

 This work is licensed under a
This work is licensed under a Abstract
Along with breast and endometrial cancers, thyroid cancer is a major component cancer in Cowden syndrome (CS). Germline variants in SDHB/C/D (SDHx) genes account for subsets of CS/CS-like cases, conferring a higher risk of breast and thyroid cancers over those with only germline PTEN mutations. To investigate whether SDHx alterations at both germline and somatic levels occur in apparently sporadic breast cancer and differentiated thyroid cancer (DTC), we analyzed SDHx genes in the following four groups: i) 48 individuals with sporadic invasive breast adenocarcinoma for germline mutation; ii) 48 (expanded to 241) DTC for germline mutation; iii) 37 pairs DTC tumor-normal tissues for germline and somatic mutation and mRNA expression levels; and iv) data from 476 patients in the Cancer Genome Atlas thyroid carcinoma dataset for validation. No germline SDHx variant was found in a pilot series of 48 breast cancer cases. As germline SDHx variants were found in our pilot of 48 thyroid cancer cases, we expanded to three series of DTC comprising a total 754 cases, and found 48 (6%) with germline SDHx variants (P<0.001 compared with 0/350 controls). In 513 tumors, we found 27 (5%) with large somatic duplications within chromosome 1 encompassing SDHC. Both papillary and follicular thyroid tumors showed consistent loss of SDHC/D gene expression (P<0.001), which is associated with earlier disease onset and higher pathological-TNM stage. Therefore, we conclude that both germline and somatic SDHx mutations/variants occur in sporadic DTC but are very rare in sporadic breast cancer, and overall loss of SDHx gene expression is a signature of DTC.
Keywords: SDHx, variant, gene expression, papillary thyroid cancer
Introduction
Succinate dehydrogenase (SDH) or complex II of the mitochondrial respiratory chain catalyzes the oxidation of succinate to fumarate in the Krebs cycle with electron transfer to the terminal acceptor ubiquinone. The SDH complex has four subunits encoded by autosomal genes SDHA, SDHB, SDHC, and SDHD (reviewed in Eng et al. (2003)). Germline homozygous or compound heterozygous mutations in SDHA result in severe neurological dysfunction, such as Leigh syndrome, a rare but fatal neurodegenerative disease (Bourgeron et al. 1995, Parfait et al. 2000). In contrast, germline heterozygous mutations in the genes encoding the SDH subunits result in hereditary pheochromocytoma–paraganglioma syndrome (Baysal et al. 2000, Neumann et al. 2002, 2004). It was noticed that a rare subset of individuals with germline SDHB or SDHD mutations in the European–American pheochromocytoma registry had renal cancers and papillary thyroid cancer (PTC) (Neumann et al. 2004, Vanharanta et al. 2004).
Cowden syndrome (CS (MIM 158350)) is an autosomal dominant heritable neoplasia syndrome. Because epithelial thyroid, breast, and renal carcinomas are component to CS (Pilarski & Eng 2004, Tan et al. 2012), we wanted to determine whether germline SDHB/C/D (SDHx) variants could also occur in PTEN (MIM 601728) mutation-negative CS/CS-like individuals. Indeed, we found 8–10% of such patients carry SDHx mutations (Ni et al. 2008, 2012). CS/CS-like individuals with germline SDHx variants have significantly higher risks of developing breast cancer and epithelial thyroid cancer compared with PTEN mutation-only carriers. Among all SDHx variant carriers with thyroid cancer in CS/CS-like cases, papillary histology is the major subtype in contrast to the over-presentation of follicular histology in PTEN mutation carriers (Ngeow et al. 2011, Ni et al. 2012).
Family history is an important risk factor for epithelial thyroid cancer: case–control studies have consistently shown that the risk to first-degree relatives of probands is three- to 11-fold, being among the highest of all cancers (Risch 2001, Hemminki et al. 2005). However, no predisposing genes have yet been detected that account for anything but a small portion of all familial and particularly sporadic cases. In 2014, 62 980 new cases of thyroid cancer will be diagnosed and more than 1890 patients will die from it, a number that is rising yearly despite aggressive multi-modal therapy (NationalCancerInstitute). It is worth noting that, in addition to the rising incident in thyroid cancer among all cancers, almost all of the increase is in the papillary histology subtype, the subtype we observed associated with SDHx variations in CS.
Taking all these observations together, we sought to address the hypothesis that alterations in SDHx at both germline and somatic levels may also occur in apparently sporadic breast cancer and differentiated thyroid cancer (DTC).
Research participants, materials and methods
Research participants
Consenting adult patients with invasive female breast adenocarcinomas seen anywhere in the Cleveland Clinic Health System were prospectively accrued. The most proximal 48 research participants diagnosed with breast cancers within this prospective series were enrolled for purposes of our current pilot study of apparently sporadic breast cancers. In parallel, a pilot of 48 then expanded to 241 consecutive patients with differentiated epithelial thyroid carcinoma visiting a multi-disciplinary thyroid tumor clinic at The Ohio State University between August 2006 and September 2007 were prospectively accrued. Individuals were eligible for an IRB-approved research study on individuals with thyroid cancer and completed detailed medical history and family history questionnaires. Participants were also asked to donate a blood sample for genetics research.
Thirty-seven thyroid tumor tissues together with paired adjacent normal tissue samples from Corporative Human Tissue Network (CHTN) are accrued via archival anonymized registry protocol.
Three hundred and fifty normal (population) controls are whites of northern and western European origin and were anonymized before storage and analysis. Informed consent was obtained for all subjects in accordance with procedures and protocols approved by the respective Human Subjects Protection Committee of each participating institution.
Genomic DNA extraction
Genomic DNA was extracted from snap-frozen tumor and adjacent normal tissues using GeneJET Genomic DNA Purification kit (Thermo Scientific, Waltham, MA, USA).
SDHx mutation and copy number variation analysis
Genomic DNA was first analyzed using high-resolution melting LightScanner technology (Idaho Technology, Inc., Salt Lake City, UT, USA), which detects nucleic acid sequence variations by changes in the melting curve. Primers to amplify a total of 20 amplicons spanning the exons, exon–intron junctions and flanking intronic regions as well as promoters of SDHB/C/D were designed using LightScanner Primer Design Software (all primers are listed in Supplementary Table 1, see section on supplementary data given at the end of this article) and optimized according to the manufacturer's instructions. Germline genomic DNA samples were amplified with LCGreen Plus (Idaho Technology, Inc.) in a final reaction volume of 10 μl with 20 μl oil overlay. The temperature cycling protocol consisted of an initial denaturation step at 95 °C for 2 min, followed by 37 cycles of denaturation at 94 °C for 30 s, optimal annealing temperature for each amplicon for 30 s, and heteroduplex formation step at 95 °C for 30 s and final hold at 25 °C. Melting curve analysis was performed on LightScanner with LightScanner software employing three steps, namely, normalization, temperature shift, and generating difference plot to cluster samples. The samples with melting curves that clustered differently from reference samples were directly sequenced for SDHB, SDHC, or SDHD, as previously reported by our laboratory (Mutter et al. 2000, McWhinney et al. 2004).
Deletion analysis using the multiplex ligation-dependent probe amplification (MLPA) assay (Schouten et al. 2002) was performed using the P158 MLPA kit (MRC-Holland, Amsterdam, The Netherlands) according to manufacturer's protocol. Genome-wide SNP array analysis was performed using Illumina HumanOmni2.5 BeadChip containing ∼2.5 million markers. Copy number variation (CNV) was called out by CNV partition plug-in in the Beadstudio Software.
RNA extraction and quantitative RT-PCR
Total RNA was extracted from peripheral blood-derived lymphoblastoid cell lines from controls and patients using the GeneJET RNA Purification kit (Thermo Scientific), according to the manufacturer's protocol, and subsequently treated with DNase I (Invitrogen). DNase-treated total RNA was reverse-transcribed into cDNA using qScript cDNA SuperMix (Quanta BioSciences, Inc., Gaithersburg, MD, USA) as specified by the manufacturer. Quantitative PCR was performed on LightCycler 480 system (Roche Diagnostics Corporation) using the TaqMan primer-probe ready mix for SDHB, SDHC, SDHD, and 18S as endogenous loading control (Life Technologies).
The Cancer Genome Atlas dataset analysis
Whole genome and/or exome sequencing (WGS/WES) .bam files were obtained through the Cancer Genome Atlas (TCGA) project via download from the Cancer Genomics Hub (https://cghub.ucsc.edu). Germline data were generated from peripheral blood samples. Patient clinical information and SNP array data were downloaded from TCGA's Data Portal (https://tcga-data.nci.nih.gov/tcga/tcgaDownload.jsp).
From the .bam sequence files, SDHx gene reads were extracted using SAMtools (Li et al. 2009). The bam2fastq software (http://gsl.hudsonalpha.org/information/software/bam2fastq) was used to revert these aligned sequences to fastq format, which were realigned against hg19 reference genome using bowtie2 (Langmead & Salzberg 2012) and the pileup file was generated using SAMtools. From the pileup file, variants were called using bcftools and the Integrative Genome Viewer (IGV) (Thorvaldsdottir et al. 2013).
The CNV data were collected from TCGA genome_wide_SNP_6 level 3 dataset, using ‘nocnv_hg19.seg’ results downloaded from TCGA's Data Portal, which removed CNV in a panel of more than 3000 blood normals from TCGA and used hg19 as the reference genome.
The gene expression analysis from the TCGA dataset was based on IlluminaHiSeq_RNASeqV2 ‘rsem.gene.normalized_results’ files downloaded from TCGA's Data Portal, in which RNA-seq reads were quantified by upper quartile normalized RNA-seq by expectation maximization (RSEM) count estimates. In order to quantify multiple gene expression as a set, we generated a variable gene_score (range from 0 to 4) by first categorizing each gene's expression as either 1 as higher than normal, or 0 as lower than normal, then adding up the binary score for each of these SDHA–D genes.
The DNA methylation data from Illumina Human Methylation 450 BeadChip array were downloaded from TCGA's Data Portal, where absolute methylation values (β-value) of all available CpG sites for each of SDHx genes were extracted for 500 thyroid tumors and 56 normal tissues. Then the average of β-values from all CpG sites within a CpG island for each gene promoter region was calculated and used for comparison. The CpG island was annotated using City of Hope CpG Island Analysis Pipeline (COHCAP) (Warden et al. 2013) based on Illumina Human Methylation 450k platform with respect to hg19.
Statistical analysis
Statistical analysis was carried out using SPSS (IBM SPSS Statistics for Macintosh, Version 21: IBM Corp., Armonk, NY, USA) with significance at P<0.05.
Results
Germline SDHx variants in apparently sporadic PTC patients but absent in apparently sporadic breast cancer patients
Breast cancer and thyroid cancer are major malignancies associated with CS/CSL; SDHx variant carriers show significantly increased prevalence of both breast and thyroid cancers compared with PTEN mutation carriers (Ni et al. 2012). Therefore, we sought to determine whether SDHx alleles also associate with apparently sporadic breast cancer cases and in apparently sporadic thyroid cancer cases. To pilot this hypothesis, we performed germline mutation scanning in 48 women (median age 45, 37–85 years) with invasive breast cancers. This sample size gave us >80% power to detect a 5% prevalence of SDHx variants. No mutation or variant was identified in these 48 breast cancer cases, and so we did not proceed to a validation series or further studies in breast cancer.
When our pilot of 48 apparently sporadic DTC samples revealed germline variation in SDHB and SDHD, we expanded our series to a total of 241 unrelated PTEN mutation negative research participants with differentiated thyroid carcinoma (Nagy et al. 2011) from The Ohio State University's (OSU) Thyroid Center. Of the 241, we found 15 (6%) with SDHB/D missense variants (P<0.001 compared with 0/350 controls), six in SDHB (Ala3Gly (n=1), and Ser163Pro (n=5)), and nine in SDHD (Gly12Ser (n=6) and His50Arg (n=3)) (Table 1). Consistent with what we observed in our CS/CSL series, the SDHD variants comprise the major proportion (9/15, 60%) of all variants.
Table 1.
Germline SDHx variants in 3 independent series of sporadic differentiated thyroid cancer samples and pooled summary
| Variation | n | |
|---|---|---|
| (A) Germline SDHx variants in PTEN mutation-negative DTC individuals (15/241, 6.2%) | ||
| SDHB (n=6) | c.8C>G, p.Arg3Gly | 1 |
| c.487T>C, p.Ser163Pro | 5 | |
| SDHD (n=9) | c.34G>A, p.Gly12Ser | 6 |
| c.149A>G, p.His50Arg | 3 | |
| (B) Germline SDHx variants in TCGA THCA individuals (28/476, 5.9%) | ||
| SDHB (n=13) | c.8C>G, p.Arg3Gly | 1 |
| c.158G>A, p.Gly53Glu | 1 | |
| c.178A>G, p.Thr60Ala | 1 | |
| c.425A>T, p.Asp142Val | 1 | |
| c.487T>C, p.Ser163Pro | 9 | |
| SDHD (n=15) | c.34G>A, p.Gly12Ser | 10 |
| c.149A>G, p.His50Arg | 5 | |
| (C) Germline SDHx variants in CHTN-paired thyroid tumor samples (5/37, 13.5%) | ||
| SDHB (n=3) | c.8C>G, p.Arg3Gly | 2 |
| c.487T>C, p.Ser163Pro | 1 | |
| SDHD (n=2) | c.149A>G, p.His50Arg | 2 |
| (D) Germline SDHx variants in pooled 754 individuals (48/754, 6.3%) | ||
| SDHB (n=22) | c.8C>G, p.Arg3Gly | 4 |
| c.158G>A, p.Gly53Glu | 1 | |
| c.178A>G, p.Thr60Ala | 1 | |
| c.425A>T, p.Asp142Val | 1 | |
| c.487T>C, p.Ser163Pro | 15 | |
| SDHD (n=26) | c.34G>A, p.Gly12Ser | 16 |
| c.149A>G, p.His50Arg | 10 |
(A) Consecutive series of apparently sporadic PTEN mutation-negative differentiated thyroid cancer (DTC) samples from OSU thyroid center; (B) TCGA thyroid cancer (THCA) samples; (C) CHTN paired thyroid tumor-normal tissue samples; (D) pooled 754 subjects from above three sources.
In order to further confirm our findings, we used the TCGA thyroid cancer (THCA) dataset composed mainly of PTC samples as a validation series for germline SDHx variation. Of all 476 TCGA PTC patients who had WGS/WES .bam files from peripheral blood-derived DNA, a total of 28 (6%) had germline SDHx variants, with 13 in SDHB (Ala3Gly: n=1, Gly53Glu: n=1, Thr60Ala n=1, Asp142Val n=1, and Ser163Pro: n=9) and 15 in SDHD (Gly12Ser: n=10 and His50Arg: n=5) (Table 1). These variants were detected in sequencing data from both blood and corresponding primary tumor samples confirming they are indeed germline. The histological type of these 28 papillary carcinomas includes four of follicular variant form, two of tall cell variant form, and 22 of classical form.
Somatic SDHx alterations in sporadic thyroid tumors
We performed mutation analysis of the SDHx genes in 37 pairs of apparently sporadic epithelial thyroid carcinomas with adjacent normal tissue from CHTN. Among these tumors, seven are follicular in histology (FTC), four follicular variant of papillary histology (FvPTC), and 26 classic papillary histology (cPTC). Missense SDHx variants were identified in five of 37 pairs of samples (SDHB Ala3Gly n=2, Ser163Pro n=1, and SDHD His50Arg n=2) (Table 1), in both tumor and paired adjacent normal samples, confirming their germline origin. The five individuals with germline SDHx variants had three cPTC and two FTC. Thus, there was an overall 6% prevalence of germline SDHB/D variants in the combined datasets comprising 754 thyroid cancer patients compared with 0/350 of our residential population controls (P<0.001) (Table 1).
No somatic intragenic SDHx variants were detected in our 37 thyroid carcinoma samples. Similarly, no somatic intragenic SDHx variants were detected in 476 PTC samples that also had matched blood samples (germline) in the TCGA dataset.
We then searched for somatic large insertions/deletions of the SDHx genes using MLPA in our 37 paired samples. Interestingly, two PTC samples (5%) showed somatic duplication of SDHC (Fig. 1A). To further confirm and investigate the size of the CNV region, we subjected both samples with SDHC somatic duplication to a genome-wide SNP-array analysis. CNV analysis based on SNP array revealed a three-copy duplication region spanning position 157 370 000–249 213 900 bases on chromosome 1, where SDHC is located (chr1: 161 314 376–161 364 751) (Fig. 1B).
Figure 1.

Somatic duplication identified in CHTN papillary thyroid carcinoma samples. (A) Representative MLPA result for two-paired tumor-normal samples: 08E_134-08E_135 as pair and 08E_132-08E_133 as pair; each column represents the relative quantification ratio for each probe included in the kit. (B) Genome-wide SNP array-based CNV analysis on chromosome 1. Left panel showed the large duplication (blue bars) by CNV partition plug-in from GenomeStudio in samples displayed in (A); right panel showed B Allele Frequency plot for the same samples for genomic region 157 000 000–250 000 000.
CNV analysis of the TCGA THCA dataset revealed 25 of 476 (5%) PTC samples also having somatic duplication (three copies) of the same SDHC gene region as we observed in our in-house CHTN tumor samples (Supplementary Table 2, see section on supplementary data given at the end of this article). It is worth noting that the somatic SDHC duplication, resulting in three copies of SDHC in the genome, did not occur in patients with germline SDHB/D variation.
Overall reduced SDHx gene expression in differentiated thyroid carcinoma samples
As a pilot, we initially checked the SDHB/C/D mRNA expression in our 37 paired sporadic thyroid tumor-normal tissue samples (seven FTC and 30 PTC). Despite the two PTC tumors with SDHC duplication showing increased SDHC mRNA expression, significantly reduced transcript expression of SDHC and SDHD was observed in the tumor samples compared with their paired normal tissue (both P<0.001). Of note, FTC samples also had decreased SDHB transcript expression, whereas there was no change in SDHB expression in the PTC samples (Fig. 2).
Figure 2.
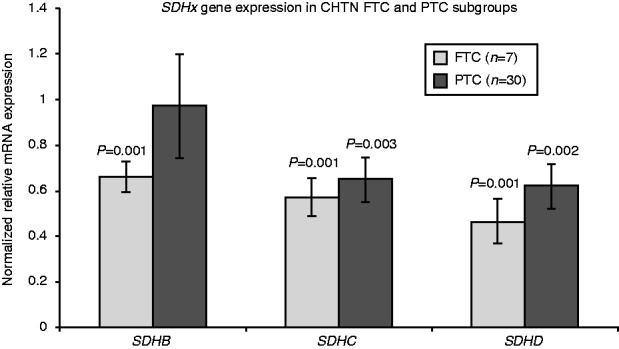
SDHx gene expression in FTC and PTC subgroups of CHTN sporadic thyroid samples. Expression was normalized for tumor sample to its paired normal tissue. Data were presented as mean±s.e.m. and P value was obtained using two-tailed Student's paired sample t-test.
To survey SDHx gene expression in TCGA THCA tumor samples, we extracted normalized RNA-seq read counts for SDHA–D and PTEN genes. Compared with available normal thyroid tissues (n=57), thyroid tumor tissue (n=484) showed significant reduction in PTEN, SDHC, and SDHD gene expression (P<0.001, Fig. 3A). Interestingly, the transcript expression of each of the SDHA/B/C/D genes has strong positive correlations with one another (Supplementary Table 3, see section on supplementary data given at the end of this article). Notably, when we divided the tumors into two groups based on PTEN gene expression status (PTEN_high and PTEN_low, compared with the average of its expression in normal tissue), to mimic the loss of PTEN as has been reported in thyroid tumors (Bruni et al. 2000, Gimm et al. 2000), we found significant reduction in SDHC and SDHD transcript expression in the PTEN_low group compared with the PTEN_high group (P<0.001 and P=0.02 respectively, Fig. 3B). In order to see if the decreased SDHC/D gene expression in tumors is caused by DNA hypermethylation, we checked the DNA methylation levels in SDHC and SDHD gene promoter regions in the TCGA dataset. There was no significant difference in these two genes' promoter methylation levels in tumors compared with normal tissues. The overall promoter methylation is low for SDHC gene (β-value=0.03) and high for SDHD gene (β-value=0.89).
Figure 3.
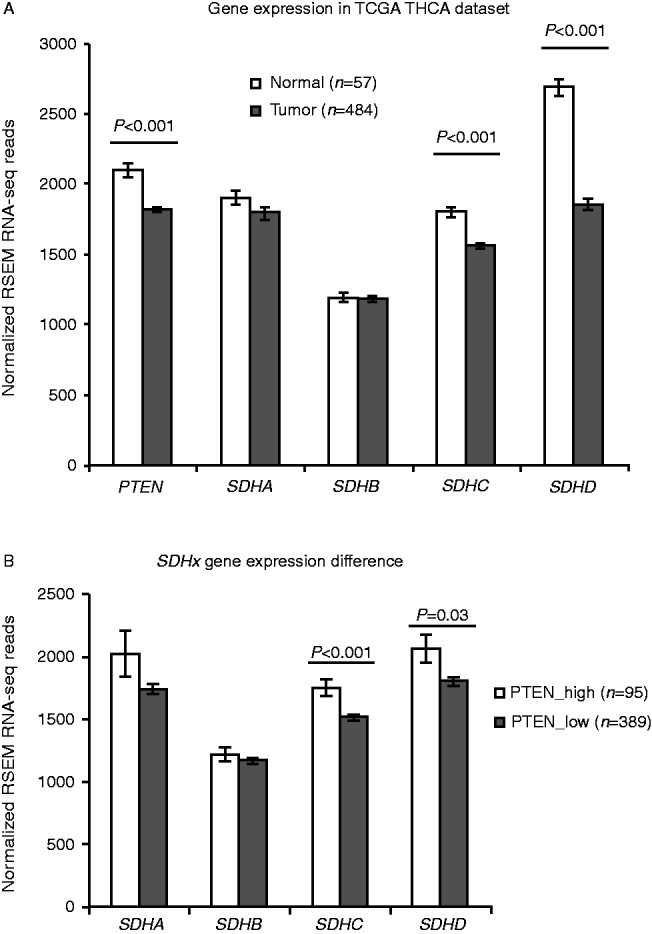
PTEN and SDHx gene expression in TCGA THCA dataset. (A) PTEN and SDHx gene expression in tumor samples compared with normal samples; (B) SDHx gene expression differences in high PTEN expression tumor group compared with low PTEN expression tumor group. P value was obtained using two-tailed Student's t-test.
Based on available information for 466 tumor samples from TCGA THCA, we then examined whether any demographic or clinical characteristics were associated with SDHx gene expression differences. Only earlier age at diagnosis and higher pTNM stage but not others were associated with lower expression of the SDHx genes overall (Table 2). Classic PTC histology was overrepresented among patients with low overall SDHx expression, compared with those with other histologic types, especially the follicular variant form of PTC. However, no association was seen with residual tumor or final vital status.
Table 2.
Demographic and clinical characteristics of TCGA THCA samples based on SDHx gene expression differences
| Gene_scorea | 0 | 1 | 2 | 3 | 4 | P valueb |
|---|---|---|---|---|---|---|
| Mean of age_at_diagnosis (n) | 44.98 (n=245) | 47.37 (n=104) | 50.14 (n=63) | 50.60 (n=25) | 52.93 (n=29) | 0.015 |
| Gender | ||||||
| Female (n=343) | 178 | 77 | 45 | 20 | 23 | 0.867 |
| Male (n=123) | 67 | 27 | 18 | 5 | 6 | |
| Pathology_T | ||||||
| T1 (n=135) | 75 (55.6%) | 22 (16.3%) | 22 (16.3%) | 7 (5.2%) | 9 (6.7%) | 0.051 |
| T2 (n=154) | 83 (53.9%) | 38 (24.7%) | 15 (9.7%) | 3 (1.9%) | 15 (9.7%) | |
| T3 (n=156) | 76 (48.7%) | 40 (25.6%) | 22 (14.1%) | 14 (9%) | 4 (2.6%) | |
| T4 (n=19) | 9 (47.4%) | 4 (21.1%) | 4 (21.1%) | 1 (5.3%) | 1 (5.3%) | |
| Pathology_N | ||||||
| N0 (n=213) | 106 (49.8%) | 38 (17.8%) | 38 (17.8%) | 16 (7.5%) | 15 (7.0%) | <0.001 |
| N1 (n=207) | 119 (57.5%) | 57 (27.5%) | 19 (9.2%) | 7 (3.4%) | 5 (2.4%) | |
| NX (n=46) | 20 (43.5%) | 9 (19.6%) | 6 (13.0%) | 2 (4.3%) | 9 (19.6%) | |
| Pathology_M | ||||||
| M0 (n=252) | 147 (58.3%) | 51 (20.2%) | 39 (15.5%) | 10 (4.0%) | 5 (2.0%) | <0.001 |
| M1 (n=8) | 8 (100%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | |
| MX (n=205) | 90 (43.9%) | 53 (25.9%) | 24 (11.7%) | 14 (6.8%) | 24 (11.7%) | |
| NA (n=1) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (100%) | 0 (0%) | |
| Pathology_stage | ||||||
| Stage I (n=264) | 145 (54.9%) | 59 (22.3%) | 37 (14.0%) | 10 (3.8%) | 13 (4.9%) | 0.002 |
| Stage II (n=0) | 0 | 0 | 0 | 0 | 0 | |
| Stage III (n=104) | 50 (48.1%) | 29 (27.9%) | 13 (11.5%) | 11 (10.6%) | 2 (1.9%) | |
| Stage IV (n=7) | 6 (85.7%) | 1 (14.3%) | 0 (0%) | 0 (0%) | 0 (0%) | |
| NA (n=91) | 44 (48.4%) | 15 (16.5%) | 14 (15.4%) | 4 (4.4%) | 14 (15.4%) | |
| Histology | ||||||
| Classic PTC (n=321) | 189 (58.9%) | 73 (22.7%) | 38 (11.8%) | 11 (3.4%) | 10 (3.1%) | <0.001 |
| FvPTC (n=99) | 36 (36.4%) | 17 (17.2%) | 16 (16.2%) | 12 (12.1%) | 18 (18.2%) | |
| TallCell PTC (n=35) | 14 (40.0%) | 12 (34.3%) | 8 (22.9%) | 1 (2.9%) | 0 (0%) | |
| Others (n=11) | 6 (54.5%) | 2 (18.2%) | 1 (9.1%) | 1 (9.1%) | 1 (9.1%) |
Gene_score (range from 0 to 4) was calculated by first categorizing each gene's expression as either 1 as higher than normal, or 0 as lower than normal, then adding up the binary score for each of these SDHA–D genes.
P value was calculated by one-way ANOVA for age_at_diagnosis with degree of freedom of 4, and by Pearson χ2 test for the rest of comparisons.
Discussion
Defects in mitochondrial function have long been shown to contribute to the development and progression of cancer. The ‘oncocytic tumors’ theory considers genes encoding proteins with mitochondrial function as putative cancer-associated genes. Supporting this were pilot observations of an increased prevalence of PTC and renal cell carcinoma, both considered oncocytic tumors, in SDHx variant carriers in CS/CS-like cases. We are aware that some of the variants we identified in our current study are reported in public databases such as dbSNP and ESP where the frequency varies among different ethnicity background population. This is the reason why we used our ethnicity-matched control as a comparison. The occurrence of these variants in patients with DTC but not in ethnicity-matched controls or sporadic breast cancer is further assurance. SDHB A3G (rs11203289) was reported in dbSNP but only in the African–American population, while our samples are derived from white individuals of European ancestry. The most frequent variants SDHB S163P (rs33927012), SDHD G12S (rs34677591), and SDHD H50A (rs11214077) have also been reported in the database. Although these relatively common (1–5% frequency) variants were computationally predicted to be functionally benign (Bayley 2011), our previous experimental data already provided molecular evidence that they could have functional impact in cellular signaling regulation (Ni et al. 2008, 2012). The reason why bioinformatics analysis of prediction fails in SDHx genes is because they are extremely well conserved throughout species (Ng & Henikoff 2006). Indeed, these very same bioinformatics tools predicted that the SDHD P81L missense mutation would be benign when this is really the North American founder mutation predisposing to familial paraganglioma (Baysal et al. 2002). With enormous numbers of variations uncovered by whole-genome sequencing, it is essential to realize that functional analysis and clinical correlations must be performed to define the true pathogenic effect of DNA variations (Ni & Eng 2011). In our 2008 study, we showed clear one-to-one correlation between the identified SDHB or SDHD variants and profound functional phenotypes such as altered reactive oxygen species (ROS) and up regulation of AKT (also known as protein kinase B) and mitogen-activated protein kinase (MAPK). The latter may explain why SDHB or SDHD variation can result in phenotypes similar to (but not identical to) Cowden and Cowden-like syndromes: the AKT and MAPK pathways are important pathways downstream of PTEN also (Ni et al. 2008). In our 2012 study, we validated that germline SDHx variants are associated with elevated thyroid cancer risks in Cowden and Cowden-like individuals. We also provided further functional evidence of these germline SDHx variants. We showed that these variants led to mitochondrial metabolite imbalance, and in turn cause stabilization of HIF1α, and decreased baseline p53 levels mediated by the noncanonical NQO1 pathway. These functional consequences are at least partially responsible for ROS generation. The cross-talk between SDH and PTEN results in multi-signaling pathways that contribute to tumorigenesis (Ni et al. 2012). A recent in vitro study specifically in thyroid cancer cell lines also showed that SDHD G12S and SDHD H50R variants lead to impaired PTEN function through alteration of its subcellular localization accompanied by resistance to apoptosis and induction of migration, mediated by Rous sarcoma protooncogene (SRC) (Yu et al. 2014). Taking all the evidence together, we believe that the variants we reported in this and previous studies are very likely associated with thyroid carcinogenesis.
The somatic 1q duplication spans almost 92 Mb, which is not reported in any structural variation or CNV databases. There are more than 100 genes, including SDHC, residing in this region. In the TCGA dataset, the 25 samples with somatic SDHC duplication had ∼1.5-fold SDHC gene expression compared with samples without duplication, indicating that the additional copy of the gene does generate transcript (Supplementary Figure 1, see section on supplementary data given at the end of this article). However, how exactly this large duplication (including SDHC and other genes) impacts tumorigenesis warrants further investigation, given that the TCGA thyroid tumor dataset as a whole showed an overall decrease of SDHC expression compared with their normal tissues.
The association of germline loss-of-function mutations in SDHx genes and loss of SDH subunit protein expression in paraganglioma–pheochromocytoma and gastrointestinal stromal tumors are well established (Neumann et al. 2002, 2004, Doyle et al. 2012, Dwight et al. 2013). At the expression level, most studies utilized immunohistochemistry to measure SDHA and SDHB protein, and showed loss of SDHB in pheochromocytoma–paraganglioma tumors (van Nederveen et al. 2009). Reduced SDHB protein expression was also associated with growth and de-differentiation of colorectal cancer cells (Zhang et al. 2013). In the study from Papathomas et al. (2014), SDHB immunonegativity was observed in renal cell carcinoma but not PTC associated with SDH-related pheochromocytoma/paraganglioma syndrome. In our study with sporadic PTC, we did not see alterations in SDHB at the mRNA level, but rather significant reductions in SDHC/D gene expression. The methylation analysis in these two genes did not show a difference in tumors compared with normal tissues, indicating the loss of the gene expression was not the result of DNA hypermethylation but most likely due to other transcriptional regulation alterations in tumor cells. As SDHC/D subunits mainly function as the anchor proteins to position the whole SDH complex into the mitochondrial inner membrane, it is likely that the loss of the structural SDHC/D subunits will affect the overall stability and integrity of complex II and lead to mitochondrial abnormalities. It has been reported that the protein assembly of the SDH complex is critical for regulation of cell death, especially the dissociation of SDHA and SDHB subunits from the membrane-anchoring proteins through pH changes or mitochondrial Ca2+ influx (Hwang et al. 2014). Unfortunately, we could not find reliable SDHC/D antibodies to measure protein expression of these two subunits in our tumor samples. The association of overall low SDHx gene expression with earlier disease onset as well as higher pTNM stage was found in the TCGA dataset but needs to be verified in an independent study. Even though we did not further investigate the SDHx expression in breast tumors, loss of SDHA or SDHB expression by immunohistochemistry has been reported in about 3% of breast cancers and low SDHA/B expression status in breast tumor cells was associated with younger age at diagnosis and low-grade histology (Kim et al. 2013).
The question of if and how SDHx alterations differ in papillary and follicular histological subtype of thyroid cancer remains to be explored in depth. Our earlier analysis revealed elevated risks of FTC due to germline PTEN pathogenic mutations and of PTC for germline SDHx alterations in CS/CS-like individuals (Ngeow et al. 2011). In this study of sporadic DTCs, germline SDHx variations were detected in both PTC and FTC cases. It is notable that SDHB transcript levels are decreased in our FTC, albeit represented by small sample size, in contrast to no change in SDHB expression in the PTC samples. As the TCGA dataset is limited by its papillary histology-focused sample collection, whether somatic SDHB expressional differences could distinguish papillary from follicular histology needs further validation with an expanded sample size. SDHB protein expression has not been examined in sporadic differentiated thyroid tumors by immunohistochemistry yet and it will be interesting to correlate protein expression with its gene expression. Previous microarray analysis reported five genes (CITED1, CLDN10, IGFBP6, CAV1, and CAV2) that collectively distinguish the two histologic types (Aldred et al. 2004). If the differential SDHB gene expression was validated, it will be noteworthy to see whether SDHB could improve the five-gene classification capability.
In conclusion, we have shown germline and somatic SDHx variants occur in sporadic DTC and overall loss of SDHx gene expression could represent a molecular signature of differentiated thyroid tumors.
Supplementary data
This is linked to the online version of the paper at http://dx.doi.org/10.1530/ERC-14-0537.
Footnotes
R T Kloos is now at Veracyte, Inc., San Francisco, California 94080, USA
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This work was funded, in part, by the National Cancer Institute P01CA124570 (to C Eng, R T Kloos, and M D Ringel), the Breast Cancer Research Foundation (to C Eng) and the William Randolf Hearst Foundations (to C Eng). Y Ni is a CoGEC Scholar funded by NCI grant R25TCA094186; S Ganapathi was a recipient of an American Cancer Society (Ohio Division) Joseph S. Silber Undergraduate Summer Research Fellowship; and J Ngeow was a recipient of the National Medical Research Council Singapore Fellowship and of the Ambrose Monell Foundation Cancer Genomic Medicine Fellowship. C Eng is the Sondra J. and Stephen R. Hardis Chair of Cancer Genomic Medicine at the Cleveland Clinic, and is an American Cancer Society Clinical Research Professor, generously funded, in part, by the F.M. Kirby Foundation.
References
- Aldred MA, Huang Y, Liyanarachchi S, Pellegata NS, Gimm O, Jhiang S, Davuluri RV, de la Chapelle A, Eng C. Papillary and follicular thyroid carcinomas show distinctly different microarray expression profiles and can be distinguished by a minimum of five genes. Journal of Clinical Oncology. 2004;22:3531–3539. doi: 10.1200/JCO.2004.08.127. [DOI] [PubMed] [Google Scholar]
- Bayley JP. Succinate dehydrogenase gene variants and their role in Cowden syndrome. American Journal of Human Genetics. 2011;88:674–675. doi: 10.1016/j.ajhg.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, Bosch A, van der Mey A, Taschner PE, Rubinstein WS, Myers EN, et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science. 2000;287:848–851. doi: 10.1126/science.287.5454.848. [DOI] [PubMed] [Google Scholar]
- Baysal BE, Willett-Brozick JE, Lawrence EC, Drovdlic CM, Savul SA, McLeod DR, Yee HA, Brackmann DE, Slattery WH, III, Myers EN, et al. Prevalence of SDHB, SDHC, and SDHD germline mutations in clinic patients with head and neck paragangliomas. Journal of Medical Genetics. 2002;39:178–183. doi: 10.1136/jmg.39.3.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgeron T, Rustin P, Chretien D, Birch-Machin M, Bourgeois M, Viegas-Pequignot E, Munnich A, Rotig A. Mutation of a nuclear succinate dehydrogenase gene results in mitochondrial respiratory chain deficiency. Nature Genetics. 1995;11:144–149. doi: 10.1038/ng1095-144. [DOI] [PubMed] [Google Scholar]
- Bruni P, Boccia A, Baldassarre G, Trapasso F, Santoro M, Chiappetta G, Fusco A, Viglietto G. PTEN expression is reduced in a subset of sporadic thyroid carcinomas: evidence that PTEN-growth suppressing activity in thyroid cancer cells mediated by p27kip1. Oncogene. 2000;19:3146–3155. doi: 10.1038/sj.onc.1203633. [DOI] [PubMed] [Google Scholar]
- Doyle LA, Nelson D, Heinrich MC, Corless CL, Hornick JL. Loss of succinate dehydrogenase subunit B (SDHB) expression is limited to a distinctive subset of gastric wild-type gastrointestinal stromal tumours: a comprehensive genotype–phenotype correlation study. Histopathology. 2012;61:801–809. doi: 10.1111/j.1365-2559.2012.04300.x. [DOI] [PubMed] [Google Scholar]
- Dwight T, Benn DE, Clarkson A, Vilain R, Lipton L, Robinson BG, Clifton-Bligh RJ, Gill AJ. Loss of SDHA expression identifies SDHA mutations in succinate dehydrogenase-deficient gastrointestinal stromal tumors. American Journal of Surgical Pathology. 2013;37:226–233. doi: 10.1097/PAS.0b013e3182671155. [DOI] [PubMed] [Google Scholar]
- Eng C, Kiuru M, Fernandez MJ, Aaltonen LA. A role for mitochondrial enzymes in inherited neoplasia and beyond. Nature Reviews. Cancer. 2003;3:193–202. doi: 10.1038/nrc1013. [DOI] [PubMed] [Google Scholar]
- Gimm O, Perren A, Weng LP, Marsh DJ, Yeh JJ, Ziebold U, Gil E, Hinze R, Delbridge L, Lees JA, et al. Differential nuclear and cytoplasmic expression of PTEN in normal thyroid tissue, and benign and malignant epithelial thyroid tumors. American Journal of Pathology. 2000;156:1693–1700. doi: 10.1016/S0002-9440(10)65040-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemminki K, Eng C, Chen B. Familial risks for nonmedullary thyroid cancer. Journal of Clinical Endocrinology and Metabolism. 2005;90:5747–5753. doi: 10.1210/jc.2005-0935. [DOI] [PubMed] [Google Scholar]
- Howlader N, Noone AM, Krapcho M, Garshell J, Miller D, Altekruse SF, Kosary CL, Yu M, Ruhl J, Tatalovich Z, Mariotto A, Lewis DR, Chen HS, Feuer EJ & Cronin KA (eds). SEER Cancer Statistics Review, 1975–2011, National Cancer Institute: Bethesda, MD, USA; http://seer.cancer.gov/csr/1975_2011/, based on November 2013 SEER data submission, posted to the SEER web site, April 2014.
- Hwang MS, Rohlena J, Dong LF, Neuzil J, Grimm S. Powerhouse down: complex II dissociation in the respiratory chain. Mitochondrion. 2014;Pt A:20–28. doi: 10.1016/j.mito.2014.06.001. [DOI] [PubMed] [Google Scholar]
- Kim S, Kim DH, Jung WH, Koo JS. Succinate dehydrogenase expression in breast cancer. SpringerPlus. 2013;2:299. doi: 10.1186/2193-1801-2-299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nature Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Genome Project Data Processing S The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWhinney SR, Pilarski RT, Forrester SR, Schneider MC, Sarquis MM, Dias EP, Eng C. Large germline deletions of mitochondrial complex II subunits SDHB and SDHD in hereditary paraganglioma. Journal of Clinical Endocrinology and Metabolism. 2004;89:5694–5699. doi: 10.1210/jc.2004-0769. [DOI] [PubMed] [Google Scholar]
- Mutter GL, Lin MC, Fitzgerald JT, Kum JB, Baak JP, Lees JA, Weng LP, Eng C. Altered PTEN expression as a diagnostic marker for the earliest endometrial precancers. Journal of the National Cancer Institute. 2000;92:924–930. doi: 10.1093/jnci/92.11.924. [DOI] [PubMed] [Google Scholar]
- Nagy R, Ganapathi S, Comeras I, Peterson C, Orloff M, Porter K, Eng C, Ringel MD, Kloos RT. Frequency of germline PTEN mutations in differentiated thyroid cancer. Thyroid. 2011;21:505–510. doi: 10.1089/thy.2010.0365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Nederveen FH, Gaal J, Favier J, Korpershoek E, Oldenburg RA, de Bruyn EM, Sleddens HF, Derkx P, Riviere J, Dannenberg H, et al. An immunohistochemical procedure to detect patients with paraganglioma and phaeochromocytoma with germline SDHB, SDHC, or SDHD gene mutations: a retrospective and prospective analysis. Lancet. Oncology. 2009;10:764–771. doi: 10.1016/S1470-2045(09)70164-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann HP, Bausch B, McWhinney SR, Bender BU, Gimm O, Franke G, Schipper J, Klisch J, Altehoefer C, Zerres K, et al. Germ-line mutations in nonsyndromic pheochromocytoma. New England Journal of Medicine. 2002;346:1459–1466. doi: 10.1056/NEJMoa020152. [DOI] [PubMed] [Google Scholar]
- Neumann HP, Pawlu C, Peczkowska M, Bausch B, McWhinney SR, Muresan M, Buchta M, Franke G, Klisch J, Bley TA, et al. Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. Journal of the American Medical Association. 2004;292:943–951. doi: 10.1001/jama.292.8.943. [DOI] [PubMed] [Google Scholar]
- Ng PC, Henikoff S. Predicting the effects of amino acid substitutions on protein function. Annual Review of Genomics and Human Genetics. 2006;7:61–80. doi: 10.1146/annurev.genom.7.080505.115630. [DOI] [PubMed] [Google Scholar]
- Ngeow J, Mester J, Rybicki LA, Ni Y, Milas M, Eng C. Incidence and clinical characteristics of thyroid cancer in prospective series of individuals with Cowden and Cowden-like syndrome characterized by germline PTEN, SDH, or KLLN alterations. Journal of Clinical Endocrinology and Metabolism. 2011;96:E2063–E2071. doi: 10.1210/jc.2011-1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni Y, Eng C. Response to bayley: functional study informs bioinformatic analysis. American Journal of Human Genetics. 2011;88:676. doi: 10.1016/j.ajhg.2011.03.020. [DOI] [Google Scholar]
- Ni Y, Zbuk KM, Sadler T, Patocs A, Lobo G, Edelman E, Platzer P, Orloff MS, Waite KA, Eng C. Germline mutations and variants in the succinate dehydrogenase genes in Cowden and Cowden-like syndromes. American Journal of Human Genetics. 2008;83:261–268. doi: 10.1016/j.ajhg.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni Y, He X, Chen J, Moline J, Mester J, Orloff MS, Ringel MD, Eng C. Germline SDHx variants modify breast and thyroid cancer risks in Cowden and Cowden-like syndrome via FAD/NAD-dependant destabilization of p53. Human Molecular Genetics. 2012;21:300–310. doi: 10.1093/hmg/ddr459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papathomas TG, Gaal J, Corssmit EP, Oudijk L, Korpershoek E, Heimdal K, Bayley JP, Morreau H, van Dooren M, Papaspyrou K, et al. Non-pheochromocytoma (PCC)/paraganglioma (PGL) tumors in patients with succinate dehydrogenase-related PCC–PGL syndromes: a clinicopathological and molecular analysis. European Journal of Endocrinology. 2014;170:1–12. doi: 10.1530/EJE-13-0623. [DOI] [PubMed] [Google Scholar]
- Parfait B, Chretien D, Rotig A, Marsac C, Munnich A, Rustin P. Compound heterozygous mutations in the flavoprotein gene of the respiratory chain complex II in a patient with Leigh syndrome. Human Genetics. 2000;106:236–243. doi: 10.1007/s004390051033. [DOI] [PubMed] [Google Scholar]
- Pilarski R, Eng C. Will the real Cowden syndrome please stand up (again)? Expanding mutational and clinical spectra of the PTEN hamartoma tumour syndrome. Journal of Medical Genetics. 2004;41:323–326. doi: 10.1136/jmg.2004.018036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risch N. The genetic epidemiology of cancer: interpreting family and twin studies and their implications for molecular genetic approaches. Cancer Epidemiology, Biomarkers & Prevention. 2001;10:733–741. [PubMed] [Google Scholar]
- Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Research. 2002;30:e57. doi: 10.1093/nar/gnf056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan MH, Mester JL, Ngeow J, Rybicki LA, Orloff MS, Eng C. Lifetime cancer risks in individuals with germline PTEN mutations. Clinical Cancer Research. 2012;18:400–407. doi: 10.1158/1078-0432.CCR-11-2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative genomics viewer (IGV): high-performance genomics data visualization and exploration. Briefings in Bioinformatics. 2013;14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanharanta S, Buchta M, McWhinney SR, Virta SK, Peczkowska M, Morrison CD, Lehtonen R, Januszewicz A, Jarvinen H, Juhola M, et al. Early-onset renal cell carcinoma as a novel extraparaganglial component of SDHB-associated heritable paraganglioma. American Journal of Human Genetics. 2004;74:153–159. doi: 10.1086/381054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warden CD, Lee H, Tompkins JD, Li X, Wang C, Riggs AD, Yu H, Jove R, Yuan YC. COHCAP: an integrative genomic pipeline for single-nucleotide resolution DNA methylation analysis. Nucleic Acids Research. 2013;41:e117. doi: 10.1093/nar/gkt242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, He X, Ni Y, Ngeow J, Eng C. Cowden syndrome-associated germline SDHD variants alter PTEN nuclear translocation through SRC-induced PTEN oxidation. Human Molecular Genetics. 2014;24:142–153. doi: 10.1093/hmg/ddu425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Wang W, Xiang B, Li N, Huang S, Zhou W, Sun Y, Wang X, Ma J, Li G, et al. Reduced succinate dehydrogenase B expression is associated with growth and de-differentiation of colorectal cancer cells. Tumour Biology. 2013;34:2337–2347. doi: 10.1007/s13277-013-0781-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.