sanderson T. H., White J. H., Ellis C. L., Gallagher K. T., Uney J., Thomas J., Maycox P. R., Mudge A. W. (2009) Prolyl oligopeptidase binds to GAP-43 and functions without its peptidase activity. Mol. Cell. Neurosci. 41, 373–382 [DOI] [PubMed] [Google Scholar]
- 55. Kaszuba K., Róg T., Danne R., Canning P., Fülöp V., Juhász T., Szeltner Z., St Pierre J. F., García-Horsman A., Männistö P. T., Karttunen M., Hokkanen J., Bunker A. (2012) Molecular dynamics, crystallography, and mutagenesis studies on the substrate gating mechanism of prolyl oligopeptidase. Biochimie 94, 1398–1411 [DOI] [PubMed] [Google Scholar]
- 56. Meuvis J., Gerard M., Desender L., Baekelandt V., Engelborghs Y. (2010) The conformation and the aggregation kinetics of α-synuclein depend on the proline residues in its C-terminal region. Biochemistry 49, 9345–9352 [DOI] [PubMed] [Google Scholar]