

Background: The significance of gingipains on the PI3K/Akt signaling pathway is poorly understood.
Results: The inactivation of PI3K and Akt was induced by P. gingivalis, which was associated with gingipains.
Conclusion: Gingipains are critical for suppressing the PI3K/Akt signaling pathway via extracellular proteolysis independent of P. gingivalis invasion.
Significance: This study shows the first evidence that the gingipains negatively regulate the PI3K/Akt signaling pathway.
Keywords: Akt PKB, Phosphatidylinositide 3-Kinase (PI 3-Kinase), Proteolysis, Signal Transduction, Virulence Factor, Porphyromonas gingivalis, Gingipain
Abstract
Porphyromonas gingivalis is a major pathogen of periodontal diseases, including periodontitis. We have investigated the effect of P. gingivalis infection on the PI3K/Akt (protein kinase B) signaling pathway in gingival epithelial cells. Here, we found that live P. gingivalis, but not heat-killed P. gingivalis, reduced Akt phosphorylation at both Thr-308 and Ser-473, which implies a decrease in Akt activity. Actually, PI3K, which is upstream of Akt, was also inactivated by P. gingivalis. Furthermore, glycogen synthase kinase 3α/β, mammalian target of rapamycin, and Bad, which are downstream proteins in the PI3K/Akt cascade, were also dephosphorylated, a phenomenon consistent with Akt inactivation by P. gingivalis. However, these events did not require direct interaction between bacteria and host cells and were independent of P. gingivalis invasion into the cells. The use of gingipain-specific inhibitors and a gingipain-deficient P. gingivalis mutant KDP136 revealed that the gingipains and their protease activities were essential for the inactivation of PI3K and Akt. The associations between the PI3K regulatory subunit p85α and membrane proteins were disrupted by wild-type P. gingivalis. Moreover, PDK1 translocation to the plasma membrane was reduced by wild-type P. gingivalis, but not KDP136, indicating little production of phosphatidylinositol 3,4,5-triphosphate by PI3K. Therefore, it is likely that PI3K failed to transmit homeostatic extracellular stimuli to intracellular signaling pathways by gingipains. Taken together, our findings indicate that P. gingivalis attenuates the PI3K/Akt signaling pathway via the proteolytic effects of gingipains, resulting in the dysregulation of PI3K/Akt-dependent cellular functions and the destruction of epithelial barriers.
Introduction
Porphyromonas gingivalis is an oral Gram-negative anaerobic bacterium that is closely correlated with chronic and asymptomatic periodontal diseases, including periodontitis and alveolar bone loss. The periodontal diseases have a potential relationship with systemic diseases such as aspiration pneumonia, atherosclerosis, and diabetes (1–3). P. gingivalis is one of the most widely studied oral pathogens at the molecular level, and its pathogenicity is attributed to various virulence factors including LPS, fimbriae, hemagglutinins, outer membrane vesicles, and three cysteine proteinases: arginine-specific gingipains A and B (RgpA and RgpB)2 and lysine-specific gingipain (Kgp) (4–7). Recent studies have demonstrated that other molecules of P. gingivalis, specifically SerB and adhesin domains, contribute to its pathogenicity as well (8–10). Gingipains are considered to be the most prominent virulence factors among the various molecules described above, and they greatly contribute to the pathogenesis of periodontal diseases (6, 11–14).
The phosphatidylinositol 3-kinase (PI3K)/Akt signaling pathway is one of the key regulators of various host cell signal transductions such as cell survival, proliferation, differentiation, endocytosis and vesicular trafficking, metabolism, and host inflammatory responses (15, 16). PI3K converts phosphatidylinositol 4,5-biphosphate (PI(4,5)P2) to phosphatidylinositol 3,4,5-triphosphate (PI(3,4,5)P3), and subsequently phosphoinositide-dependent protein kinase 1 (PDK1) and Akt can translocate to the plasma membrane via their pleckstrin homology domain (17–20). Akt is phosphorylated at Thr-308 by PDK1 (19) and at Ser-473 by the mammalian target of rapamycin (mTOR) complex 2 to become fully activated (21). Active Akt is a central node in cell signaling downstream of PI3K by various cellular stimuli, and it modulates the substrates and downstream molecules Bad, caspase-9, glycogen synthase kinase 3 (GSK3) α/β, mTOR, and forkhead transcription factors (22, 23). However, the dysregulation of Akt activity underlies the pathophysiological properties of a variety of diseases, including type 2 diabetes and cancer (22, 24, 25). The PI3K/Akt signaling pathway has also been implicated in bacterial infectious diseases. Interestingly, bacteria and their virulence factors have been shown to regulate this pathway positively or negatively during infection (26–29). P. gingivalis activates the PI3K/Akt signaling pathway, which is linked to cell survival (30, 31) and immune responses (32). Among several known virulence factors of P. gingivalis, both LPS and fimbriae regulate immune responses through PI3K/Akt activation in monocytes/macrophages (5, 33–35) and human gingival fibroblasts (36). Gingipains have the potential to control both cell survival and apoptosis via the PI3K/Akt pathway (37). However, recent reports by other research groups have shown that P. gingivalis infection in the liver inactivates Akt and alters hepatic glycogen synthesis, leading to potential progression of diabetes (38), and that P. gingivalis LPS also represses mucin synthesis and Akt activation (39).
In light of these observations, the PI3K/Akt signaling pathway can be concluded to have pivotal roles in infectious diseases; however, the significance of this pathway has not been sufficiently clarified in P. gingivalis-mediated periodontal diseases. Here, we investigated the effect and role of P. gingivalis infection and gingipains on the PI3K/Akt signaling pathway in human gingival epithelial cells. We found that P. gingivalis infection caused the attenuation of the PI3K/Akt pathway, which was strongly associated with the activities of the gingipains, but P. gingivalis invasion was not required for the attenuation. Our studies revealed a unique function of gingipains as potent negative effectors of the PI3K/Akt signaling pathway.
EXPERIMENTAL PROCEDURES
Cells, Antibodies, and Inhibitors
The human gingival epithelial Ca9-22 cell line was obtained from the Culture Collection of Health Science Research Resources Bank, Japan Health Sciences Foundation, and the cells were grown in MEMα (Wako) containing 10% FCS. Human primary gingival epithelial cells (HGEP; HGEPp.05. lot ES1208166) were obtained from CELLnTec and maintained in CnT-24 culture medium (CELLnTec). Cells were cultured at 37 °C in a humidified atmosphere of 5% CO2 in air. The antibodies against GAPDH (no. sc-25778) and Bad (no. sc-8044) were purchased from Santa Cruz Biotechnology. Caveolin-1 (no. 610406) and β-catenin (no. 610153) were purchased from BD Biosciences. The tag antibodies against HA (no. MMS-101R) and DYKDDDDK (no. 018-22381) were purchased from Covance and Wako, respectively. PI3K p85 (no. 06-195) was purchased from Millipore. All phosphorylation-specific antibodies and their nonphosphorylated specific controls used in this study were purchased from Cell Signaling Technology, except for the antibodies described above. Cytochalasin D (CytD) (037–17561) and methyl-β-cyclodextrin (MCD) (320–84252) were obtained from Wako. KYT-1 (4395-v) and KYT-36 (4396-v), which are inhibitors of Rgp and Kgp, respectively, were purchased from the Peptide Institute.
Infection of Human Host Cells with P. gingivalis
P. gingivalis wild-type strain ATCC3327 (WT) and the gingipains-deficient mutant strain KDP136 (rgpA, rgpB, and kgp) (40) were used in this study. Before challenging the host cells, P. gingivalis strains were cultured on TSA/BHI agar plates containing hemin, menandione, and l-cysteine (41) under anaerobic conditions in an atmosphere of 10% CO2, 10% H2, and 80% N2 at 36 °C for 24–48 h. P. gingivalis was incubated with the host cells at a multiplicity of infection (MOI) of 100 for the indicated times.
Western Blotting
Ca9-22 and HGEP cells were infected with P. gingivalis at an MOI of 100 at 37 °C for the indicated times. The infected cells were lysed, and the cell lysates were run in SDS-PAGE using the indicated gel concentrations and then transferred to PVDF membranes for Western blotting. The target proteins were probed with the primary antibodies listed above, and subsequently goat anti-rabbit (1:1000) or goat anti-mouse HRP-conjugated (1:1000) secondary antibodies (Dako) were used for detection. The proteins were detected using ECL Western blotting detection reagents (GE Healthcare), according to the manufacturer's instructions and scanned using an ImageQuant LAS 4000 mini. The band densities were quantified using ImageJ software.
Immunofluorescence Analysis
Ca9-22 and HGEP cells, seeded at 3.0 × 105 cells per well in 6-well plates were incubated in serum-free medium for 24 h. The cells were infected with or without P. gingivalis at an MOI of 100 at 37 °C for 2 h. After incubation, the cells were fixed at room temperature for 10 min in PBS containing 4% paraformaldehyde. The fixed cells were treated for 5 min with 0.1% Triton X-100 for membrane permeabilization and blocked with 4% BSA in PBS for 30 min. The permeabilized cells were incubated with Ser(P)-473 Akt (1:50) and then incubated with anti-rabbit antibodies conjugated with Alexa Fluor 488 (1:500, Invitrogen) as secondary antibody in TBS containing 1% BSA and 0.5 μg/ml DAPI for 1 h. The samples were analyzed using a Cellomics ArrayScan VTI HCS reader (Thermo Scientific) to quantify relative fluorescence intensities for the phospho-Akt positive cells versus control cells.
Cell Culture Insert System and Stimulation by P. gingivalis Culture Supernatant (Transwell Assay)
Ca9-22 cells (1.0 × 105 cells/well in 24-well plates) were incubated with serum-free medium for 24 h. After incubation, the cells were treated by challenging them with P. gingivalis at an MOI of 300 through cell culture inserts for 24-well plates (BD Falcon), or the cells were incubated with 300 μl of P. gingivalis culture supernatant for the indicated times, which was prepared by incubation of P. gingivalis (4.0 × 107 bacteria/tube) in 400 μl of MEMα under anaerobic conditions for 24 h; the suspension was then filtered through a 0.45-μm PTFE filter (Sartorius Minisart SRP Syringe Filters). The cells treated under each condition were lysed and analyzed with SDS-PAGE and Western blotting using the antibodies for Akt (1:1000) and Ser(P)-473 Akt (1:1000).
Kinase Assay for PI3K and Akt
The activities of PI3K and Akt were measured using the PI3K Activity ELISA Pico Kit (Echelon Biosciences) and a modification of the nonradioactive Akt kinase assay kit (Cell Signaling Technology), respectively, according to the manufacturer's instructions. In brief, Ca9-22 and HGEP cells were infected with or without P. gingivalis at an MOI of 100 at 37 °C for 4 h. The cells were then lysed in a buffer containing 1 mm leupeptin, 10 mm N-α-tosyl-l-lysyl-chloromethyl-ketone, 1 mm KYT-1, and 1 mm KYT-36. For the determination of PI3K activity, PI3K was isolated by immunoprecipitation using an anti-PI3K p85 regulatory subunit antibody, and the activity of the PI3K p110 catalytic subunit that coimmunoprecipitated with PI3K p85 was assessed by measurement of PI(3,4,5)P3 generated from a PI(4,5)P2 substrate solution by ELISA. With regard to Akt activity, the immobilized Akt antibody slurry was used to immunoprecipitate Akt from cell lysates via gentle agitation for 4 h at 4 °C followed by spinning at 10,000 × g in a centrifuge. The pellets were resuspended in 40 μl of kinase buffer containing 200 mm ATP and 1 μg of GSK3 fusion protein and incubated for 30 min at 30 °C to phosphorylate GSK3. The phosphorylation of GSK3 fusion proteins was evaluated by Western blotting using phospho-GSK3α/β (Ser-21/9) antibodies (1:1000).
P. gingivalis Invasion Assay
Ca9-22 cells were treated with or without CytD (37 °C for 60 min) or MCD (37 °C for 60 min) at the indicated concentrations. After treatment with inhibitors, the cells were infected with P. gingivalis at an MOI of 100 at 37 °C for 2 h under an atmosphere of 5% CO2. After infection, external nonadherent bacteria were removed by washing with PBS at least twice, and external adherent bacteria were killed with 200 μg/ml metronidazole and 300 μg/ml gentamycin in MEMα for 1 h. The cells treated with the two antibiotics were washed with PBS three times, and lysed by adding 1 ml of sterile water and incubated at 37 °C. The lysed extracts were diluted to 1:100 and 1:1000, and the diluted samples including internal surviving bacteria were seeded and cultured anaerobically on blood agar plates for 7–10 days. The colonies of surviving P. gingivalis were counted and averaged from each of the diluted samples and quantified as the ratio of colony-forming units of P. gingivalis in each condition.
Expression of Bad, Akt, and PI3K p110α
Ca9-22 cells were transiently transfected with pEF4C-His empty vector, pEF4C-His-human Bad, p3×FLAG-CMV, p3×FLAG-CMV-wild-type Akt (WT-Akt), or p3×FLAG-CMV-myr-Akt (constitutively active form of Akt) using Lipofectamine 2000 (Invitrogen). In brief, following incubation for 5 h, the medium was replaced, and fresh medium containing 10% FCS was added, followed by 24 h of incubation. The levels of proteins expressed from each gene were determined by Western blotting using the specific antibodies described above. The transfected cells were infected with P. gingivalis at 37 °C for the indicated times and then were lysed. The lysates were analyzed with SDS-PAGE and Western blotting with the indicated antibodies as shown in each figure. Alternatively, the constitutively active form of PI3K p110α (myr-p110α) tagged with HA was expressed by infecting Ca9-22 cells with retroviral vectors encoding these proteins. Briefly, the retroviral vectors pBABE-puro and pBABE-puro-PI3K myr-p110α (Addgene plasmid 12522) were co-transfected into human embryonic kidney 293T cells (20 × 105 cells) by using the retrovirus packaging kit Ampho (Takara), according to the manufacturer's instructions. The retroviral vectors produced from 293T cells were harvested 48 h after transfection and filtered through 0.45-μm pore size filters (Sartorius) to eliminate any remaining 293T cells. On the day prior to infection, Ca9-22 cells (1 × 105 cells) were seeded in six-well plates. The viral supernatant was added at a ratio of 1:1 to the culture medium in the presence of 8 μg/ml Polybrene, and the cells were spun at 1300 rpm for 120 min to increase the infection rate. The infected cells were incubated with the retroviruses for 24 h. A second infection was performed following the same protocol on the next day. After an additional 24 h of recovery in normal medium, the cells were passaged and selected using 1 μg/ml puromycin.
Pulldown Assay for Biotin-labeled Membrane Proteins
Proteins on the plasma membrane of Ca9-22 cells (30 × 105 cells) were biotin-labeled in PBS containing 0.5 mm CaC12 and 0.5 mm MgC12 (PBS-CaMg), pH 8.6, at 4 °C for 30 min using an ECL protein biotinylation module (GE Healthcare), according to the manufacturer's instructions. After incubation, the biotin-labeled cells were solubilized with lysis buffer (50 mm Tris-HCl, pH 7.5, containing 150 mm NaCl, 20% glycerol, 1% Triton X-100, 1 mm Na3VO4, 50 mm NaF, 1 μg/ml leupeptin, 1 μg/ml aprotinin, and 1 mm PMSF) and incubated on ice for 30 min. The lysates were centrifuged at 20,000 × g for 20 min, and the supernatants were transferred to new tubes on ice. The supernatants were then mixed with streptavidin-Sepharose beads (GE Healthcare) and incubated for at least 1 h. The streptavidin-Sepharose beads were collected by centrifugation and washed with lysis buffer three times. The precipitants were analyzed with SDS-PAGE and Western blotting using the indicated primary antibodies.
Extraction of Membrane Fraction
Ca9-22 cells were infected with P. gingivalis WT or KDP136 for 4 h and subsequently harvested with a cell scraper, washed with cold PBS, and utilized for preparation of membrane fractions using a subcellular protein fractionation kit for cultured cells (Thermo Scientific) according to the manufacturer's instructions. Briefly, the cells were collected and lysed, and the insoluble fraction was treated with membrane extraction buffer. The membrane fraction prepared using ME buffer contains the plasma membrane, mitochondria, and endoplasmic reticulum/Golgi membrane components but not nuclear membrane. The membrane fraction was analyzed using SDS-PAGE and Western blotting to detect the indicated proteins by specific antibodies.
Statistical Analysis
To establish the significance of the results, the Student's t test was used for numerical data. Fisher's exact test or χ2 test was used for categorical data as appropriate. A p value of less than 0.05 was considered statistically significant.
RESULTS
P. gingivalis Dephosphorylates Akt at Thr-308 and Ser-473
The PI3K/Akt signaling pathway is activated in response to insulin and growth factors (e.g. EGF), and this subsequently affects its downstream substrates, which regulate physiological functions such as survival/apoptosis and metabolism. To determine whether P. gingivalis has an influence on the PI3K/Akt signaling pathway, Ca9-22 cells were incubated with P. gingivalis. We unexpectedly observed that live P. gingivalis, but not heat-killed P. gingivalis, induced dephosphorylation of Akt at Thr-308 and Ser-473 in an MOI-dependent and infection time-dependent manner (Fig. 1, A and B). The expression levels of total Akt remained constant over all time points, indicating that the decrease in Akt phosphorylation was due not to changes in the expression levels of Akt but to changes in its phosphorylation levels induced by P. gingivalis infection. Consistent with this result, immunofluorescence analysis showed that Akt dephosphorylation at Ser-473 occurred in P. gingivalis-infected cells, but not in control cells (Fig. 1C).
FIGURE 1.

P. gingivalis dephosphorylates Akt at Thr-308 and Ser-473. A and B, Ca9-22 cells were infected with heat-killed or live P. gingivalis. A, MOI dependence of Akt dephosphorylation for 2 h. B, infection time dependence of Akt dephosphorylation at an MOI of 100. After incubation, the cells were lysed and analyzed with SDS-PAGE and Western blotting probed with the indicated antibodies. The graphs below indicate the ratio of phospho-Akt at Thr-308 (solid bar) and Ser-473 (open bar) to total Akt by Western blotting. C, Ca9-22 cells were infected with or without P. gingivalis at an MOI of 100 for 2 h. The infected cells were fixed with 4% paraformaldehyde in PBS, and immunofluorescence was evaluated as described under “Experimental Procedures.” The graph indicates the relative fluorescence intensity for the phospho-Akt positive cells compared with control cells. D and E, Ca9-22 cells in 24-well plates were incubated with P. gingivalis at an MOI of 300 for up to 8 h through a cell culture insert system (D), and Ca9-22 cells were incubated for 120 min with P. gingivalis culture supernatant prepared as described under “Experimental Procedures” (E). After incubation, the cells were lysed and analyzed with SDS-PAGE and Western blotting probed with the indicated antibodies. The graphs indicate the ratio of Ser(P)-473 Akt/total Akt on Western blotting. These results were independently demonstrated on three separate occasions. Statistical significance: *, p < 0.05.
To further characterize the potential mechanisms of P. gigivalis-induced Akt dephosphorylation, P. gingivalis infection and the addition of culture supernatant from P. gingivalis was performed in cells in a Transwell assay to assess the requirement for direct interaction between bacteria and host cells. As shown in Fig. 1 (D and E), Akt dephosphorylation induced by P. gingivalis did not require a direct interaction of the epithelial cells with bacteria; P. gingivalis cell-free culture supernatant was sufficient for Akt dephosphorylation. These results in Fig. 1 indicate that P. gingivalis-induced Akt dephosphorylation was caused by P. gingivalis-secreted proteins.
P. gingivalis Induces Akt Inactivation and Subsequent Dephosphorylation of Downstream Proteins of Akt
To confirm the kinase activity of Akt, the dephosphorylation of which was induced by P. gingivalis, we used an in vitro assay to evaluate the Akt protein and the phosphorylation of Akt substrates in the PI3K/Akt signaling pathway. Akt protein was precipitated with anti-Akt antibodies from cell extracts of P. gingivalis-infected cells and then incubated with a fusion protein containing the region corresponding to Akt-targeted residues surrounding GSK3α/β (Ser-21/9). Akt precipitated from noninfected cells phosphorylated the GSK3 fusion protein, whereas Akt from the infected cells did not (Fig. 2A). In addition, the experiment using anti-Ser(P)/Thr(P) Akt substrate antibody revealed that the phosphorylation levels of a number of Akt substrates were directly affected by P. gingivalis infection within 4 h (Fig. 2B). These affected Akt substrates may include GSK3α/β, Bad, and caspase-9. To clarify this point, we examined the influence of P. gingivalis on the phosphorylation levels of GSK3α/β, mTOR, and Bad. As shown in Fig. 2C, the phosphorylation levels of GSK3α/β at Ser-21/9, which is a direct substrate of Akt, and mTOR at Ser-2448, which is a downstream protein of Akt, decreased until 8 h postinfection. This result is consistent with Akt dephosphorylation and subsequent inactivation of its kinase activity (Fig. 2C). The Ser-136 residue of Bad, a pro-apoptotic factor and direct Akt substrate, was also dephosphorylated by P. gingivalis in Ca9-22 cells (Fig. 2D). These results indicate that P. gingivalis-induced Akt dephosphorylation at Thr-308 and Ser-473 results in the inactivation of Akt kinase activity in P. gingivalis-infected Ca9-22 gingival epithelial cells.
FIGURE 2.

P. gingivalis induces Akt inactivation and the dephosphorylation of the downstream proteins in the PI3K/Akt signaling pathway. A, Ca9-22 cells were infected with or without P. gingivalis at an MOI of 100 for 4 h. Cell lysates were incubated with an anti-Akt monoclonal antibody immobilized with Sepharose beads for 4 h. Akt kinase assay was performed as described under “Experimental Procedures.” B, Ca9-22 cells were infected with or without P. gingivalis at an MOI of 100 for 4 h and prepared for whole cell lysates. The lysates were analyzed with SDS-PAGE and Western blotting using anti-Ser(P)/Thr(P) Akt substrate antibody. C and D, Ca9-22 cells were infected with P. gingivalis at an MOI of 100 for up to 8 h (C), and Ca9-22 cells were transfected with 0.2 μg of pEF4C-His-Bad. After transfection, Ca9-22 cells expressing His-Bad were infected with P. gingivalis at an MOI of 100 for 6 h (D). The infected cells were lysed and analyzed with SDS-PAGE and Western blotting probed with the indicated antibodies, respectively (C and D). These results were independently demonstrated three times. cont., control; IP, immunoprecipitation.
P. gingivalis-induced Akt Inactivation Is Independent of P. gingivalis Invasion
P. gingivalis is capable of invading gingival epithelial cells and fibroblasts (13, 42, 43), and the virulence factors produced by P. gingivalis including outer membrane vesicles, which enter into host cells (44), and SerB, which is a serine phosphatase and contributes to P. gingivalis invasion (45), also interacts with host proteins in gingival epithelial cells. Lipid rafts are also involved in P. gingivalis invasion (42, 46). To investigate whether Akt dephosphorylation is caused by invading P. gingivalis or endocytosed virulence factors, we employed CytD and MCD. CytD is an inhibitor of actin polymerization, which is mainly involved in macropinocytosis, whereas MCD disrupts the integrity of cholesterol-enriched microdomains such as lipid rafts and caveolae, resulting in inhibition of lipid raft- and caveolae-dependent endocytosis. Following treatment with each inhibitor, Ca9-22 cells were infected with P. gingivalis at an MOI of 100, and internalized P. gingivalis was estimated by the number of colony-forming units produced from water-lysed Ca9-22 cells. Invasion of P. gingivalis into Ca9-22 cells was decreased to ∼20% of control by treatment of CytD and MCD (Fig. 3A). However, these inhibitors did not prevent Akt dephosphorylation by P. gingivalis under the same conditions (Fig. 3B). These data indicate that invading P. gingivalis cells or endocytosed virulence factors from P. gingivalis are not associated with Akt dephosphorylation by P. gingivalis.
FIGURE 3.
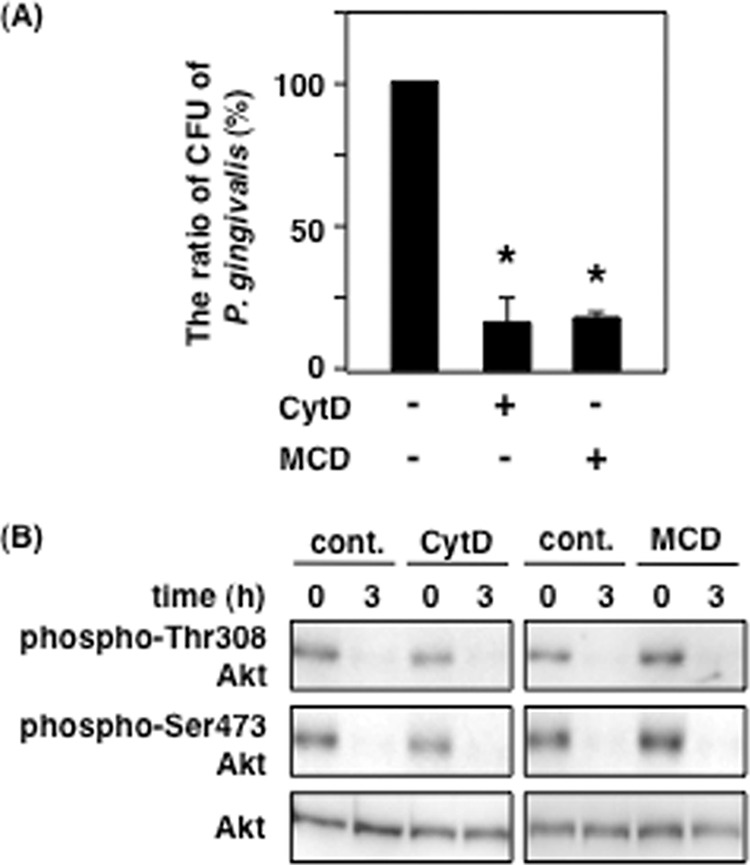
Effect of P. gingivalis invasion on P. gingivalis-induced Akt inactivation. Ca9-22 cells were treated with or without 10 μm CytD or 1 mm MCD for 1 h. A, the treated cells were infected with P. gingivalis at an MOI of 100 for 2 h. P. gingivalis invasion assay was performed using the antibiotics treatment as described under “Experimental Procedures.” B, the treated cells were infected with P. gingivalis at an MOI of 100 for 3 h and then lysed. The lysates were analyzed with SDS-PAGE and Western blotting using the antibodies for total Akt and phosphorylated Akt at Thr-308 and Ser-473. These results were independently demonstrated three times. Statistical significance: *, p < 0.05. cont., control.
Gingipains Are Involved in the Inhibition of Akt Signaling by P. gingivalis
P. gingivalis-induced Akt inactivation was independent of P. gingivalis invasion as shown in Fig. 3. We next suspected that gingipains, which are cysteine proteases, could be responsible for dephosphorylation of Akt because they are well known as major virulence factors of P. gingivalis. To demonstrate this possibility, we inhibited the proteolytic activity of gingipains using the gingipain-specific inhibitors KYT-1 and KYT-36. We observed that neither KYT-1 nor KYT-36 alone was sufficient to inhibit P. gingivalis-induced Akt dephosphorylation in Ca9-22 cells, but the combination of KYT-1 and KYT-36 did inhibit P. gingivalis-induced Akt dephosphorylation (Fig. 4A). To ascertain whether gingipains are associated with P. gingivalis-induced Akt dephosphorylation, we compared Akt dephosphorylation with both wild-type P. gingivalis (WT-Pg) and gingipain-deficient KDP136. Although WT-Pg decreased the phosphorylation levels of Akt as heretofore described, KDP136 did alter its phosphorylation for up to 240 min (Fig. 4B). As for the Akt downstream proteins GSK3α/β and mTOR, although both of them were also dephosphorylated by WT-Pg, infection with KDP136 and treatment with gingipain-specific inhibitors showed no significant differences in their phosphorylation (Fig. 4C). These results indicate that gingipains play a crucial role in the dephosphorylation of Akt, GSK3α/β, and mTOR in P. gingivalis infection and that their proteolytic activity is indispensably involved in this event.
FIGURE 4.

Rgp and Kgp play a critical role in Akt dephosphorylation and inhibit the PI3K/Akt signaling pathway. A, Ca9-22 cells were treated with the inhibitors KYT-1 and KYT-36, followed by P. gingivalis infection at an MOI of 100 for 2 h. The cell lysates were analyzed with Western blotting by using primary antibodies for total Akt and phospho-Akt at Thr-308 and Ser-473. The lower panel indicates the ratio of phospho-specific Akt/total Akt by Western blotting. B, Ca9-22 cells were infected with P. gingivalis WT or KDP136 in an infection time-dependent manner for up to 240 min. The cell lysates were analyzed with Western blotting using the indicated antibodies. The lower panel indicates the ratio of phospho-specific Akt/total Akt by Western blotting. C, Ca9-22 cells were treated with or without a mixture of 5 μm KYT-1 and 5 μm KYT-36 for 1 h prior to P. gingivalis infection and infected with P. gingivalis WT or KDP136 at MOI of 100 for 6 h. The infected cells were lysed and analyzed with Western blotting using the indicated antibodies. These results were independently demonstrated three times. Statistical significance: *, p < 0.05.
Gingipains Cleave Membrane Proteins Coupled to PI3K p85α
Based on the results of Figs. 3 and 4, we hypothesized that gingipains act from the outside, not inside, of epithelial cells and cleave membrane proteins, resulting in inactivation of PI3K and dephosphorylation of Akt. To confirm this hypothesis, total membrane proteins of Ca9-22 cells were labeled with biotin and precipitated with streptavidin-conjugated Sepharose beads in an attempt to detect PI3K regulatory subunit p85α bound to membrane proteins. As shown in Fig. 5A, the biotinylated membrane proteins were precipitated from uninfected Ca9-22 cells. In cells infected with P. gingivalis, PI3K p85α did not co-precipitate from Ca9-22 cells infected for 4 h with WT-Pg, as compared with control and KDP136-infected cells at all time points (Fig. 5B). In contrast, β-catenin and integrin-linked kinase (47), which bind to E-cadherin and integrins, respectively, were unaffected under all conditions (Fig. 5B). In Fig. 5C, we used CytD to examine the possibility of an intracellular action by P. gingivalis invasion on the disruption of p85α-membrane protein associations. CytD did not inhibit the disruption induced by WT-Pg. These results suggest that gingipains from WT-Pg disrupt the interaction between membrane proteins and PI3K p85α by their extracellular action.
FIGURE 5.

Interaction between PI3K p85α and membrane proteins is disrupted by gingipains. A, all membrane proteins in Ca9-22 cells were labeled with biotin for 30 min, and the cells were lysed for a pulldown assay as described under “Experimental Procedures.” The pulldown samples were then analyzed by SDS-PAGE and Western blotting using the antibodies for the respective membrane proteins. B and C, untreated Ca9-22 cells (B) and 10 μm CytD-treated Ca9-22 cells (C) were infected with or without P. gingivalis WT or KDP136 at an MOI of 100 for 4 h. After infection, the cells were labeled for 30 min, lysed, and run in a pulldown assay. The precipitants were analyzed by SDS-PAGE and Western blotting using the indicated antibodies. These results were independently demonstrated three times. cont., control; WCL, whole cell lysate; ILK, integrin-linked kinase.
Inactivation of PI3K Is Caused by Gingipains
Based on the results shown in Fig. 5, we hypothesized that PI3K activity might be affected by gingipains. We challenged Ca9-22 cells with WT-Pg and KDP136, and immunoprecipitates of PI3K under each condition were used for assessing PI3K activity. WT-Pg, but not control and KDP136, decreased the activity of PI3K by ∼70% (Fig. 6A). Similarly, PI3K activity was decreased by 60–70% in the presence of CytD during WT-Pg infection compared with control (Fig. 6B). In Fig. 6 (A and B), regulatory subunit p85α and catalytic subunit p110α of PI3K were detected in equal amounts in all conditions. These data indicate that PI3K activity is affected by WT-Pg infection, and gingipains are closely associated with the decrease in PI3K activity via an extracellular interaction.
FIGURE 6.

The kinase activity of PI3K is decreased by gingipains from P. gingivalis. A, Ca9-22 cells were infected with or without P. gingivalis WT or KDP136 at an MOI of 100 for 4 h. After infection, the cells were lysed and immunoprecipitated using an anti-p85α polyclonal antibody for 6 h. B, Ca9-22 cells were treated with 10 μm CytD and were infected with or without P. gingivalis WT at an MOI of 100 for 4 h. The activity of PI3K was measured as described under “Experimental Procedures.” These results were independently demonstrated three times. Statistical significance: *, p < 0.05. cont., control.
Gingipains Alter the Localization of PDK1 and Akt at the Plasma Membrane
We demonstrated a gingipain-dependent decrease in PI3K activity in Fig. 6. Next, we examined whether gingipains affect the localization of PDK1 and Akt at the plasma membrane, where they bind PI(3,4,5)P3 through their pleckstrin homology domain. As shown in Fig. 7A, 4 h after infection there was no significant change in total protein level in whole cell lysates under the indicated conditions (lower panels). WT-Pg remarkably reduced the distribution of PDK1 in the membrane fraction compared with control and KDP136-infected cells (upper panels in Fig. 7A). Interestingly, we also found that the distribution of p85α, p110α, and Akt increased in the membrane fraction extracted from WT-Pg-infected cells compared with noninfected and KDP136-infected cells (upper panels in Fig. 7A). In the cells treated with CytD, WT-Pg decreased the level of PDK1 and increased the level of Akt in the membrane fraction to similarly those of untreated cells infected by WT-Pg (upper panels in Fig. 7B). The total protein expression level was not changed (lower panels in Fig. 7, A and B). Caveolin-1 also served as a protein loading control in both Fig. 7 (A and B). Here we demonstrate that gingipain-induced PI3K inactivation changed the localization of PDK1 and Akt at the plasma membrane, suggesting that the changes are strongly implicated in a decrease in Akt phosphorylation because of the low level of PDK1 localization and subsequent blockade of the Akt signaling cascade.
FIGURE 7.
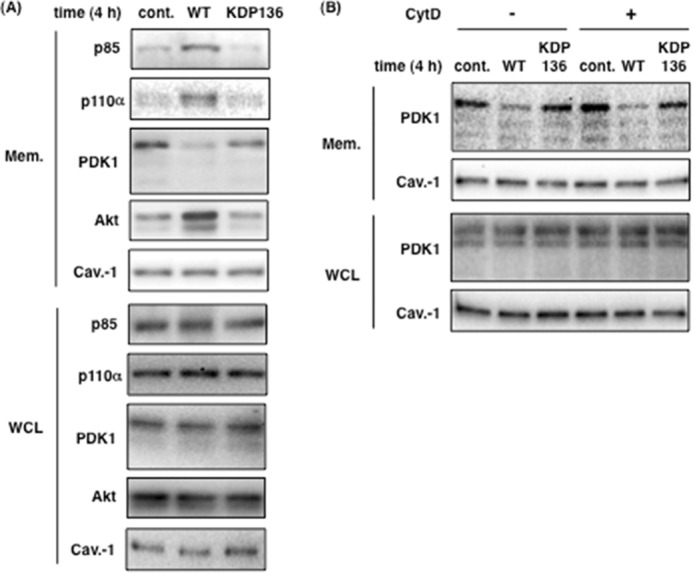
Gingipains affect the localization of PDK1 and Akt at the plasma membrane. A, membrane fractions (Mem.) were prepared from Ca9-22 cells infected with or without P. gingivalis WT or KDP136 at an MOI of 100 for 4 h as described under “Experimental Procedures.” B, Ca9-22 cells were infected with or without P. gingivalis WT or KDP136 at an MOI of 100 for 4 h after treatment with 10 μm CytD, and membrane fractions were extracted. The extracts and whole cell lysates (WCL) were analyzed by SDS-PAGE and Western blotting using the indicated antibodies. Caveolin-1 (Cav.-1) was used as a marker of membrane fractions. cont., control.
Effect of Constitutively Active PI3K p110α and Akt on the Localization of PDK1 and Akt, and Akt Signaling in P. gingivalis-infected Cells
To support the above results, we tested the effect of expression of constitutively active forms of PI3K p110α and Akt on the localization of PDK1 and Akt and the phosphorylation of GSK3α/β and mTOR by P. gingivalis infection. For this purpose, we utilized genes encoding a myristoylation signal to produce constitutively activated proteins independently of all upstream signaling events. We showed that the level of PDK1 and Akt in the membrane fraction was unchanged from 0 to 4 h post infection in the cells expressing myr-p110α, whereas the localization of PDK1 was decreased, and that of Akt was increased by P. gingivalis infection in the cells transfected with empty vector (Fig. 8A). In myr-p110α-expressing cells, the phosphorylation levels of Akt, GSK3α/β, and mTOR were sustained during infection compared with empty vector-transfected control cells (Fig. 8B). The expression of WT-Akt or myr-Akt also blocked the dephosphorylation of GSK3α/β and mTOR by P. gingivalis infection (Fig. 8C). Based on these observations, we support that gingipains reduce the kinase activity of PI3K and affect the translocation of PDK1 and Akt at the plasma membrane and the phosphorylation level of the downstream proteins on the PI3K/Akt signaling pathway by extracellular interaction between gingipains and Ca9-22 cell surface during P. gingivalis infection.
FIGURE 8.
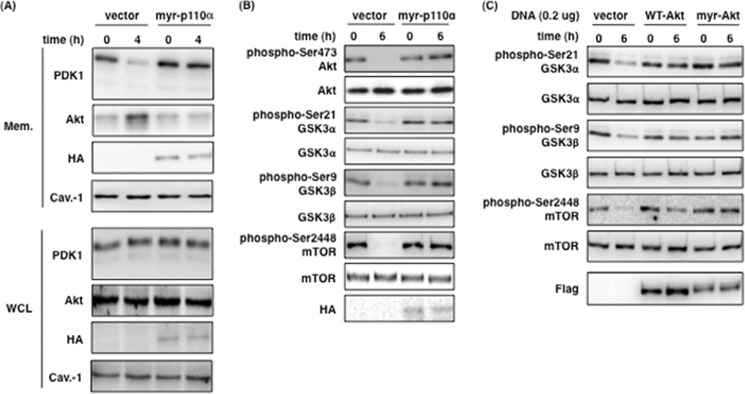
Effect of constitutively active form of PI3K p110α and Akt on the localization of PDK1 and Akt, and the Akt signaling pathway. A, Ca9-22 cells transfected with the empty vector or the myr-p110α expression vector were infected with P. gingivalis WT for 4 h. After infection, membrane fractions (Mem.) and whole cell lysates (WCL) were extracted and analyzed by SDS-PAGE and Western blotting using the indicated antibodies. B, Ca9-22 cells were transfected with the empty vector or the myr-p110α expression vector. All cells were infected with P. gingivalis WT at an MOI of 100 for 6 h, and then cell lysates were analyzed by SDS-PAGE and Western blotting using the indicated antibodies. C, Ca9-22 cells were transfected with empty vector or expression vectors for WT-Akt and myr-Akt and infected with P. gingivalis WT at an MOI of 100 for 6 h, and then cell lysates were analyzed by SDS-PAGE and Western blotting using the indicated antibodies. These results were independently demonstrated three times.
Dysregulation of the PI3K/Akt Signaling Pathway by Gingipains in Human Primary Gingival Epithelial Cells
All of the experiments described above were performed with a cultured cancer cell line. We next investigated whether these signaling events also occur after P. gingivalis infection in HGEP cells. Western blotting, immunofluorescence analysis, and Akt kinase assay revealed that Akt was dephosphorylated and inactivated by infection with WT-Pg, but not KDP136, in HGEP cells (Fig. 9, A–C). We also examined the changes in phosphorylation levels of GSK3, mTOR, and Bad by P. gingivalis infection in HGEP cells. Infection with WT-Pg, but not KDP136, decreased the phosphorylation of GSK3α/β, mTOR, and Bad in HGEP cells (Fig. 9D). Next, we demonstrated that PI3K activity was decreased by ∼70% by gingipains during P. gingivalis infection (Fig. 9E). To evaluate PI(3,4,5)P3 production, PDK1 translocation to the plasma membrane was examined, and these results indicate that there was a negligible amount of PDK1 in the membrane fraction in WT-Pg-infected cells compared with control and KDP136-infected cells (Fig. 9F). Taken together, these data show that alteration of the PI3K/Akt signaling pathway by gingipains occurs in HGEP cells in a manner similar to that in Ca9-22 cells.
FIGURE 9.

Gingipains attenuate the PI3K/Akt signaling pathway in HGEP cells infected with P. gingivalis. HGEP cells were infected with or without P. gingivalis WT or KDP136 at an MOI of 100 for 4 h (A and C–F) or 2 h (B). After infection, the cells were lysed or fixed, and then the samples were analyzed by SDS-PAGE and Western blotting (A and D), immunostaining for phosphorylated Akt (B), in vitro kinase assay for Akt (C) and PI3K (E), and membrane extraction for translocation of PDK1 (F) as described under “Experimental Procedures.” The antibodies were total protein- and phospho-specific as indicated. These results were independently demonstrated three times. Statistical significance: *, p < 0.05. cont., control; IP, immunoprecipitation.
DISCUSSION
In this study, we reveal a novel mechanism of attenuation of the PI3K/Akt signaling pathway by gingipains, the cysteine proteases of P. gingivalis. The proteolytic effects of gingipains play an essential role in the dysfunction of PI3K and changes in the Akt signaling pathway during P. gingivalis infection, independent of invasion (Fig. 10). The results elucidate a new role for gingipains in disturbing cell functions regulated by the PI3K/Akt signaling pathway, including cell survival and growth, apoptosis, endocytosis, and metabolism.
FIGURE 10.
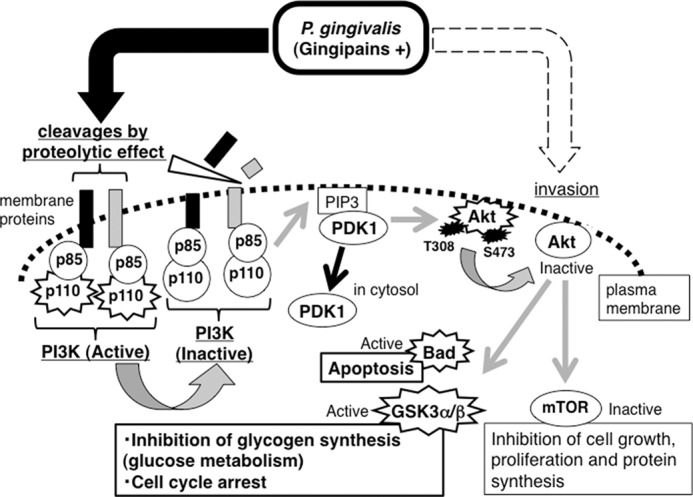
The molecular mechanism of attenuation of the PI3K/Akt signaling pathway by gingipains. Gingipains (RgpA, RgpB, and Kgp) cleave the membrane proteins associated with PI3K p85α, which is the regulatory subunit of PI3K, resulting in inactivation of PI3K and inhibition of PDK1 translocation to the plasma membrane. Hence, Thr-308 of Akt is not phosphorylated by PDK1, which is required for Akt activation. Consequently the phosphorylation levels and activation status of the Akt downstream proteins including GSK3α/β, mTOR, and Bad are impacted. PIP3, PI(3,4,5)P3.
Here, we demonstrate that P. gingivalis attenuates PI3K and Akt and decreases the phosphorylation levels of the Akt downstream proteins GSK3, mTOR, and Bad, in gingival epithelial cells. However, other groups have reported the activation of PI3K and Akt by P. gingivalis infection (30, 31, 43, 48). These apparently contradictory results may be due to differences in cell type and experimental conditions such as infection time, strains, and culture conditions of P. gingivalis. For example, Chang et al. (43) have shown that high glucose treatment of P. gingivalis increases expression of fimA mRNA. Moreover, phenotypic differences might exist in the gingival epithelial cells used here, such as in the expression level and pattern of receptors and adaptor proteins in the PI3K/Akt signaling pathway, compared with the cells used in other studies. In addition, it is plausible that these cells may have originated from a different area of gingival tissue and/or from individuals of various health conditions and ages.
We demonstrate the possibility that inactivation of PI3K and Akt by P. gingivalis is not required for its direct interaction with cell surface (Fig. 1, D and E). Furthermore, our experiments using CytD and MCD show that this inactivation is independent of P. gingivalis invasion into gingival epithelial cells and intracellular interaction with virulence factors such as gingipains, SerB (45), and outer membrane vesicles (49) produced by P. gingivalis (Figs. 3 and 6). We also clarified the inactivation of Akt and the alteration of phosphorylation levels of its substrate proteins including Bad, GSK3, and mTOR (Figs. 2 and 8). Inhibitors of gingipains and infection with the gingipain-null mutant KDP136 revealed that gingipains are closely associated with P. gingivalis infection-mediated Akt dephosphorylation (Fig. 4). Thus, the alteration of the PI3K/Akt signaling pathway by gingipains from P. gingivalis have several effects on PI3K/Akt-dependent cellular functions as discussed below. First, the dephosphorylation of Bad resulting from gingipain-induced Akt inactivation, which leads to increased activity of Bad, may contribute to apoptosis during P. gingivalis infection, as suggested by a previous report (37). GSK3 is known to regulate glycogen synthesis in response to insulin. Recently, Ishikawa et al. (38) indicated that P. gingivalis has an effect on glycogen synthesis in liver via the Akt/GSK3β signaling pathway. Our results support that gingipains are implicated in the attenuation of the PI3K/Akt/GSK3β pathway and decrease in glycogen synthesis and glucose metabolism. It may be worth noting in the current study, however, that all experiments were performed using gingival epithelial cells and not hepatocytes, which are highly specialized for the biosynthesis of glycogen. We also show that mTOR is dephosphorylated by gingipains. The PI3K/Akt/mTOR signaling pathway has a role in endocytosis (50, 51) and internalization of bacteria such as Salmonella typhimurium, Helicobacter pylori, Streptococcus pneumoniae, Staphylococcus aureus (52–55), and autophagy (56–58). Some reports have shown a causal relationship between the Akt/mTOR signaling pathway and P. gingivalis and gingipains (59, 60), suggesting that gingipains might cause a decrease in mTOR function via attenuation of the PI3K/Akt signaling pathway.
Here we describe a novel mechanism by which gingipain-mediated inactivation of PI3K is associated with the disruption of complex formation between PI3K p85α and membrane proteins by wild-type P. gingivalis infection, but not the gingipain-deficient mutant KDP136, leading to alteration of the localization of PDK1 and Akt and decrease in the phosphorylation and activity of Akt. At first, we were wondering whether the amount of PI(3,4,5)P3 was reduced by gingipain-induced PI3K inactivation, leading to decrease in PDK1 localization at the plasma membrane. However, Akt was apparently increased by P. gingivalis infection. Therefore, we consider that there is little change in the amount of PI(3,4,5)P3 at the plasma membrane during infection, although the turnover and newly synthesis of PI(3,4,5)P3 might be decreased by gingipain-mediated PI3K inactivation. Furthermore, it is likely that residual levels of PI(3,4,5)P3 are sufficient to interact with Akt, which is mainly nonphosphorylated form and/or the form after working in cytosol. Consequently, we suspect the possibility that Akt occupies the residual PI(3,4,5)P3, and Akt accumulated at the plasma membrane is ready and waiting for the phosphorylation by PDK1. This mean brings us to the point that the accumulation of Akt prevents PDK1 translocation to the plasma membrane via the pleckstrin homology domain of PDK1. Actually, based on the data in Figs. 7 and 8, the level of PDK1 at the plasma membrane is sufficiently low to phosphorylate Akt at Thr-308 in WT-Pg-infected cells compared with in control cells and in KDP136-infected cells, which fail to normally transmit the signals from PDK1 to Akt. We evidently show that gingipains reduce the PI3K activity, PDK1 translocation at the plasma membrane, and the phosphorylation level of Akt and the downstream proteins on Akt signaling. These observations are supported by expression of constitutively active forms of PI3K p110α and Akt (Fig. 8).
According to the results in Fig. 5B, β-catenin and integrin-linked kinase were detected, and E-cadherin and integrin β4 remain unaffected by gingipains within 4 h of infection. Although EGFR and integrin β1 are cleaved, we confirmed that EGFR does not associate with PI3K p85α in the absence of serum and growth factors (data not shown). We suggest that gingipains cleave one or more membrane proteins that bind PI3K p85α, resulting in reduction in both PI3K activity and phosphorylation of Akt. We are currently attempting to characterize the as yet unidentified membrane proteins that bind to PI3K p85α. The PI3K/Akt signaling pathway is intrinsically regulated by PTEN (phosphatase and tensin homologue deleted on chromosome 10) and protein serine/threonine phosphatases, such as protein phosphatases 2A, 2B, and 1 (28, 61, 62). However, our results using siRNA for PTEN and protein phosphatase inhibitors (data not shown) suggest virtually no association between PI3K/Akt and these phosphatases. Moreover, there was little change in the interaction between Akt and C-terminal modulator protein by P. gingivalis infection (data not shown) (63, 64). It is therefore likely that gingipain-induced attenuation of PI3K and Akt is mediated by interference with homeostatic PI3K signaling via cleavage of membrane proteins in complexes with PI3K p85α.
In conclusion, our data provide strong evidence that gingipains are a critical virulence factor for disrupting cellular functions regulated by the PI3K/Akt signaling pathway in gingival epithelial cells and almost certainly cause the damage to the gingival epithelial barrier. These effects promote the invasion and colonization of gingival tissue by P. gingivalis and can lead to long term infection. Future investigations will clarify additional functions of gingipains and the significance of the PI3K/Akt signaling pathway in P. gingivalis infection.
This work was supported by grants-in-aid for scientific research from the Ministry of Education, Culture, Sports, Science and Technology of Japan, by the Ryobi Teien Memory Foundation, by the Takeda Science Foundation, and by the Institute for Fermentation, Osaka.
- Rgp
- arginine-specific gingipain
- Kgp
- lysine-specific gingipain
- GSK3
- glycogen synthase kinase 3
- mTOR
- mammalian target of rapamycin
- PDK1
- phosphoinositide-dependent protein kinase 1
- HGEP
- human primary gingival epithelial
- CytD
- cytochalasin D
- MCD
- methyl-β-cyclodextrin
- MOI
- multiplicity of infection
- PI(3,4,5)P3
- phosphatidylinositol 3,4,5-triphosphate.
REFERENCES
- 1. Iacopino A. M., Cutler C. W. (2000) Pathophysiological relationships between periodontitis and systemic disease: recent concepts involving serum lipids. J. Periodontol. 71, 1375–1384 [DOI] [PubMed] [Google Scholar]
- 2. Lalla E., Papapanou P. N. (2011) Diabetes mellitus and periodontitis: a tale of two common interrelated diseases. Nat. Rev. Endocrinol. 7, 738–748 [DOI] [PubMed] [Google Scholar]
- 3. Bansal M., Khatri M., Taneja V. (2013) Potential role of periodontal infection in respiratory diseases: A review. J. Med. Life 6, 244–248 [PMC free article] [PubMed] [Google Scholar]
- 4. Frazer L. T., O'Brien-Simpson N. M., Slakeski N., Walsh K. A., Veith P. D., Chen C. G., Barr I. G., Reynolds E. C. (2006) Vaccination with recombinant adhesins from the RgpA-Kgp proteinase-adhesin complex protects against Porphyromonas gingivalis infection. Vaccine 24, 6542–6554 [DOI] [PubMed] [Google Scholar]
- 5. Hajishengallis G., Wang M., Liang S. (2009) Induction of distinct TLR2-mediated proinflammatory and proadhesive signaling pathways in response to Porphyromonas gingivalis fimbriae. J. Immunol. 182, 6690–6696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nakao R., Takashiba S., Kosono S., Yoshida M., Watanabe H., Ohnishi M., Senpuku H. (2014) Effect of Porphyromonas gingivalis outer membrane vesicles on gingipain-mediated detachment of cultured oral epithelial cells and immune responses. Microbes Infect. 16, 6–16 [DOI] [PubMed] [Google Scholar]
- 7. Takii R., Kadowaki T., Baba A., Tsukuba T., Yamamoto K. (2005) A functional virulence complex composed of gingipains, adhesins, and lipopolysaccharide shows high affinity to host cells and matrix proteins and escapes recognition by host immune systems. Infect. Immun. 73, 883–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bainbridge B., Verma R. K., Eastman C., Yehia B., Rivera M., Moffatt C., Bhattacharyya I., Lamont R. J., Kesavalu L. (2010) Role of Porphyromonas gingivalis phosphoserine phosphatase enzyme SerB in inflammation, immune response, and induction of alveolar bone resorption in rats. Infect. Immun. 78, 4560–4569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Naito M., Sakai E., Shi Y., Ideguchi H., Shoji M., Ohara N., Yamamoto K., Nakayama K. (2006) Porphyromonas gingivalis-induced platelet aggregation in plasma depends on Hgp44 adhesin but not Rgp proteinase. Mol. Microbiol. 59, 152–167 [DOI] [PubMed] [Google Scholar]
- 10. Fujita Y., Nakayama M., Naito M., Yamachika E., Inoue T., Nakayama K., Iida S., Ohara N. (2014) Hemoglobin receptor protein from Porphyromonas gingivalis induces interleukin-8 production in human gingival epithelial cells through stimulation of the mitogen-activated protein kinase and NF-κB signal transduction pathways. Infect. Immun. 82, 202–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kadowaki T., Takii R., Yamatake K., Kawakubo T., Tsukuba T., Yamamoto K. (2007) A role for gingipains in cellular responses and bacterial survival in Porphyromonas gingivalis-infected cells. Front. Biosci. 12, 4800–4809 [DOI] [PubMed] [Google Scholar]
- 12. Fitzpatrick R. E., Wijeyewickrema L. C., Pike R. N. (2009) The gingipains: scissors and glue of the periodontal pathogen, Porphyromonas gingivalis. Future Microbiol. 4, 471–487 [DOI] [PubMed] [Google Scholar]
- 13. Kinane J. A., Benakanakere M. R., Zhao J., Hosur K. B., Kinane D. F. (2012) Porphyromonas gingivalis influences actin degradation within epithelial cells during invasion and apoptosis. Cell Microbiol. 14, 1085–1096 [DOI] [PubMed] [Google Scholar]
- 14. Inaba H., Kuboniwa M., Sugita H., Lamont R. J., Amano A. (2012) Identification of signaling pathways mediating cell cycle arrest and apoptosis induced by Porphyromonas gingivalis in human trophoblasts. Infect. Immun. 80, 2847–2857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Song G., Ouyang G., Bao S. (2005) The activation of Akt/PKB signaling pathway and cell survival. J. Cell Mol. Med. 9, 59–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Engelman J. A., Luo J., Cantley L. C. (2006) The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 7, 606–619 [DOI] [PubMed] [Google Scholar]
- 17. Currie R. A., Walker K. S., Gray A., Deak M., Casamayor A., Downes C. P., Cohen P., Alessi D. R., Lucocq J. (1999) Role of phosphatidylinositol 3,4,5-trisphosphate in regulating the activity and localization of 3-phosphoinositide-dependent protein kinase-1. Biochem. J. 337, 575–583 [PMC free article] [PubMed] [Google Scholar]
- 18. McManus E. J., Collins B. J., Ashby P. R., Prescott A. R., Murray-Tait V., Armit L. J., Arthur J. S., Alessi D. R. (2004) The in vivo role of PtdIns(3,4,5)P3 binding to PDK1 PH domain defined by knockin mutation. EMBO J. 23, 2071–2082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Anderson K. E., Coadwell J., Stephens L. R., Hawkins P. T. (1998) Translocation of PDK-1 to the plasma membrane is important in allowing PDK-1 to activate protein kinase B. Curr. Biol. 8, 684–691 [DOI] [PubMed] [Google Scholar]
- 20. Bayascas J. R., Wullschleger S., Sakamoto K., García-Martínez J. M., Clacher C., Komander D., van Aalten D. M., Boini K. M., Lang F., Lipina C., Logie L., Sutherland C., Chudek J. A., van Diepen J. A., Voshol P. J., Lucocq J. M., Alessi D. R. (2008) Mutation of the PDK1 PH domain inhibits protein kinase B/Akt, leading to small size and insulin resistance. Mol. Cell Biol. 28, 3258–3272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sarbassov D. D., Guertin D. A., Ali S. M., Sabatini D. M. (2005) Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101 [DOI] [PubMed] [Google Scholar]
- 22. Fayard E., Tintignac L. A., Baudry A., Hemmings B. A. (2005) Protein kinase B/Akt at a glance. J. Cell Sci. 118, 5675–5678 [DOI] [PubMed] [Google Scholar]
- 23. Manning B. D., Cantley L. C. (2007) AKT/PKB signaling: navigating downstream. Cell 129, 1261–1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Stambolic V., Woodgett J. R. (2006) Functional distinctions of protein kinase B/Akt isoforms defined by their influence on cell migration. Trends Cell Biol. 16, 461–466 [DOI] [PubMed] [Google Scholar]
- 25. Carnero A. (2010) The PKB/AKT pathway in cancer. Curr. Pharm. Des. 16, 34–44 [DOI] [PubMed] [Google Scholar]
- 26. Pendaries C., Tronchère H., Arbibe L., Mounier J., Gozani O., Cantley L., Fry M. J., Gaits-Iacovoni F., Sansonetti P. J., Payrastre B. (2006) PtdIns5P activates the host cell PI3-kinase/Akt pathway during Shigella flexneri infection. EMBO J. 25, 1024–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sauvonnet N., Lambermont I., van der Bruggen P., Cornelis G. R. (2002) YopH prevents monocyte chemoattractant protein 1 expression in macrophages and T-cell proliferation through inactivation of the phosphatidylinositol 3-kinase pathway. Mol. Microbiol. 45, 805–815 [DOI] [PubMed] [Google Scholar]
- 28. Wiles T. J., Dhakal B. K., Eto D. S., Mulvey M. A. (2008) Inactivation of host Akt/protein kinase B signaling by bacterial pore-forming toxins. Mol. Biol. Cell 19, 1427–1438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nakayama M., Hisatsune J., Yamasaki E., Isomoto H., Kurazono H., Hatakeyama M., Azuma T., Yamaoka Y., Yahiro K., Moss J., Hirayama T. (2009) Helicobacter pylori VacA-induced inhibition of GSK3 through the PI3K/Akt signaling pathway. J. Biol. Chem. 284, 1612–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yilmaz O., Jungas T., Verbeke P., Ojcius D. M. (2004) Activation of the phosphatidylinositol 3-kinase/Akt pathway contributes to survival of primary epithelial cells infected with the periodontal pathogen Porphyromonas gingivalis. Infect. Immun. 72, 3743–3751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yao L., Jermanus C., Barbetta B., Choi C., Verbeke P., Ojcius D. M., Yilmaz O. (2010) Porphyromonas gingivalis infection sequesters pro-apoptotic Bad through Akt in primary gingival epithelial cells. Mol. Oral Microbiol. 25, 89–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Blair P., Rex S., Vitseva O., Beaulieu L., Tanriverdi K., Chakrabarti S., Hayashi C., Genco C. A., Iafrati M., Freedman J. E. (2009) Stimulation of Toll-like receptor 2 in human platelets induces a thromboinflammatory response through activation of phosphoinositide 3-kinase. Circ. Res. 104, 346–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Martin M., Schifferle R. E., Cuesta N., Vogel S. N., Katz J., Michalek S. M. (2003) Role of the phosphatidylinositol 3 kinase-Akt pathway in the regulation of IL-10 and IL-12 by Porphyromonas gingivalis lipopolysaccharide. J. Immunol. 171, 717–725 [DOI] [PubMed] [Google Scholar]
- 34. Park S. Y., Park da J., Kim Y. H., Kim Y., Choi Y. W., Lee S. J. (2011) Schisandra chinensis alpha-iso-cubebenol induces heme oxygenase-1 expression through PI3K/Akt and Nrf2 signaling and has anti-inflammatory activity in Porphyromonas gingivalis lipopolysaccharide-stimulated macrophages. Int. Immunopharm. 11, 1907–1915 [DOI] [PubMed] [Google Scholar]
- 35. Park Y. D., Kim Y. S., Jung Y. M., Lee S. I., Lee Y. M., Bang J. B., Kim E. C. (2012) Porphyromonas gingivalis lipopolysaccharide regulates interleukin (IL)-17 and IL-23 expression via SIRT1 modulation in human periodontal ligament cells. Cytokine 60, 284–293 [DOI] [PubMed] [Google Scholar]
- 36. Gutiérrez-Venegas G., Contreras-Sánchez A. (2013) Luteolin and fisetin inhibit the effects of lipopolysaccharide obtained from Porphyromonas gingivalis in human gingival fibroblasts. Mol. Biol. Rep. 40, 477–485 [DOI] [PubMed] [Google Scholar]
- 37. Urnowey S., Ansai T., Bitko V., Nakayama K., Takehara T., Barik S. (2006) Temporal activation of anti- and pro-apoptotic factors in human gingival fibroblasts infected with the periodontal pathogen, Porphyromonas gingivalis: potential role of bacterial proteases in host signalling. BMC Microbiol. 6, 26. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 38. Ishikawa M., Yoshida K., Okamura H., Ochiai K., Takamura H., Fujiwara N., Ozaki K. (2013) Oral Porphyromonas gingivalis translocates to the liver and regulates hepatic glycogen synthesis through the Akt/GSK-3beta signaling pathway. Biochim. Biophys. Acta 1832, 2035–2043 [DOI] [PubMed] [Google Scholar]
- 39. Slomiany B. L., Slomiany A. (2011) Ghrelin protects against the detrimental consequences of Porphyromonas gingivalis-induced Akt inactivation through S-nitrosylation on salivary mucin synthesis. Int. J. Inflam. 2011, 807279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shi Y., Ratnayake D. B., Okamoto K., Abe N., Yamamoto K., Nakayama K. (1999) Genetic analyses of proteolysis, hemoglobin binding, and hemagglutination of Porphyromonas gingivalis: construction of mutants with a combination of rgpA, rgpB, kgp, and hagA. J. Biol. Chem. 274, 17955–17960 [DOI] [PubMed] [Google Scholar]
- 41. Nakayama K., Kadowaki T., Okamoto K., Yamamoto K. (1995) Construction and characterization of arginine-specific cysteine proteinase (Arg-gingipain)-deficient mutants of Porphyromonas gingivalis: evidence for significant contribution of Arg-gingipain to virulence. J. Biol. Chem. 270, 23619–23626 [DOI] [PubMed] [Google Scholar]
- 42. Saito A., Kokubu E., Inagaki S., Imamura K., Kita D., Lamont R. J., Ishihara K. (2012) Porphyromonas gingivalis entry into gingival epithelial cells modulated by Fusobacterium nucleatum is dependent on lipid rafts. Microb. Pathog. 53, 234–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chang L. C., Kuo H. C., Chang S. F., Chen H. J., Lee K. F., Lin T. H., Huang T. Y., Choe C. S., Lin L. T., Chen C. N. (2013) Regulation of ICAM-1 expression in gingival fibroblasts infected with high-glucose-treated P. gingivalis. Cell Microbiol. 15, 1722–1734 [DOI] [PubMed] [Google Scholar]
- 44. Furuta N., Takeuchi H., Amano A. (2009) Entry of Porphyromonas gingivalis outer membrane vesicles into epithelial cells causes cellular functional impairment. Infect. Immun. 77, 4761–4770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Moffatt C. E., Inaba H., Hirano T., Lamont R. J. (2012) Porphyromonas gingivalis SerB-mediated dephosphorylation of host cell cofilin modulates invasion efficiency. Cell Microbiol. 14, 577–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tsuda K., Furuta N., Inaba H., Kawai S., Hanada K., Yoshimori T., Amano A. (2008) Functional analysis of α5β1 integrin and lipid rafts in invasion of epithelial cells by Porphyromonas gingivalis using fluorescent beads coated with bacterial membrane vesicles. Cell Struct. Funct. 33, 123–132 [DOI] [PubMed] [Google Scholar]
- 47. Persad S., Attwell S., Gray V., Mawji N., Deng J. T., Leung D., Yan J., Sanghera J., Walsh M. P., Dedhar S. (2001) Regulation of protein kinase B/Akt-serine 473 phosphorylation by integrin-linked kinase: critical roles for kinase activity and amino acids arginine 211 and serine 343. J. Biol. Chem. 276, 27462–27469 [DOI] [PubMed] [Google Scholar]
- 48. Mao S., Park Y., Hasegawa Y., Tribble G. D., James C. E., Handfield M., Stavropoulos M. F., Yilmaz O., Lamont R. J. (2007) Intrinsic apoptotic pathways of gingival epithelial cells modulated by Porphyromonas gingivalis. Cell Microbiol. 9, 1997–2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Furuta N., Tsuda K., Omori H., Yoshimori T., Yoshimura F., Amano A. (2009) Porphyromonas gingivalis outer membrane vesicles enter human epithelial cells via an endocytic pathway and are sorted to lysosomal compartments. Infect. Immun. 77, 4187–4196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Peña-Llopis S., Vega-Rubin-de-Celis S., Schwartz J. C., Wolff N. C., Tran T. A., Zou L., Xie X. J., Corey D. R., Brugarolas J. (2011) Regulation of TFEB and V-ATPases by mTORC1. EMBO J. 30, 3242–3258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. MacGurn J. A., Hsu P. C., Smolka M. B., Emr S. D. (2011) TORC1 regulates endocytosis via Npr1-mediated phosphoinhibition of a ubiquitin ligase adaptor. Cell 147, 1104–1117 [DOI] [PubMed] [Google Scholar]
- 52. Forsberg M., Blomgran R., Lerm M., Särndahl E., Sebti S. M., Hamilton A., Stendahl O., Zheng L. (2003) Differential effects of invasion by and phagocytosis of Salmonella typhimurium on apoptosis in human macrophages: potential role of Rho-GTPases and Akt. J. Leukoc. Biol. 74, 620–629 [DOI] [PubMed] [Google Scholar]
- 53. Allen L. A., Allgood J. A., Han X., Wittine L. M. (2005) Phosphoinositide3-kinase regulates actin polymerization during delayed phagocytosis of Helicobacter pylori. J. Leukoc. Biol. 78, 220–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Agarwal V., Hammerschmidt S. (2009) Cdc42 and the phosphatidylinositol 3-kinase-Akt pathway are essential for PspC-mediated internalization of pneumococci by respiratory epithelial cells. J. Biol. Chem. 284, 19427–19436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wang J. H., Zhang K., Wang N., Qiu X. M., Wang Y. B., He P. (2013) Involvement of phosphatidylinositol 3-kinase/Akt signaling pathway in β1 integrin-mediated internalization of Staphylococcus aureus by alveolar epithelial cells. J. Microbiol. 51, 644–650 [DOI] [PubMed] [Google Scholar]
- 56. Lee C. H., Lin S. T., Liu J. J., Chang W. W., Hsieh J. L., Wang W. K. (2014) Salmonella induce autophagy in melanoma by the downregulation of AKT/mTOR pathway. Gene. Ther. 21, 309–316 [DOI] [PubMed] [Google Scholar]
- 57. Laplante M., Sabatini D. M. (2009) mTOR signaling at a glance. J. Cell Sci. 122, 3589–3594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Quy P. N., Kuma A., Pierre P., Mizushima N. (2013) Proteasome-dependent activation of mammalian target of rapamycin complex 1 (mTORC1) is essential for autophagy suppression and muscle remodeling following denervation. J. Biol. Chem. 288, 1125–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Cho T. J., Wee S. W., Woo V. H., Choi J. I., Kim S. J., Shin H. I., Lee J. H., Park H. R. (2014) Porphyromonas gingivalis-induced autophagy suppresses cell proliferation through G1 arrest in oral cancer cells. Arch. Oral Biol. 59, 370–378 [DOI] [PubMed] [Google Scholar]
- 60. Stafford P., Higham J., Pinnock A., Murdoch C., Douglas C. W., Stafford G. P., Lambert D. W. (2013) Gingipain-dependent degradation of mammalian target of rapamycin pathway proteins by the periodontal pathogen Porphyromonas gingivalis during invasion. Mol. Oral Microbiol. 28, 366–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Park C. H., Kim Y. S., Kim Y. H., Choi M. Y., Yoo J. M., Kang S. S., Choi W. S., Cho G. J. (2008) Calcineurin mediates AKT dephosphorylation in the ischemic rat retina. Brain Res. 1234, 148–157 [DOI] [PubMed] [Google Scholar]
- 62. Yang W. L., Wu C. Y., Wu J., Lin H. K. (2010) Regulation of Akt signaling activation by ubiquitination. Cell Cycle 9, 487–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Maira S. M., Galetic I., Brazil D. P., Kaech S., Ingley E., Thelen M., Hemmings B. A. (2001) Carboxyl-terminal modulator protein (CTMP), a negative regulator of PKB/Akt and v-Akt at the plasma membrane. Science 294, 374–380 [DOI] [PubMed] [Google Scholar]
- 64. Zhou Q., Leeman S. E., Amar S. (2009) Signaling mechanisms involved in altered function of macrophages from diet-induced obese mice affect immune responses. Proc. Natl. Acad. Sci. U.S.A. 106, 10740–10745 [DOI] [PMC free article] [PubMed] [Google Scholar]