

Background: CsyB from Aspergillus oryzae catalyzes the condensation of two β-ketoacyl units.
Results: Crystal structures of CsyB revealed a novel pocket for accommodating the acetoacetyl-CoA starter.
Conclusion: CsyB catalyzes the remarkable one-pot condensation of two β-ketoacyl units within a single active site.
Significance: Structure-function analyses of CsyB provide insights into molecular bases for polyketide coupling reactions.
Keywords: Biosynthesis, Enzyme, Enzyme Mechanism, Polyketide, Structural Biology
Abstract
The acylalkylpyrone synthase CsyB from Aspergillus oryzae catalyzes the one-pot formation of the 3-acyl-4-hydroxy-6-alkyl-α-pyrone scaffold from acetoacetyl-CoA, fatty acyl-CoA, and malonyl-CoA. This is the first type III polyketide synthase that performs not only the polyketide chain elongation but also the condensation of two β-ketoacyl units. The crystal structures of wild-type CsyB and its I375F and I375W mutants were solved at 1.7-, 2.3-, and 2.0-Å resolutions, respectively. The crystal structures revealed a unique active site architecture featuring a hitherto unidentified novel pocket for accommodation of the acetoacetyl-CoA starter in addition to the conventional elongation/cyclization pocket with the Cys-His-Asn catalytic triad and the long hydrophobic tunnel for binding the fatty acyl chain. The structures also indicated the presence of a putative nucleophilic water molecule activated by the hydrogen bond networks with His-377 and Cys-155 at the active site center. Furthermore, an in vitro enzyme reaction confirmed that the 18O atom of the H218O molecule is enzymatically incorporated into the final product. These observations suggested that the enzyme reaction is initiated by the loading of acetoacetyl-CoA onto Cys-155, and subsequent thioester bond cleavage by the nucleophilic water generates the β-keto acid intermediate, which is placed within the novel pocket. The second β-ketoacyl unit is then produced by polyketide chain elongation of fatty acyl-CoA with one molecule of malonyl-CoA, and the condensation with the β-keto acid generates the final products. Indeed, steric modulation of the novel pocket by the structure-based I375F and I375W mutations resulted in altered specificities for the chain lengths of the substrates.
Introduction
The structurally simple type III polyketide synthases (PKSs)4 utilize a single active site to catalyze iterative condensations of CoA thioesters and cyclization of the poly-β-keto intermediates to generate various natural product scaffolds with remarkable biological activities (1–3). The acylalkylpyrone synthase CsyB from Aspergillus oryzae is the first type III PKS that catalyzes not only the polyketide chain elongation but also the one-pot condensation of two β-ketoacyl units to produce the 3-acyl-4-hydroxy-6-alkyl-α-pyrone (AcAP) scaffold (see Fig. 1A) (4–7). Thus, CsyB accepts acetoacetyl-CoA, fatty acyl-CoA, and malonyl-CoA as substrates and performs the decarboxylative condensation of malonyl-CoA with fatty acyl-CoA to produce the β-ketoacyl diketide, which then reacts with acetoacetyl-CoA to yield 3-acetyl-4-hydroxy-6-alkyl-α-pyrone (2), a putative precursor of csypyrone B (see Fig. 1A). In addition, the in vitro enzyme reaction also yields dehydroacetic acid (3-acetyl-4-hydroxy-6-methyl-α-pyrone) (1) and 3-acyl-4-hydroxy-6-alkyl-α-pyrone (3) by the coupling of two molecules of acetoacetyl-CoA and the β-ketoacyl diketide intermediate, respectively.
FIGURE 1.

Reaction schemes of the synthesis of polyketides. A, acylalkylpyrones from fatty acyl-CoA, malonyl-CoA, and acetoacetyl-CoA by CsyB. B, triketide alkylpyrones from C12–C20 fatty acyl-CoA and two molecules of malonyl-CoA by PKS18. C, pentaketide alkylresorcylic acid from C4–C20 fatty acyl-CoA and four molecules of malonyl-CoA by ORAS. D, bisdemethoxycurcumin from two molecules of 4-coumaroyl-CoA and one molecule of malonyl-CoA by CUS.
The A. oryzae CsyB shares 26% amino acid sequence identity with the Mycobacterium tuberculosis alkylpyrone synthase PKS18 (8) and 37% identity with the Neurospora crassa 2′-oxoalkylresorcylic acid synthase (ORAS) (9), which produce tri-/tetraketide alkylpyrones and pentaketide alkylresorcylic acids by iterative condensations of fatty acyl-CoA with two/three molecules and four molecules of malonyl-CoA, respectively (Figs. 1, B and C, and 2). Previous crystallographic studies of M. tuberculosis PKS18 (8) and N. crassa ORAS (9) revealed a long hydrophobic tunnel for binding the fatty acyl chain. This tunnel extends from the active site to the protein surface and is primarily generated by subtle changes of the backbone dihedral angles in the core of the protein. Thus, the functional diversity of the type III PKS enzymes is considered to be derived from slight modulations of the active site architecture (8–18).
FIGURE 2.

Comparison of the primary sequences of A. oryzae CsyB and other type III PKSs. AoCsyB, A. oryzae PCS; AoCsyA, A. oryzae CsyA; NcORAS, N. crassa ORAS; OsCUS, O. sativa CUS; MtPKS18, M. tuberculosis PKS18. The secondary structures of CsyB are also delineated: α helices (blue rectangles), β strands (orange arrows), and loops (red, bold lines) are diagrammed. The catalytic triad of Cys-His-Asn is highlighted red. The residues thought to be important for the steric modulation of the active site in a number of divergent type III PKSs are highlighted in blue.
To clarify the structural details of the remarkable one-pot formation of the AcAP scaffold and to further understand the structure-function relationships of the type III PKS enzymes, we now report the crystal structures of the wild type and mutants of A. oryzae CsyB. The crystal structures revealed the unique active site architecture of CsyB featuring a previously unidentified novel pocket for the accommodation of the acetoacetyl-CoA starter in addition to the conventional elongation/cyclization pocket with the Cys-His-Asn catalytic triad (10) and the long hydrophobic fatty acyl-binding tunnel as observed in M. tuberculosis PKS18 (8) and N. crassa ORAS (9). Furthermore, the presence of a putative nucleophilic water molecule activated by the hydrogen bond networks at the active site center suggested an unusual mechanism for the CsyB-catalyzed one-pot formation of the AcAP scaffold.
EXPERIMENTAL PROCEDURES
Materials
Fatty acyl-CoA, acetoacetyl-CoA, malonyl-CoA, and H218O were obtained from Sigma-Aldrich. Acyl diketide-NACs with various lengths were synthesized according to the published method (19, 20). Oligonucleotides were obtained from Eurofins Genomics.
Compound Analysis
Online LC-ESIMS spectra were measured with an Agilent Technologies HPLC 1100 series coupled to a Bruker Daltonics esquire4000 ion trap mass spectrometer fitted with an ESI source. HRESIMS spectra were measured with an Agilent 1100 series HPLC-microTOF mass spectrometer (JEOL) using electrospray ionization.
Compound Information
The UV and MS data of characterized compounds are as follows. 3-Butyryl-4-hydroxy-6-propyl-2H-pyran-2-one (3a): LC-ESIMS (positive): Rt = 19.8 min. MS, m/z 225 [M + H]+, 207, 167, 155. UV: λmax 311 nm. HRMS (ESI): found for [C12H15O4]− 223.10052; calcd. 223.09703. 6-Butyl-4-hydroxy-3-pentanoyl-2H-pyran-2-one (3b). LC-ESIMS (positive): Rt = 22.9 min. MS, m/z 253 [M + H]+, 235, 181, 169. UV: λmax 312 nm. HRMS (ESI): found for [C14H19O4]− 251.12979; calcd. 251.12833. 3-Hexanoyl-4-hydroxy-6-pentyl-2H-pyran-2-one (3c). LC-ESIMS (positive): Rt = 24.9 min. MS, m/z 281 [M + H]+, 263, 195, 183. UV: λmax 312 nm. HRMS (ESI): found for [C16H23O4]− 279.16399; calcd. 279.15963. 3-Heptanoyl-6-hexyl-4-hydroxy-2H-pyran-2-one (3d). LC-ESIMS (positive): Rt = 27.0 min. MS, m/z 309 [M + H]+, 291, 209, 197. UV: λmax 312 nm. HRMS (ESI): found for [C18H27O4]− 307.19303; calcd. 307.19093. 6-Heptyl-4-hydroxy-3-octanoyl-2H-pyran-2-one (3e). LC-ESIMS (positive): Rt = 28.5 min. MS, m/z 337 [M + H]+, 319, 223, 211. UV: λmax 312 nm. HRMS (ESI): found for [C20H31O4]− 335.22673; calcd. 335.22223. 4-Hydroxy-3-nonanoyl-6-octyl-2H-pyran-2-one (3f). LC-ESIMS (positive): Rt = 32.1 min. MS, m/z 365 [M + H]+, 347, 237, 225. UV: λmax 312 nm. HRMS (ESI): found for [C22H35O4]− 363.25843; calcd. 363.25353.
Structure Determination
Expression, purification, and crystallization of the recombinant A. oryzae CsyB were performed as reported previously (21). The initial phases of the CsyB structure were determined by molecular replacement using the N. crassa ORAS structure (Protein Data Bank code 3EUT) as the search model. Molecular replacement was performed with MOLREP in the CCP4 suite (22, 23). The structure was modified manually with Coot (24) and refined with PHENIX (25). The final crystal data and intensity statistics are summarized in Table 1. A structural similarity search was performed using the Dali program (26). The cavity volumes were calculated with the program CASTP. All crystallographic figures were prepared with PyMOL (Schrödinger, LLC). The refined model includes residues 14–389 in chains A and B, respectively, and 695 molecules of water.
TABLE 1.
Data collection, phasing, and refinement statistics
| CsyB WT | CsyB I375F | CsyB I375W | |
|---|---|---|---|
| Unit cell parameter | |||
| Space group | P21 | P21 | P21 |
| a, b, c (Å) | 70.0, 104.8, 73.5 | 69.9, 104.8, 73.2 | 70.1, 104.6, 73.7 |
| β (°) | 114.4 | 114.3 | 114.4 |
| Resolution range (Å) | 50.00–1.71 (1.81–1.71) | 50.00–2.30 (2.44–2.30) | 50–2.00 (2.12–2.00) |
| Completeness (%) | 96.3 (95.3) | 99.5 (99.1) | 99.6 (99.1) |
| I/σI (%) | 17.3 (5.3) | 22.3 (5.5) | 13.6 (3.8) |
| Rmergea (%) | 5.2 (26.3) | 5.3 (26.8) | 7.7 (35.5) |
| Redundancy | 3.8 (3.9) | 3.8 (3.7) | 3.8 (3.7) |
| No. of observed reflections | 388,694 (62,153) | 161,189 (25,112) | 247,674 (38,739) |
| No. of unique reflections | 101,115 (16,084) | 42,426 (6,766) | 65,058 (10,375) |
| Refinement | |||
| Resolution (Å) | 39.5-1.71 | 41.2-2.30 | 41.2-2.00 |
| Overall Rwork (%) | 17.5 | 17.3 | 17.3 |
| Overall Rfree (%) | 19.9 | 22.3 | 20.4 |
| Total atoms | 6,523 | 6,139 | 6,476 |
| No. of protein atoms | 5,730 | 5,746 | 5,723 |
| No. of waters | 695 | 393 | 655 |
| No. of ligands | 96 | 96 | |
| Average B-factors (Å2) | |||
| Protein atoms | 22.8 | 32.3 | 17.5 |
| Waters | 32.9 | 33.9 | 26.3 |
| Ligands | 34.0 | 32.1 | |
| r.m.s.d.b from ideal | |||
| Bond length (Å) | 0.007 | 0.007 | 0.007 |
| Bond angles (°) | 1.150 | 1.038 | 1.068 |
a Rmerge = ΣhklΣi|Ii(hkl) − I(hkl)|/ΣhklΣiIi(hkl) where I(hkl) is the intensity of reflection hkl, Σhkl is the sum over all reflections, and Σi is the sum over i measurements of reflection hkl.
b Root mean square deviation.
Site-directed Mutagenesis, Expression, and Purification
The plasmids expressing the mutants of A. oryzae CsyB (C123A, C123S, A265V, A265F, I375F, I375W, H377P, and H377F) were constructed with a QuikChange site-directed mutagenesis kit (Stratagene) according to the manufacturer's protocol by using the following pairs of primers (mutated codons are underlined): C123A (5′-CATTTGGTGGCTGTAACGGGCACGAATACAG-3′ and 5′-GGATGCGCTGTATTCGTGCCCGTTACAGCC-3′), C123S (5′-CATTTGGTGGCTGTAACGTCCACGAATACAG-3′ and 5′-GGATGCGCTGTATTCGTGGACGTTACAGCC-3′), A265V (5′-CAAAGGGTATCACGTGATTATTGACAAAC-3′ and 5′-GTTTGTCAATAATCACGTGATACCCTTTG-3′), A265F (5′-CAAAGGGTATCACTTTATTATTGACAAAC-3′ and 5′-GTTTGTCAATAATAAAGTGATACCCTTTG-3′), I375F (5′-GGTCCTGGCAGCTTTTGGCCATGGCATC-3′ and 5′-GATGCCATGGCCAAAAGCTGCCAGGACC-3′), I375W (5′-GGTCCTGGCAGCTTGGGGCCATGGCATC-3′ and 5′-GATGCCATGGCCCCAAGCTGCCAGGACC-3′), H377P (5′-GGCAGCTATCGGCCCGGGCATCACGCTGG-3′ and 5′-CCAGCGTGATGCCCGGGCCGATAGCTGCC-3′), and H377F (5′-GGCAGCTATCGGCTTTGGCATCACGCTGG-3′ and 5′-CCAGCGTGATGCCAAAGCCGATAGCTGCC-3′). The mutant proteins were expressed and purified according to the same procedure used for the wild-type CsyB and were concentrated to 10 mg/ml in 20 mm Tris-HCl (pH 7.5) buffer containing 200 mm NaCl and 2 mm DTT.
Enzyme Assay of CsyB and Mutants
The enzyme assay and product identification were performed as reported previously (6) except for the use of an elution program with a linear gradient of CH3CN from 20 to 100% in H2O containing 0.05% TFA for 30 min that was maintained at 100% for 20 min further to separate the reaction products. The H218O labeling experiment was performed under the same conditions for the wild-type enzyme except for the replacement of H2O with H218O.
Kinetic Parameters of CsyB
A standard reaction contained various chain lengths of diketide-NAC (concentration varied between 0.5 and 1.25 mm) and the purified enzyme in 50 mm potassium phosphate buffer (pH 8.0) in a total volume of 100 μl. After preincubation at 37 °C for 5 min, the reactions were initiated by adding the purified enzyme and then further incubated at 37 °C for 20 min before quenching with 20 μl of 6 m HCl and 80 μl of methanol. The decrease of substrate was quantified by HPLC analysis using a COSMOSIL C18-MS-II column (4.6 × 250 mm; Nacalai Tesque) eluted isocratically with 40% acetonitrile containing 0.05% TFA at a flow rate of 1.0 ml/min. Steady-state kinetic parameters were determined from Lineweaver-Burk plots.
Crystallization and Structure Determination of the I375F and I375W Mutants
The I375F mutant crystals were grown at 20 °C with a 5 mg/ml protein solution in 100 mm PIPES-NaOH (pH 6.5) buffer containing 6% (w/v) PEG 4,000 and 250 mm lithium chloride by using the sitting drop vapor diffusion method. The I375W mutant crystals complexed with CoASH were obtained in 100 mm PIPES-NaOH (pH 6.5) buffer containing 6% (w/v) PEG 4,000, 250 mm lithium chloride, and 2 mm CoASH with a 4.5 mg/ml protein solution. Both crystals were independently transferred into the reservoir solution with 18% (v/v) glycerol as a cryoprotectant and then flash cooled at 100 K in a nitrogen gas stream. Both x-ray diffraction data sets were collected at NW12 of Photon Factory-AR (wavelength, 1.0000 Å) by using an ADSC Quantum 210r charge-coupled device detector. The final model of CsyB I375F consists of residues 13–389 of chain A, residues 13–364 and 366–389 of chain B, and 393 molecules of water. The final model of CsyB I375W consists of residues 14–361 and 366–389 of chain A, residues 13–389 of chain B, and 655 molecules of water. Crystal data and intensity statistics are summarized in Table 1. Both crystals belonged to space group P21, and the unit cell dimensions were very similar to those of wild-type CsyB. The initial phases of the CsyB mutant structures were determined by molecular replacement using the wild-type CsyB structure as the search model. Molecular replacement and refinement were performed in the same manner as for the wild-type enzyme. Refinement statistics are given in Table 1. The coordinates and structure factors have been deposited in the Protein Data Bank under codes 3WXY, 3WXZ, and 3WY0 for the A. oryzae CsyB CoASH-complexed structure, CsyB I375F mutant, and CsyB I375W CoASH-complexed structure, respectively.
Molecular Modeling
The three-dimensional models of the enzyme-bound intermediates were generated by the Chem3D Ultra 13 program (CambridgeSoft). The intermediate models were manually swapped with the catalytic Cys-155 in the CsyB structure by using Coot, and the energy minimization calculation by simulated annealing with PHENIX was then performed. The parameters of the intermediates for the energy minimization calculation were obtained by the PRODRG server.
RESULTS
Overall Structure of A. oryzae CsyB
The crystal structure of the wild-type CsyB as a complex structure with CoASH was solved by the molecular replacement method and refined to 1.7-Å resolution (Table 1). The asymmetric unit contained two nearly identical monomers with root mean square deviations of 0.1 Å for the Cα atoms. The overall structure of the homodimeric CsyB revealed the conservation of the αβαβα thiolase fold as observed in other type III PKSs (Fig. 3A) (1–3). The catalytic triad of Cys-155, His-310, and Asn-343 is buried deep within each monomer and sits at the intersection of a 16-Å-long CoA-binding tunnel and a large internal cavity in a location and orientation very similar to those of the previously reported plant and microbial type III PKSs (8–18). The CoA-binding tunnel is connected to the protein surface, thus facilitating the loading of the substrate into the catalytic center (Fig. 3B). The overall structure of CsyB is thus highly homologous to those of other type III PKSs, including N. crassa ORAS (9), Gerbera hybrida 2-pyrone synthase (11), and Oryza sativa CUS (15) with root mean square deviations of 1.8, 1.9, and 2.0 Å, respectively, for the Cα atoms.
FIGURE 3.
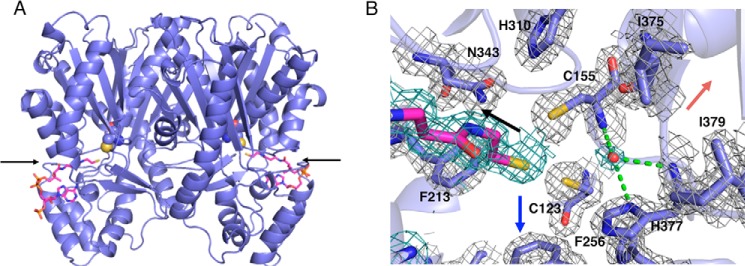
Overall structures of A. oryzae CsyB and close-up view of its active site. A, overall structure of A. oryzae CsyB wild type. The structure is represented by a schematic model. The catalytic Cys-155 and the substrate entrance are represented by a Corey-Pauling-Koltun model and an arrow, respectively. The CoASH molecules are magenta stick models. B, The Fo − Fc density map of the active site residues, CoASH, and putative nucleophilic water molecule in monomer A contoured at +1.0 σ. The density of CoA and water molecules are represented as dark green mesh. The water molecule and the hydrogen bonds are indicated with a red sphere and green dotted lines, respectively. Black, red, and blue arrows indicate the orientation of CoA-binding tunnel, novel pocket, and fatty acyl chain-binding tunnel, respectively.
In A. oryzae CsyB, the conserved active site “gatekeeper” Phe-265 (in CHS numbering) is characteristically substituted with Ala-265 (Figs. 2 and 4A) (1, 2). A similar large-to-small substitution is also observed in the previously reported curcuminoid-producing O. sativa CUS, which catalyzes the coupling reaction of a β-ketoacyl diketide intermediate with coumaroyl-CoA (15). As in the case of CUS, we propose that this substitution not only expands the active site entrance but also facilitates the movement of the enzyme-bound β-ketoacyl intermediate. Indeed, when Ala-265 was replaced with Phe or Val, both mutants almost completely lost enzyme activity (Fig. 5). Conversely, the active site Ser-338 (in CHS numbering), a neighbor of the catalytic Cys-155, is substituted with a hydrophobic Ile-345 (Figs. 2 and 4A). The hydrophobic substitution is also observed in several type III PKSs, including the alkylpyrone-producing M. tuberculosis PKS18 (8) and methylpyrone-producing G. hybrida 2-pyrone synthase, which is thought to be important for modulation of the catalytic activity (11). Most importantly, Pro-375 (in CHS numbering), which is conserved in all type III PKSs, is uniquely substituted with His-377, which plays an important role in the construction of the active site architecture as discussed later (Fig. 4, D–F). Moreover, another residue, His-128, corresponding to Met-137 of CHS, protrudes into the other monomer by the formation of a cis-peptide bond between His-128 and Pro-129 on a loop. However, in contrast to many of the typical type III PKSs, His-128 is not involved in the formation of part of the active site wall as observed in M. tuberculosis PKS18 (8), N. crassa ORAS (9), and O. sativa CUS (15). Finally, CsyB has the second Cys-123 in the active site; however, when it was mutated to Ser or Ala, no significant change in the AcAP-producing activity was observed, indicating that the residue is not essential for the enzyme activity.
FIGURE 4.

Comparison of the active site structures of A. oryzae CsyB and other type III PKSs. A–C, comparison of the active site cavity of A. oryzae CsyB (A), N. crassa ORAS (B), and M. tuberculosis PKS18 (C). The C16 fatty acid molecule in N. crassa ORAS is shown as a green stick model. D–F, close-up views of the novel pocket of A. oryzae CsyB (D) and corresponding regions of N. crassa ORAS (E) and M. tuberculosis PKS18 (F). The arrows indicate the entrance of cavity. The elongation/cyclization pocket, the downward expanding hydrophobic tunnel, and the CsyB-specific novel pocket described under “Results” are colored pink, green, and blue, respectively. The residues mutated in this study are labeled in red. G–J, close-up views of the electronic hydrogen bond networks of A. oryzae CsyB (G), P. sylvestris stilbene synthase (H), O. sativa CUS (I), and R. palmatum benzalacetone synthase (J). Catalytic residues are highlighted by a red square. The water molecules and the hydrogen bonds are indicated with red spheres and green dotted lines, respectively.
FIGURE 5.
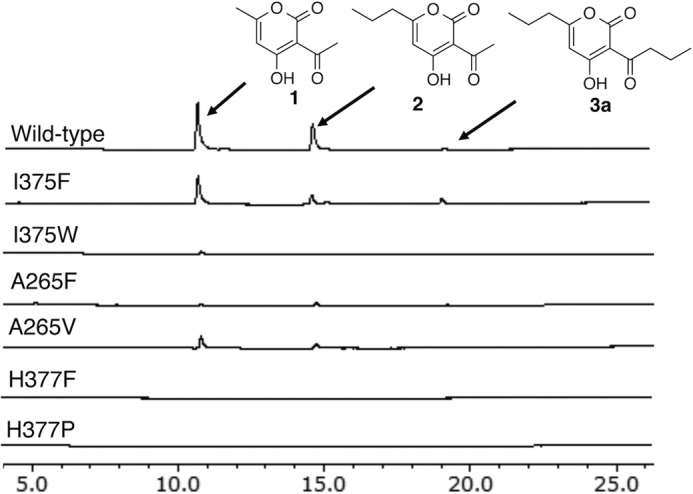
The results of mutagenesis analyses. HPLC elution profiles of enzyme reaction products of the wild-type CsyB and its I375F, I375W, A265F, A265V, H377F, and H377P mutant enzymes are shown.
Active Site Architecture of A. oryzae CsyB
The crystal structures revealed that the active site of A. oryzae CsyB is composed of three sections (Fig. 4A): (i) the conventional elongation/cyclization pocket with the Cys-His-Asn catalytic triad, which is connected to the substrate entrance through the 16-Å-long CoA-binding tunnel (10); (ii) the downward expanding hydrophobic tunnel for binding the medium-length (C2–C15) fatty acyl chain, which extends from the active site center to the protein surface, as also observed in the alkylpyrone-producing M. tuberculosis PKS18 (8) and the alkylresorcylic acid-producing N. crassa ORAS (9); and (iii) the CsyB-specific novel pocket for the accommodation of the acetoacetyl-CoA starter or the short-chain (C4–C7) β-ketoacyl diketide unit as discussed below.
Notably, most of the amino acid residues constituting the elongation/cyclization pocket are superimposable in nearly identical positions in the plant and microbial type III PKS enzymes (Fig. 4A). Conversely, in the elongation/cyclization pocket, Ile-254 (in CHS numbering) is characteristically altered to Ala-254 in CsyB. As a result of this large-to-small substitution, the size of the elongation/cyclization pocket of CsyB (581 Å3) is 1.2 times larger than those of M. sativa CHS (485 Å3) and N. crassa ORAS (489 Å3).
The fatty acyl chain-binding tunnel of CsyB (∼12 Å long) is significantly shorter than those of the longer (C12–C20) fatty acyl chain-accepting M. tuberculosis PKS18 (17 Å) and N. crassa ORAS (20 Å) (Fig. 4, A–C). These tunnels are formed by subtle conformational changes in the regions corresponding to residues 204–209 in PKS18 and 185–190 in ORAS as compared with the region of residues 193–198 in the plant CHS, respectively. In CsyB, this conformational change is also observed in the corresponding region of residues 189–193; however, the presence of the bulky side chains of the CsyB-specific Leu-34 and Trp-74 make the tunnel shorter than those of PKS18 and ORAS (Fig. 4, A–C). As a result, the estimated total cavity volume of CsyB (1,056 Å3) is smaller than those of M. tuberculosis PKS18 (1,682 Å3) and N. crassa ORAS (1,495 Å3), but it is large enough to accommodate the fatty acyl chain (C2–C15) of the substrate and the diketide intermediate, which is incorporated into the 6-alkyl moiety of the AcAP scaffold.
The CsyB-specific novel additional pocket, which is adjacent to the conventional elongation/cyclization pocket, is about 8 Å long and consists of hydrophobic residues, including Ala-254, Ile-271, Leu-274, Ile-375, and Leu-381 (Figs. 4D and 6A). The backbone torsion angle of Ile-375 (−86°, 126°) is slightly shifted by a φ angle of −43° and a ψ angle of +14° in comparison with that of Phe-373 (−129°, 140°), and the side chain of Ile-375 protrudes toward Thr-293. In contrast, the Phe-373 residue in CHS and the corresponding residues in other type III PKSs protrude in a totally different direction toward Phe-371 in CHS (Fig. 4, D–F).
FIGURE 6.

Close-up views of the novel pockets of CsyB and its mutants. A, CsyB wild type. B, CsyB I375F. C, CsyB I375W. D, the substrate specificities of wild-type CsyB and the I375F mutant for various lengths of diketide-NAC substrates.
Putative Nucleophilic Water Molecule at the Catalytic Center
In the previously reported crystal structures of the plant type III PKSs, such as the stilbene-producing Pinus sylvestris stilbene synthase (12), the curcuminoid-producing O. sativa CUS (15), and the benzalacetone-producing Rheum palmatum benzalacetone synthase (16), thioesterase-like electronic hydrogen bond networks and activated water molecules (Fig. 4, G–J) play critical roles in the enzyme reactions. For example, P. sylvestris stilbene synthase utilizes the nucleophilic water molecule activated by the so-called “aldol switch” hydrogen bond network neighboring the catalytic Cys (Fig. 4H), for thioester bond cleavage of the enzyme-bound intermediate to produce the stilbene scaffold (12). Conversely, in O. sativa CUS (15) and R. palmatum benzalacetone synthase (16), the cleavage of the thioester bond of the enzyme-bound intermediate by the activated water molecules (Fig. 4, I and J) terminates the polyketide chain elongation at the diketide stage to generate the β-keto diketide acid intermediates. The crystal structure of A. oryzae CsyB revealed the presence of a similar putative nucleophilic water molecule, which forms hydrogen bond networks with the CsyB-specific His-377 and the main chain of the catalytic Cys-155 at the active site center (Figs. 3B and 4G). When His-377 was changed to Pro or Phe, both mutants completely lost enzyme activity (Fig. 5). These observations suggested that His-377 plays important roles in the activation of a water molecule and the thioester bond cleavage of the enzyme-bound intermediate. However, only the presence of the putative nucleophilic water molecule at the active site center does not exclude the possibility that the CsyB-catalyzed enzyme reaction proceeds without the involvement of the water molecule. For example, the coupling of the enzyme-bound β-ketoacyl intermediate with the “β-ketoacyl-CoA” but not with the “β-keto acid” would afford the AcAP scaffold (Fig. 7). In this case, the oxygen atom from the water molecule is not incorporated into the final product. To test this possibility, we performed the enzyme reaction in the presence of H218O using the C5-diketide-NAC as a substrate. As a result, the obtained product 3b afforded parent ion peaks [M + H]+ at m/z 253 and m/z 255 in addition to m/z 251, indicating the incorporation of two 18O atoms into the product (Fig. 8A). The percentage yields of m/z 251, m/z 253, and m/z 255 were 24, 45, and 31%, respectively (Table 2). Furthermore, when the 18O-labeled 3b was incubated in H216O, the m/z 255 peak was dramatically decreased, and when unlabeled 3b was incubated in H218O, the m/z 253 peak newly appeared after 5 h (Fig. 8B).
FIGURE 7.

A scheme of acylalkylpyrone formed from β-keto acid (Α) and β-ketoacyl-CoA (B). The β-keto acid is produced by cleavage of enzyme-bound intermediate using an activated water molecule. Red highlighted atoms are derived from a water molecule.
FIGURE 8.

Comparisons of HR-ESI-MS spectra. A, 3b produced by CsyB in 80% H218O and the products of further incubation with 18O-labeled 3b in H216O without enzyme. B, 3b produced by CsyB in H216O and the products of further incubation with non-labeled 3b in H218O without enzyme. C, the structure of 3b and m/z of 18O-incorporated 3b.
TABLE 2.
Extent of 18O incorporation in 3b under different conditions
| m/z | Enzyme reaction in H216O | Enzyme reaction in H218O | Incubation of non-labeled 3b in H218O | Incubation of 18O-labeled 3b in H216O |
|---|---|---|---|---|
| % | % | % | % | |
| 251 (0 18O) | 100 | 24 | 30 | 45 |
| 253 (1 18O) | 0 | 45 | 70 | 52 |
| 255 (2 18O) | 0 | 31 | 0 | 3 |
Steric Modulation of the CsyB-specific Novel Additional Pocket
To further understand the intimate structural details of the CsyB-catalyzed enzyme reaction, we performed site-directed mutagenesis of the CsyB-specific Ile-375, which is the crucial residue for the creation of the novel pocket for the acetoacetyl-CoA starter binding. As a result, the I375W mutant lost enzyme activity, whereas the I375F mutant maintained activity comparable with that of the wild-type CsyB (Fig. 5).
The diffraction quality protein crystals of the I375F and I375W mutants were obtained in conditions similar to those of the wild-type CsyB, and the apo crystal structure of I375F mutant and the crystal structure of I375W mutant with CoASH were solved at 2.3- and 2.0-Å resolution, respectively. The overall structures of I375F and I375W mutants were nearly identical to that of the wild type with root mean square deviations of 0.2 and 0.4 Å, respectively. Interestingly, the hydrogen bond network-activated water molecule is also conserved in the structures of both mutants. The total active site cavity volume of the I375F mutant was increased from 1,056 to 1,135 Å3. This is due to the loss of the Cγ-methyl group of isoleucine and the subtle molecular movement of the Cα atoms of Cys-278 toward the opposite side of the novel pocket by the replacement of the Cδ-methyl group of isoleucine with the bulkier aromatic ring (Fig. 6B). In contrast, the cavity size of the I375W mutant was decreased to 983 Å3. The backbone torsion angle of Trp-375 (−72°, 134°) in comparison with that of Ile-375 (−86°, 126°) is slightly shifted by a φ angle of −14° and a ψ angle of −8°, and the side chain of Trp-375 inclines toward the novel pocket. Furthermore, Ile-253 and Ala-254 protrude toward the novel pocket generated by the large movement of residues 247–255 in the loop region. In addition to the small-to-large substitution, the conformational change and the movement of the loop region decreased the size of the active site pocket of the I375W mutant (Fig. 6C).
The expansion of the CsyB-specific novel additional pocket by the I375F substitution suggested that the substrate and product specificities of the enzyme reaction may be altered. To test this hypothesis, we performed the enzyme reactions using various lengths of β-ketoacyl diketide-NAC thioesters as substrates. The wild-type CsyB accepts two molecules of a short-chain (C4–C7) β-ketoacyl diketide-NAC and catalyzes coupling reactions to yield the single products 3b–3d with various lengths of the alkyl and acyl chains (Fig. 6D). Interestingly, the I375F mutant with the expanded pocket accepted all of the (C4–C9) β-ketoacyl diketide-NAC substrates to yield the unnatural novel products 3b–3f (Fig. 6D). The steady-state kinetics values of CsyB wild type and I375F mutant for C4–C6 β-ketoacyl diketide-NAC substrates are summarized in Table 3. The kinetic parameters for the reactions of C7–C9 β-ketoacyl diketide-NAC substrates were not determined because of too low efficacy of these reactions. The steady-state kinetics values of CsyB I375F for β-ketoacyl diketide-NAC were ∼1.2-fold better than those of CsyB wild type.
TABLE 3.
Steady-state kinetic parameters of CsyB wild-type and I375F mutant enzymes
| Substrate | Km | kcat | kcat/Km |
|---|---|---|---|
| mm | min−1 | s−1 m−1 | |
| A. oryzae CsyB WT | |||
| C4-diketide-NAC | 6.4 ± 1.2 | 27.5 ± 5.8 | 72.3 |
| C5-diketide-NAC | 3.1 ± 1.6 | 17.0 ± 6.6 | 90.2 |
| C6-diketide-NAC | 6.8 ± 1.6 | 42.7 ± 8.1 | 104.9 |
| A. oryzae CsyB I375F | |||
| C4-diketide-NAC | 7.1 ± 2.6 | 37.2 ± 11.1 | 87.5 |
| C5-diketide-NAC | 1.4 ± 0.6 | 8.7 ± 2.5 | 92.9 |
| C6-diketide-NAC | 4.5 ± 1.6 | 32.9 ± 5.4 | 122.2 |
DISCUSSION
In this study, we solved the crystal structures of the novel type III PKS CsyB from A. oryzae and its mutants. The crystal structures revealed a unique active site architecture featuring a hitherto unidentified novel pocket in addition to the conventional elongation/cyclization pocket with the Cys-His-Asn catalytic triad and the 12-Å-long hydrophobic tunnel for binding the fatty acyl chain. Furthermore, the hydrogen bond networks among a water molecule, the CsyB-specific His-377, and the main chain of the catalytic Cys-155 were observed at the active site center. The site-directed mutagenesis of His-377 suggested that this residue is crucial for the enzyme activity, presumably in the activation of a water molecule and the thioester bond cleavage of the enzyme-bound intermediate.
On the basis of these findings, we propose that the CsyB-catalyzed one-pot formation of the AcAP scaffold is initiated by the loading of acetoacetyl-CoA (or a short-chain β-ketoacyl diketide unit) onto the catalytic Cys-155 at the active site. The subsequent reorientation of the enzyme-bound β-ketoacyl unit (Fig. 9A) and the thioester bond cleavage by the nucleophilic water molecule activated by the hydrogen bond networks with His-377 and Cys-155 generate the “first” β-ketoacyl unit, which is placed within the novel additional pocket (Fig. 9B). The fatty acyl-CoA (or the second acetoacetyl-CoA) is then loaded onto the free catalytic Cys-155, and decarboxylative condensation with malonyl-CoA yields the enzyme-bound “second” β-ketoacyl diketide unit (Fig. 9, C and D). The polyketide chain elongation of the fatty acyl substrate is terminated at this diketide stage by a nucleophilic attack from the activated methylene of the first β-ketoacyl diketide unit and the thioester bond cleavage (Fig. 9E). The subsequent formation of the lactone generates the final AcAP products (Fig. 9, F and G).
FIGURE 9.

Proposed mechanism for the CsyB enzymatic reaction. A–F, three-dimensional models of reorientation of the diketide intermediate covalently bound to the catalytic Cys155 (A), hydrolysis of the first β-ketoacyl diketide unit (B), loading of the fatty acyl-CoA onto the catalytic Cys-155 (C), proton abstraction from the stored β-ketoacyl diketide unit (D), coupling reaction between two β-ketoacyl units (E), and lactamization to produce the final products (F). G, schematic representation of the proposed mechanism.
Notably, the in vitro enzyme reaction also yields dehydroacetic acid (3-acetyl-4-hydroxy-6-methyl-α-pyrone) (1) and 3-acyl-4-hydroxy-6-alkyl-α-pyrone (3) by the coupling of two molecules of acetoacetyl-CoA and the short-chain β-ketoacyl diketide intermediate, respectively. Two crucial points of the CsyB-catalyzed enzyme reaction are the termination of the fatty acyl chain elongation at the diketide stage and the generation of the second β-ketoacyl diketide unit for the final coupling reaction. Notably, triketide or tetraketide pyrone by-products are not formed by the condensations of fatty acyl-CoA with two or three molecules of malonyl-CoA.
Interestingly, the in vitro H218O incorporation experiment indicated that one of the two 18O atoms is enzymatically incorporated into the product most likely by the nucleophilic cleavage of the thioester bond of the Cys-bound intermediate to generate the β-keto acid as the intermediate (whereas the other 18O atom is exchangeable and spontaneously incorporated into the molecule). These results thus excluded the possibility of the β-ketoacyl-CoA intermediate but suggested that CsyB catalyzes the coupling of the β-keto acid intermediate and the enzyme-bound β-ketoacyl unit (Fig. 7A). This is the first direct evidence that a type III PKS utilizes a water molecule to produce the β-keto acid intermediate during the enzyme reaction.
The small novel pocket created by the CsyB-specific residues, including His-377 and Ile-375, is about 8 Å and has enough space to accept short-chain acyl diketide acids (Figs. 4D and 6A). Indeed, wild-type CsyB accepts up to C7-diketide-NAC as the substrate, producing α-pyrones with varying chain length at the C-3 position. Furthermore, the in vitro analyses of I375F and I375W mutants along with their crystal structures revealed that the elimination of the pocket causes loss of the coupling activity, whereas its expansion results in broader substrate specificity for the extender substrates (Fig. 6D). These results indicated that CsyB utilizes this novel pocket for accommodation of the acetoacetyl-CoA starter (or a short-chain β-ketoacyl diketide unit), and the size of pocket determines the chain length of the β-ketoacyl diketide that is incorporated into the 3-acyl moiety of the AcAP scaffold.
To the best of our knowledge, O. sativa CUS is the only type III PKS that catalyzes the one-pot condensation of a β-ketoacyl diketide intermediate with an enzyme-bound acyl substrate (Fig. 1D), but this is not the coupling of “two” β-ketoacyl units. In this case, CUS accepts two molecules of 4-coumaroyl-CoA and one molecule of malonyl-CoA as substrates. The previous crystallographic studies revealed that CUS utilizes a similar activated water molecule to generate a 4-coumaroyl diketide acid intermediate, which is then kept within the active site, and the subsequent “decarboxylative” condensation with the second coumaroyl unit yields the linear curcuminoid scaffold (15).
In contrast, CsyB catalyzes the coupling of two β-ketoacyl units in a “non-decarboxylative” manner. Similar coupling reactions were reported for the non-homologous bacterial ketosynthases involved in the biosynthesis of 2,5-dialkylpyrones and 2,5-dialkylresorcinols (27–30). For example, CorB and MxnB from the myxobacteria Corallococcus coralloides and Myxococcus fulvus catalyze the coupling of two β-ketoacyl diketide intermediates to produce the α-pyrone antibiotics corallopyronin and myxopyronin, respectively (27, 28). Furthermore, in photopyrone biosynthesis in Photorhabdus luminescens, PpyS produces a signaling pyrone molecule in a similar manner (29). Conversely, the bacterial ketosynthase DarB catalyzes the coupling of a β-ketoacyl diketide intermediate and an α,β-unsaturated acyl precursor, thereby generating the dialkylcyclohexanedione scaffold, which is further converted to 2,5-dialkylresorcinols by the aromatase DarA in Chitinophaga pinensis (30). Although the fungal CsyB does not share any sequence similarities with these bacterial ketosynthases, the model structure of DarB predicted the presence of a CsyB-like additional pocket in the active site (30). This suggests that these non-homologous bacterial ketosynthases also utilize similar active site architectures to catalyze their coupling reactions.
In conclusion, the crystallographic studies of the fungal A. oryzae CsyB provided the structural basis for the remarkable one-pot formation of the AcAP scaffold by the condensation of two β-ketoacyl units. These findings provide further strategies toward expanding the catalytic repertoire of the versatile type III PKS enzymes and producing structurally divergent, biologically active novel polyketides for drug discovery.
This work was supported in part by a grant-in-aid for scientific research from the Ministry of Education, Culture, Sports, Science and Technology, Japan (to T. M., H. M., and I. A.).
The atomic coordinates and structure factors (codes 3WXY, 3WXZ, and 3WY0) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- PKS
- polyketide synthase
- AcAP
- 3-acyl-4-hydroxy-6-alkyl-α-pyrone
- ORAS
- 2′-oxoalkylresorcylic acid synthase
- CHS
- chalcone synthase
- CUS
- curcuminoid synthase
- NAC
- N-acetylcysteamine
- ESI
- electrospray ionization
- HR
- high resolution
- Rt
- retention time.
REFERENCES
- 1. Abe I., Morita H. (2010) Structure and function of the chalcone synthase superfamily of plant type III polyketide synthases. Nat. Prod. Rep. 27, 809–838 [DOI] [PubMed] [Google Scholar]
- 2. Austin M. B., Noel J. P. (2003) The chalcone synthase superfamily of type III polyketide synthases. Nat. Prod. Rep. 20, 79–110 [DOI] [PubMed] [Google Scholar]
- 3. Katsuyama Y., Horinouchi S. (2010) in Comprehensive Natural Products II (Mander L., Liu H. W., eds) Vol. 1, pp. 147–170, Elsevier Oxford [Google Scholar]
- 4. Seshime Y., Juvvadi P. R., Kitamoto K., Ebizuka Y., Fujii I. (2010) Identification of csypyrone B1 as the novel product of Aspergillus oryzae type III polyketide synthase CsyB. Bioorg. Med. Chem. 18, 4542–4546 [DOI] [PubMed] [Google Scholar]
- 5. Hashimoto M., Ishida S., Seshime Y., Kitamoto K., Fujii I. (2013) Aspergillus oryzae type III polyketide synthase CsyB uses a fatty acyl starter for the biosynthesis of csypyrone B compounds. Bioorg. Med. Chem. Lett. 23, 5637–5640 [DOI] [PubMed] [Google Scholar]
- 6. Hashimoto M., Koen T., Takahashi H., Suda C., Kitamoto K., Fujii I. (2014) Aspergillus oryzae CsyB catalyzes the condensation of two β-ketoacyl-CoAs to form 3-acetyl-4-hydroxy-6-alkyl-α-pyrone. J. Biol. Chem. 289, 19976–19984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hashimoto M., Nonaka T., Fujii I. (2014) Fungal type III polyketide synthases. Nat. Prod. Rep. 31, 1306–1317 [DOI] [PubMed] [Google Scholar]
- 8. Sankaranarayanan R., Saxena P., Marathe U. B., Gokhale R. S., Shanmugam V. M., Rukmini R. (2004) A novel tunnel in mycobacterial type III polyketide synthase reveals the structural basis for generating diverse metabolites. Nat. Struct. Mol. Biol. 11, 894–900 [DOI] [PubMed] [Google Scholar]
- 9. Goyal A., Saxena P., Rahman A., Singh P. K., Kasbekar D. P., Gokhale R. S., Sankaranarayanan R. (2008) Structural insights into biosynthesis of resorcinolic lipids by a type III polyketide synthase in Neurospora crassa. J. Struct. Biol. 162, 411–421 [DOI] [PubMed] [Google Scholar]
- 10. Ferrer J. L., Jez J. M., Bowman M. E., Dixon R. A., Noel J. P. (1999) Structure of chalcone synthase and the molecular basis of plant polyketide biosynthesis. Nat. Struct. Biol. 6, 775–784 [DOI] [PubMed] [Google Scholar]
- 11. Jez J. M., Austin M. B., Ferrer J., Bowman M. E., Schröder J., Noel J. P. (2000) Structural control of polyketide formation in plant-specific polyketide synthases. Chem. Biol. 7, 919–930 [DOI] [PubMed] [Google Scholar]
- 12. Austin M. B., Bowman M. E., Ferrer J. L., Schröder J., Noel J. P. (2004) An aldol switch discovered in stilbene synthases mediates cyclization specificity of type III polyketide synthases. Chem. Biol. 11, 1179–1194 [DOI] [PubMed] [Google Scholar]
- 13. Austin M. B., Izumikawa M., Bowman M. E., Udwary D. W., Ferrer J. L., Moore B. S., Noel J. P. (2004) Crystal structure of a bacterial type III polyketide synthase and enzymatic control of reactive polyketide intermediates. J. Biol. Chem. 279, 45162–45174 [DOI] [PubMed] [Google Scholar]
- 14. Morita H., Kondo S., Oguro S., Noguchi H., Sugio S., Abe I., Kohno T. (2007) Structural insight into chain-length control and product specificity of pentaketide chromone synthase from Aloe arborescens. Chem. Biol. 14, 359–369 [DOI] [PubMed] [Google Scholar]
- 15. Morita H., Wanibuchi K., Nii H., Kato R., Sugio S., Abe I. (2010) Structural basis for the one-pot formation of the diarylheptanoid scaffold by curcuminoid synthase from Oryza sativa. Proc. Natl. Acad. Sci. U.S.A. 107, 19778–19783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Morita H., Shimokawa Y., Tanio M., Kato R., Noguchi H., Sugio S., Kohno T., Abe I. (2010) A structure-based mechanism for benzalacetone synthase from Rheum palmatum. Proc. Natl. Acad. Sci. U.S.A. 107, 669–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Morita H., Yamashita M., Shi S. P., Wakimoto T., Kondo S., Kato R., Sugio S., Kohno T., Abe I. (2011) Synthesis of unnatural alkaloid scaffolds by exploiting plant polyketide synthase. Proc. Natl. Acad. Sci. U.S.A. 108, 13504–13509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mori T., Shimokawa Y., Matsui T., Kinjo K., Kato R., Noguchi H., Sugio S., Morita H., Abe I. (2013) Cloning and structure-function analyses of quinolone- and acridone-producing novel type III polyketide synthases from Citrus microcarpa. J. Biol. Chem. 288, 28845–28858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gilbert I. H., Ginty M., O'Neill J. A., Simpson T. J., Staunton J., Willis C. L. (1995) Synthesis of β-keto and α,β-unsaturated N-acetylcysteamine thioesters. Bioorg. Med. Chem. Lett. 5, 1587–1590 [Google Scholar]
- 20. Oguro S., Akashi T., Ayabe S., Noguchi H., Abe I. (2004) Probing biosynthesis of plant polyketides with synthetic N-acetylcysteamine thioesters. Biochem. Biophys. Res. Commun. 325, 561–567 [DOI] [PubMed] [Google Scholar]
- 21. Yang D., Mori T., Matsui T., Hashimoto M., Morita H., Fujii I., Abe I. (2014) Expression, purification and crystallization of a fungal type III polyketide synthase that produces the csypyrones. Acta Crystallogr. F Struct. Biol. Commun. 70, 730–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vagin A., Teplyakov A. (2010) Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 66, 22–25 [DOI] [PubMed] [Google Scholar]
- 23. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 25. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D. Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Holm L., Sander C. (1995) Dali: a network tool for protein structure comparison. Trends Biochem. Sci. 20, 478–480 [DOI] [PubMed] [Google Scholar]
- 27. Erol O., Schäberle T. F., Schmitz A., Rachid S., Gurgui C., El Omari M., Lohr F., Kehraus S., Piel J., Müller R., König G. M. (2010) Biosynthesis of the myxobacterial antibiotic corallopyronin A. Chembiochem 11, 1253–1265 [DOI] [PubMed] [Google Scholar]
- 28. Sucipto H., Wenzel S. C., Müller R. (2013) Exploring chemical diversity of α-pyrone antibiotics: molecular basis of myxopyronin biosynthesis. Chembiochem 14, 1581–1589 [DOI] [PubMed] [Google Scholar]
- 29. Brachmann A. O., Brameyer S., Kresovic D., Hitkova I., Kopp Y., Manske C., Schubert K., Bode H. B., Heermann R. (2013) Pyrones as bacterial signaling molecules. Nat. Chem. Biol. 9, 573–578 [DOI] [PubMed] [Google Scholar]
- 30. Fuchs S. W., Bozhüyük K. A., Kresovic D., Grundmann F., Dill V., Brachmann A. O., Waterfield N. R., Bode H. B. (2013) Formation of 1,3-cyclohexanediones and resorcinols catalyzed by a widely occurring ketosynthase. Angew. Chem. Int. Ed. Engl. 52, 4108–4112 [DOI] [PubMed] [Google Scholar]