

 This work is licensed under a
This work is licensed under a Summary
Xanthogranulomatous hypophysitis (XGH) is a very rare form of pituitary hypophysitis that may present both clinically and radiologically as a neoplastic lesion. It may either be primary with an autoimmune aetiology and can occur in isolation or as a part of autoimmune systemic disease or secondary as a reactive degenerative response to an epithelial lesion (e.g. craniopharyngioma (CP), Rathke's cleft cyst, germinoma and pituitary adenomas) or as a part of a multiorgan systemic involvement such as tuberculosis, sarcoidosis or granulomatosis. It may also present with a variation of symptoms in children and adults. Our case series compares the paediatric and adult presentations of XGH and the differential diagnoses considered in one child and two adult patients, highlighting the wide spectrum of this condition. Endocrine investigations suggested panhypopituitarism in all three patients and imaging revealed a suprasellar mass compressing the optic chiasm suggestive of CP or Rathke's cleft cyst in one patient and non-functioning pituitary macroadenoma in two patients. Magnetic resonance imaging (MRI) demonstrated mixed signal intensities on T1- and T2-weighted sequences. Following endoscopic transsphenoidal surgery, histological analysis revealed necrotic material with a xanthogranulomatous reaction confirming XGH in two patients and a necrobiotic granulomatous chronic inflammatory infiltrate with neutrophils in one patient, which is not typical of current descriptions of this disorder. This case series describes the wide spectrum of XGH disease that is yet to be defined. Mixed signal intensities on T1- and T2-weighted MRI sequences may indicate XGH and diagnosis is confirmed by histology. Histological variation may indicate an underlying systemic process.
Learning points
XGH is a rare form of pituitary hypophysitis with a wide clinical and histological spectrum and can mimic a neoplastic lesion.
XGH primarily presents with growth arrest in children and pubertal arrest in adolescents. In adults, the presentation may vary.
A combination of hypopituitarism and mixed signal intensity lesion on MRI is suggestive of XGH and should be considered in the differential diagnosis of sellar lesions.
Radical surgery is the treatment of choice and carries an excellent prognosis with no recurrence.
Background
Pituitary hypophysitis is a rare, inflammatory condition that may present both clinically and radiologically as a neoplastic lesion (1). Clinically, it is associated with headaches and visual disturbance due to the local mass effect and compression of the optic chiasm. Pituitary dysfunction secondary to inflammatory infiltration is common.
Hypophysitis is histologically divided into the following five distinct categories: lymphocytic, granulomatous, xanthomatous, necrotising and xanthogranulomatous hypophysitis (XGH), with the latter being a mixture of the xanthomatous and granulomatous subtypes (2). They are also classified as either adenohypophysitis or infundibuloneurohypophysitis based on their anatomical location and areas of infiltration (2). Lymphocytic hypophysitis is the most common type (1); XGH is rare and characterised by cholesterol clefts, haemosiderin deposits, multinucleated giant cells, macrophage accumulation and fibrous proliferation (3). Its pathogenesis is unclear and aetiologically it can either be a primary (most common) or a secondary hypophysitis. Primary hypophysitis is autoimmune in origin and can occur in isolation or as a part of systemic disease such as polyglandular autoimmune syndromes or IgG4 systemic disease (4). Secondary hypophysitis is thought to occur as a consequence of local inflammation caused by lesions including craniopharyngioma (CP)/Rathke's cleft cyst or as a part of systemic diseases such as tuberculosis, sarcoidosis, Wegener's granulomatosis or syphilis (4) (5).
In this case series, we describe three patients with XGH of different aetiologies, demonstrating variable clinical presentation depending on the age of diagnosis and the spectrum of histological presentation of XGH.
Case presentation
Patient 1
A 14-year-old female was initially referred with an 18-month history of lethargy, reduced appetite, abdominal pain, bloating and nausea. She developed a headache at a later stage that was not associated with vomiting, visual symptoms or neurological deficit. Her significant concerns at presentation were primary amenorrhoea and lack of pubertal progression reporting no progressive breast development since 11 years. On examination her height was 151 cm (−1.73 SDS) and weight was 33 kg (−3.36 SDS) with a BMI of 14 kg/m2 (−3.03 SDS). Her pubertal assessment showed her to be at Tanner breast stage 3, with no axillary or pubic hair. A previously performed thyroid function test had revealed a low free thyroxine (T4) (8.9 pmol/l) and an inappropriately low TSH (2.3 IU/l) suggestive of central hypothyroidism. Subsequent dynamic pituitary function tests confirmed anterior hypopituitarism (Table 1) and she was commenced on hydrocortisone (10 mg/m2) and levothyroxine (50 μg daily) replacement. Steroid replacement unmasked her background diabetes insipidus both clinically and biochemically (Table 1) and she was commenced on desmopressin 100 μg twice daily. Her TW3 bone age was delayed by 2 years. Formal visual assessment showed bitemporal quadrantanopia worse on the left than on the right. Cranial magnetic resonance imaging (MRI) revealed a suprasellar mass compressing the optic chiasm with a relatively uniform high-signal intensity on T1- and T2-weighted sequences (Fig. 1A and B). Differential diagnoses considered based on these findings were pituitary macroadenoma, CP or a Rathke's cleft cyst. Endoscopic transsphenoidal surgery was performed in order to preserve her vision and obtain a formal diagnosis. Histopathology of the resected lesion showed reactive changes with necrotic material and cholesterol clefts suggestive of xanthogranuloma of the sellar region. Haemosiderin was observed and, although multinucleate cells were around some cholesterol clefts, these were not prominent (Table 2 and Fig. 4A). Gram stain (for bacteria), Warthin–Starry stain (for treponemes) and Ziehl–Neelsen stain (for TB) were all negative. Histology did not demonstrate evidence of vasculitis or granuloma formation, therefore ruling out sarcoidosis and Wegener's granulomatosis as the other biochemical tests were not performed.
Table 1.
Combined pituitary and dynamic function tests at diagnosis
| Serum | Patient 1 | Patient 2 | Patient 3 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 0 min | 20 min | 60 min | 120 min | Impression | 0 min | 30 min | 0 min | 30 min | ||
| Anterior pituitary | ||||||||||
| GNRH stimulation test | ||||||||||
| FSH | 1.4 | 1.5 | 2.0 | – | 3.9 | – | 1.0 | – | ||
| LH | 0.1 | 0.3 | 0.6 | – | 0.7 | – | 0.4 | – | ||
| Oestradiol | a | – | – | – | 18 | – | – | – | ||
| SHBG | 123.5 | – | – | – | – | – | – | – | ||
| Testosterone | – | – | – | – | – | – | 0.8 | – | ||
| With Tanner stage 3 breast development, this GNRH test suggests pubertal arrest | ||||||||||
| GH stimulation test | ||||||||||
| GH | 0.4 | 0.2 | 0.4 | 0.4 | – | – | 0.2 | – | ||
| IGF1 | 77 | – | – | – | 48 | – | 96 | – | ||
| Low IGF1 along with peak GH <5.5 suggests growth hormone deficiency | ||||||||||
| Synacthen test | ||||||||||
| Cortisol | 158 | 282 | 360 | – | b 55 | b 229 | 68 (random) | |||
| ACTH | 10.5 (0900 h) | – | – | – | b 5.8 (0000 0.25 h) | – | – | – | ||
| Adrenal insufficiency with a relatively normal ACTH | ||||||||||
| TRH test | ||||||||||
| TSH | 2.65 | 8.68 | 13.15 | – | 1.8 | – | 0.36 | – | ||
| FT4 | 8.3 | – | – | – | 8.7 | – | 9.4 | – | ||
| FT3 | 3.73 | – | – | – | 2.7 | – | – | – | ||
| Hypothalamic response to TRH suggests central hypothyrodism | ||||||||||
| Prolactin | 1034 | – | – | – | Raised | 2207 | – | 424 | – | |
| Posterior pituitary | ||||||||||
| Plasma osmolality | 300 | – | – | – | b 296 | – | 300 | – | ||
| Urine osmolality | 301 | – | – | – | b 99 | – | 349 | – | ||
| Sodium | 145 | – | – | – | Normal c | b 144 | – | 135 | – | |
| Others c | ||||||||||
| Plasma HbA1c | – | – | – | – | 109 | – | – | – | ||
| Plasma random glucose | – | – | – | – | 9.5 | – | – | – | ||
| Bone age | delay by 2 years | |||||||||
Unrecordable.
Post-surgery.
Patient 2 had diabetes insipidus.
Figure 1.
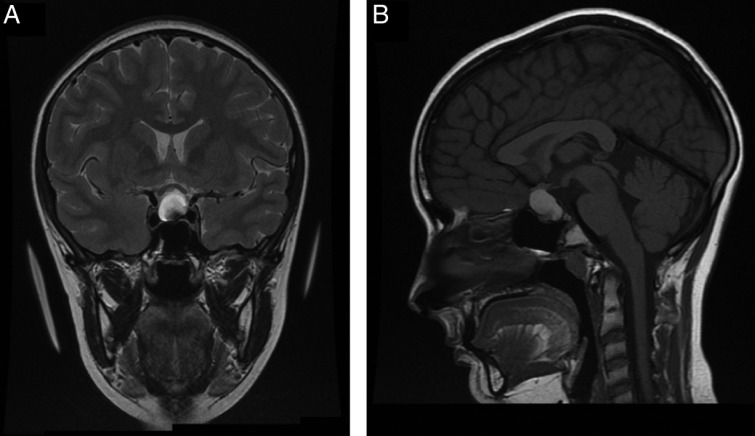
(A and B) MRI image of patient 1 demonstrating the pituitary lesion pre-surgery.
Table 2.
Summary of histological features
| Case | Necrosis | Cholesterol clefts | Fibrosis | Haemosiderin | Vague granuloma | Multinucleate giant cells a | Prominent foamy macrophage | Neutrophils | Eosinophils |
|---|---|---|---|---|---|---|---|---|---|
| Patient 1 | + | + | + | + | − | − | − | − | − |
| Patient 2 | + | + | + | + | − | + | − | − | − |
| Patient 3 | |||||||||
| Biopsy 1 | + | − | + | + | + | + | + | + | + |
| Biopsy 2 | + | − | + | − | − | + | + | + | + |
Separate to cholesterol clefts.
Figure 4.
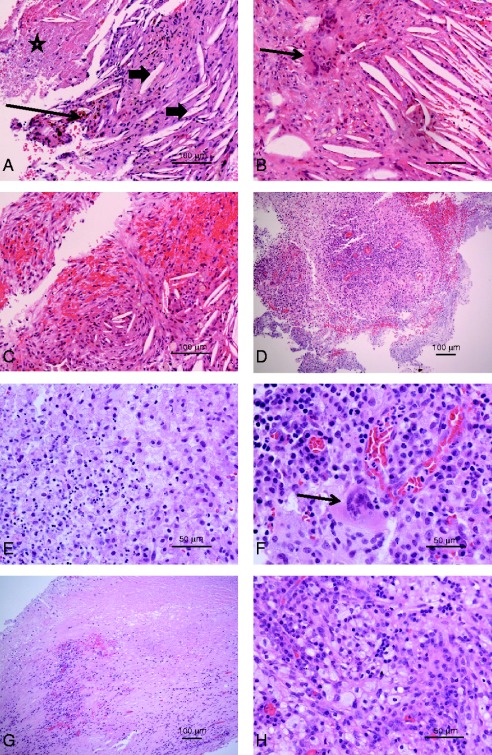
(A) Patient 1. Haematoxylin and eosin-stained section showing numerous cholesterol clefts (arrowheads). Brown, haemosiderin pigment is present (arrow) and there is an area of necrosis (star). Scale bar, 100 μm. (B and C) Patient 2. In addition to cholesterol clefts, there is an aggregate of multinucleate giant cells (arrow, B). In C, the cholesterol clefts are associated with new fibrosis. Scale bars, 100 μm. (D, E and F) Patient 3, first biopsy. An area of lymphoplasmacytic cells surrounded by paler macrophages, with a loose, vague granulomatous appearance (D, scale bar, 100 μm). In areas, foamy macrophages are admixed with neutrophils (E, scale bar, 50 μm), while F shows a somewhat foamy multinucleate macrophage (arrow) in a background of lymphocytes, histiocytes and plasma cells (scale bar, 50 μm). (G and H) Patient 3, second biopsy. Low-power view (G) showing dense fibrous tissues with a lymphoplasmacytic infiltrate. There is an area of necrosis towards the top of the field (scale bar, 100 μm). At high power (H), there is a prominent foamy macrophage infiltrate with admixed lymphocytes and histiocytes.
She made an excellent recovery following hormone replacement and surgery. She has shown remarkable catch up growth (height velocity 9.36 cm/year) following growth hormone (0.7 mg/m2 per day) treatment. She is due to commence oestrogen replacement. An MRI scan performed 6 months post-surgery demonstrated significant decompression of the chiasm and a thin line of pituitary tissue in the base of the pituitary fossa.
Patient 2
A 21-year-old woman with a past medical history of poorly controlled type 1 diabetes mellitus (HbA1c=11.2%) was investigated at her local hospital after reporting amenorrhoea for 3 years following the birth of her child. In addition, she gave a history of lethargy and frontal headaches. Endocrinological investigations demonstrated anterior pituitary dysfunction with adrenal insufficiency, hypothyroidism, growth hormone deficiency, hypogonadotropic hypogonadism and hyperprolactinaemia with a prolactin level of 2207 mIU/l (Table 1). She was therefore commenced on hydrocortisone (11.8 mg/m2), T4 75 μg daily and cabergoline 0.25 μg twice weekly, which markedly improved her symptoms; she continued on oestrogen and progesterone hormone replacement therapy. She subsequently reported having a severe, sudden onset headache with transient loss of vision suggestive of pituitary apoplexy. Cranial MRI scan revealed an 18×14×19 mm pituitary lesion, with suprasellar extension and superior displacement of the optic chiasm (Fig. 2A and B). Despite compression of the optic tract, visual fields and visual acuity were unaffected. She was subsequently referred to the regional pituitary specialist team and a diagnosis of a non-functioning pituitary macroadenoma was made. She underwent endoscopic transsphenoidal surgery. Histological analysis of the resected lesion demonstrated areas of cholesterol cleft formation associated with multinucleate giant cells and numerous macrophages, highlighted by immunohistochemistry to CD68. Areas of necrotic tissue, fragments of fibrous tissue, and the residual compressed adenohypophysis were also present (Fig. 4B and C). Gram stain (for bacteria), Warthin–Starry stain (for treponemes) and Ziehl–Neelsen stain (for TB) were all negative. Histology did not demonstrate evidence of vasculitis or granuloma formation, therefore ruling out sarcoidosis and Wegener's granulomatosis as the other biochemical tests were not performed.
Figure 2.
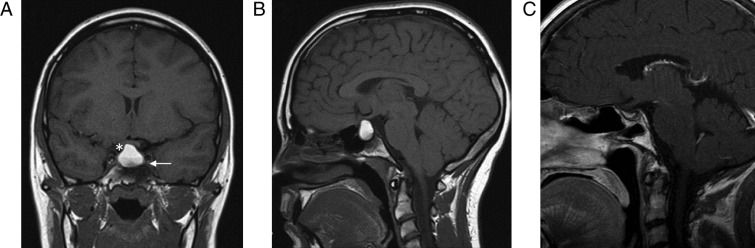
(A and B) MRI image of patient 2 demonstrating the pituitary lesion pre-surgery. (C) MRI image of patient 2 demonstrating the pituitary lesion post-surgery.
She made a full post-operative recovery and continued on hydrocortisone, T4 and HRT. She developed diabetes insipidus post-surgery. An MRI scan performed 6 months following surgery demonstrated normal pituitary gland, stalk and chiasm with no residual lesion (Fig. 2C).
Patient 3
A 67-year-old man was diagnosed with hypopituitarism following an episode of collapse and intermittent temporal headaches. His pituitary MRI at the time of presentation revealed an enlarged gland with cystic appearance consistent with an adenoma. He was commenced on replacement therapy with T4 50 μg daily, hydrocortisone 10.5 mg/m2 and testosterone 50 mg daily initially followed by testosterone enanthate 1000 mg injections every 3 months. He developed partial diabetes insipidus 6 months after diagnosis, which was confirmed by an abnormal water deprivation test with urine osmolality increasing only to 346 mOsm/kg with a paired serum osmolality of 302 mmol/kg; post-DDAVP urine osmolality was 707 mOsm/kg. He had a past medical history of rheumatoid arthritis diagnosed at 53 years, hypertension and bladder outflow obstruction. A transurethral resection of prostate (TURP), at the age of 56, showed granulomatous changes consistent with the development of rheumatoid nodules within the prostate. His prostate-specific antigen (PSA) levels were monitored relating to his testosterone replacement. A sudden rise in PSA at the age of 70 led to a diagnosis of prostate cancer, which was managed with testosterone blockage and radical radiotherapy. He subsequently developed osteoporosis and was treated with bisphosphonates, calcium and vitamin D supplements.
After 1 year, he presented with an apoplectic event associated with the sudden onset of a vomiting episode followed by a significant headache similar to the one he experienced at the time of his initial presentation. Following that he felt extremely weak and lethargic and continued to experience worsening severe headaches. A repeat MR scan showed a significant enlargement of the pituitary mass, elevating the optic chiasm more on the right. The chiasm was oedematous and enlarged along with the proximal portion of the right optic nerve. Following gadolinium contrast, the tumour had a heterogeneous pattern of enhancement with a low-signal centre suggesting central necrosis; there was, however, no evidence of intratumoural haemorrhage. Visual examination was normal. Histological examination of the mass removed by the transsphenoidal approach revealed a florid, mixed inflammatory infiltrate composed predominantly of lymphocytes and plasma cells with some admixed neutrophils and eosinophils. No well-formed granuloma was observed although loose areas of epithelial macrophage aggregates were present in a vaguely granulomatous manner and occasional multinucleate giant cells, partly foamy (Touton-like), were observed (Fig. 4D, E and F). No adenoma or other neoplastic elements were observed and special stains for bacteria, fungi and mycobacteria were negative. Epithelial cell markers were negative. A histological diagnosis of hypophysitis with a mixed inflammatory cell infiltrate was made.
Persistence of severe right temporal and orbital pain for a year led to further transsphenoidal surgery; the histology findings showed a dense inflammatory infiltrate of lymphocytes, plasma cells and histiocytes, with scattered polymorphs (Fig. 3A and B). There were also xanthomatous and multinucleate macrophages, but without well-formed granuloma (Fig. 4G and H). In view of the progressive nature of this inflammatory process with sclerosis and lymphoplasmacytic infiltrate, immunohistochemical staining for IgG4 was performed, which showed IgG4-positive plasma cells (>5%), raising possibility of a systemic IgG4-related process. However, the proportion of IgG4-positive cells was not as high as that mentioned in some of the previous reports (often 80%) and the serum IgG4 level was also not raised, hence a primary IgG4-related sclerosing disease was thought unlikely. He showed good response to high-dose steroids with prednisolone and immunosuppressive therapy with methotrexate.
Figure 3.
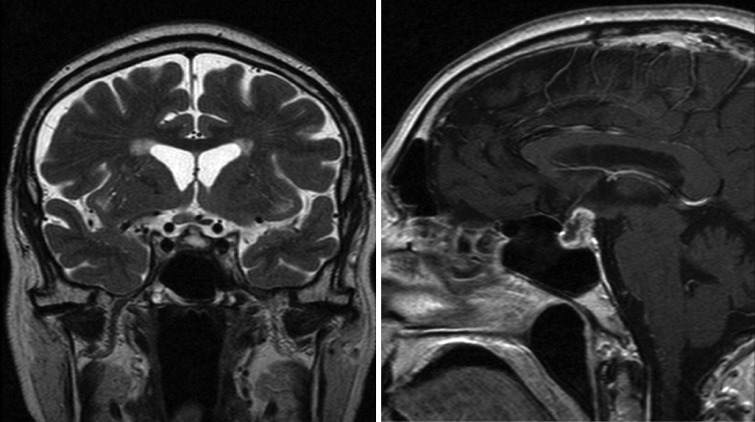
(A and B) MRI image of patient 3 demonstrating the pituitary lesion post-surgery.
Investigation
Investigations performed on all three patients are summarised in Table 1 and reference values in Table 3. All three patients demonstrated evidence of anterior hypopituitarism at the time of diagnosis. Steroid replacement unmasked diabetes insipidus in patient 1. Patients 2 and 3 developed diabetes insipidus post surgery.
Table 3.
Reference values of the various tests carried out
| Tests | Reference ranges |
|---|---|
| Prolactin (mIU/l) | 102–496 |
| Cortisol (nmol/l) | 198–720 |
| ACTH (ng/l) | 0000 h <15; 0900 h <46 |
| Serum osmolality (mmol/kg) | 275–295 |
| Urine osmolality (mOsmol/kg) | 100–800 |
| Serum sodium (mmol/l) | 133–144 |
| HbA1c (mmol/mol) | 19–48 |
| Random glucose (mmol/l) | <11.1 |
| ACE (IU/l) | 12–68 |
| Follicular phase | |
| FSH (IU/l) | 3.5–13.0 |
| LH (IU/l) | 2.4–13.0 |
| Oestradiol (pmol/l) | 46–607 |
| Ovulation phase | |
| FSH (IU/l) | 4.7–22.0 |
| LH (IU/l) | 14.0–96.0 |
| Oestradiol (pmol/l) | 315–1828 |
| Luteal phase | |
| FSH (IU/l) | 1.7–7.7 |
| LH (IU/l) | 1.0–11.0 |
| Oestradiol (pmol/l) | 161–774 |
| Post-menopausal | |
| FSH (IU/l) | 26–135 |
| LH (IU/l) | 7.7–59 |
| Oestradiol (pmol/l) | 18.4–201 |
| Patient 1 | |
| TSH (IU/l) | 0.5–3.6 |
| FT4 (pmol/l) | 10.0–16.9 |
| FT3 (pmol/l) | 3.4–6.5 |
| IGF1 (μg/ml) | 284–713 |
| Patient 2 | |
| TSH (IU/l) | 0.27–4.2 |
| FT4 (pmol/l) | 12.0–22.0 |
| FT3 (pmol/l) | 3.1–6.8 |
| IGF1 (μg/ml) | 115–340 |
| Patient 3 | |
| TSH (IU/l) | 0.35–4.5 |
| FT4 (pmol/l) | 10.3–21.9 |
| FT3 (pmol/l) | 3.1–6.8 |
| IGF1 (μg/ml) | 52–227 |
All three patients underwent endoscopic transphenoidal resection of the mass followed by hormone replacement therapy with good effect. Patient 3, in view of his underlying autoimmune systemic involvement was also treated with high dose steroids and immunosuppressants with good effect.
Patients 1 and 2 made an excellent recovery following surgery and hormone replacement. Patient 3 developed recurrence one year post surgery. He showed good response to high dose steroids and immunosuppressants along with hormone replacement.
Discussion
Hypophysitis is a rare, inflammatory condition of the pituitary that mimics neoplastic lesions resulting in anterior and posterior pituitary dysfunction. Of the different subtypes, lymphocytic hypophysitis is the most common subtype and is associated with autoimmune disease (5). XGH is the least common subtype and is reported to occur predominantly in young adults (6) (7). In this case series, we report three patients (one child and two adults) with XGH resulting from different aetiologies. XGH can present as i) a separate entity as described in our first patient, ii) a secondary reaction to a local process such as apoplexy as described in our second patient and iii) progression of inflammation of a systemic condition with an autoimmune aetiology as described in our third patient. While the pathologies of patients 1 and 2 were similar, case 3 showed some distinctive features, namely a lack of cholesterol clefts, more mixed inflammatory cells and more prominent foamy macrophages. Vague granulomatous inflammation, differing from the aggregates of multinuclear macrophages seen with (and separate from) cholesterol clefts, was also a feature in patient 3. As with other lesions of the pituitary gland, the clinical presentation of XGH is age dependent, particularly in relation to the impact on growth hormone and gonadotrophin secretion resulting in short stature and pubertal arrest in children.
Thus far, only six cases of XGH have been reported in detail in children between the age of 5 and 12 years (7) (8). Of the six children, four out of six had DI as an isolated endocrine dysfunction pre-operatively; one child presented with short stature. In our case series, we observed more pan-pituitary involvement with the younger patient than has been reported previously. The difference in presentation between paediatric and adult patients reflects the functional impact of pituitary hormone deficiency. Our paediatric patient presented with growth failure and pubertal arrest, whereas our adult patients presented with variable symptoms and signs relating to anterior and posterior pituitary dysfunction. Interestingly, the consistent presentation in these patients was frontal headache. XGH does not have sex preponderance and has been reported previously in both sexes across all age groups (7).
The rarity of this lesion is perhaps in part due to the relatively recent discovery of this inflammatory subtype of hypophysitis. Tashiro et al. were the first group to report the entity of XGH in 2002 following the retrospective analysis of 31 hypophysitis cases, revealing two specimens consisting of multinucleated giant cells, epithelioid cells and lymphocytic infiltration, consistent with granulomatous hypophysitis. In addition, cholesterol clefts (xanthomatous cells) were present within these tissue samples and thus the new histological category of XGH was created (2). Subsequently, a few cases of XGH have been reported with typical histopathological findings of cholesterol clefts, haemosiderin deposits, multinucleated giant cells and only small epithelial cell clusters (7) (8) (9) (10). Infiltration of both the adeno- and neuro-hypophysis explains the clinical correlation with headaches, anterior pituitary dysfunction and diabetes insipidus observed in previous patients and our reported cases with XGH (2) (7) (11) (12) (13). Cases of isolated anterior pituitary dysfunction, as reported in our case series (patient 2), have also been described previously (14). In previous case reports, tumour size and the extent of infiltration do not appear to correlate with endocrine dysfunction; however, patients diagnosed with XGH are reported to present with more extensive hypopituitarism when compared with those with non-XGH (14). Xanthogranulomatous lesions are more commonly found in other parts of the body including the skin, skull, choroid plexus, orbit and gallbladder (6). Lesions may be present in more than one location as demonstrated by a previous coincidental finding of xanthogranuloma of the skin and pituitary in a 64-year-old male, which may give further clues to the diagnosis of pituitary XGH (14).
The pathogenesis and histogenesis of XGH still remain unclear. Pituitary XGH may develop spontaneously as described in our first patient or as a secondary reaction to haemorrhage, tissue necrosis or severe inflammation following leak of cystic contents into the surrounding tissue in pre-existing lesions such as CPs, adenomas or Rathke's cleft cysts (6) (14) (15) (16) (21). This may explain the origin of the lesion in our second patient as she did experience an episode suggestive of apoplexy, which may have triggered xanthogranulomatous infiltration. Zada et al.have recently reviewed the developmental relationships and differentiating features of various epithelial lesions of the sellar region including CP, Rathke's cleft cyst, xanthogranuloma, and dermoid and epidermoid cysts. They highlighted that XGH represents approximately 34% of the suspected cases of CP and the recurrence rate is unknown. The lesion is mainly composed of xanthogranulomatous component with minimal squamous epithelium (17).
XGH can also arise as part of a systemic condition with autoimmune aetiology that presents with different findings in relation to XGH on histological examination as described in our third patient. A differential diagnosis of IgG4 disease was considered in view of the progressive nature of the inflammatory process with histological findings of pituitary lymphoplasmacytic infiltrate and sclerosis with an increased proportion of IgG4-positive plasma cells (>5%) raising the possibility of IgG4-related sclerosing disease. Previous reports have shown that increased numbers of IgG4-positive plasma cells are a common finding in orbital xanthogranuloma as part of a primary xanthogranulomatous process (18) (19). An elevated proportion of IgG4 plasma cells have also been found in some tissues in association with rheumatoid disease, though data for pituitary are lacking. Hence, the presence of IgG4 plasma cells in our patient may be a result of xanthogranulomatous change or secondary to rheumatoid arthritis. The xanthomatous macrophage component of this patient differed from our other reported patients, perhaps reflecting a histological variation in XGH, though the spectrum of the XGH disease is yet to be defined.
Cholesterol deposits typically show as T1 high- or T2 high-signal intensities on MRI, whereas the haemosiderin appears isointense on T1 and hypointense on T2 images. Granulation tissue, however, appears hypointense on both T1 and T2 respectively (14). Given the complex architecture of xanthogranulomatous lesions, the MRI appearance is usually of mixed signal intensities on T1- and T2-weighted sequences consistent with the MRI findings in our patients (7) (8) (14). CP may also contain cholesterol clefts that appear hyperintense on MRI (10), therefore making it difficult to radiologically differentiate from XGH. However, the coexistence of haemosiderin deposits and absence of abundant epithelial cells should aid in delineating these two lesions by MRI.
Evidence suggests radical surgery as the treatment modality of choice for XGH resulting in complete remission without any recurrence (6) (17) (20). Post-operative obesity is not encountered in XGH, as changes are usually limited to the anterior hypothalamus when compared with CP, where the posterior hypothalamus is predominantly involved (20). The transsphenoidal approach to surgery may also reduce the risk of complications (20). Though the 5-year overall survival rate has been reported as 1.00±0.00 in both XGH and CP, the event-free survival rate in terms of relapse or residual tumour progression is much higher in XGH (1.00±0.0) when compared with CP (0.52±0.05) (20).
Conclusion
Xanthogranulomatous inflammatory lesions of the pituitary are rare causes of pituitary dysfunction and can mimic neoplastic lesions. The clinical presentation in children and adults may vary in relation to the extent of the pituitary damage and clinical presentation in relation to the hormone pathways affected. Our case series highlights that a combination of hypopituitarism and a mixed signal intensity lesion on MRI is suggestive of XGH and should therefore be considered in the differential diagnosis of sellar lesions as surgical intervention carries an excellent prognosis with no recurrence. The histological variation in XGH lesions may require further classification in relation to the underlying disease process.
Patient consent
The authors confirm that a written consent was obtained from all the three patients towards the publication of their case histories and images. A written informed consent was obtained from patients 2 and 3 and patient 1's guardian for publication of the submitted article and the accompanying images.
Author contribution statement
J S Gopal-Kothandapani and V Bagga drafted the article. S B Wharton and D J Connolly, critically reviewed the article. S Sinha and P J Dimitri reviewed the article critically, and revised and approved the final version.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This research did not receive any specific grant from any funding agency in the public, commercial or not-for-profit sector.
References
- 1.Aste L, Bellinzona M, Meleddu V, Farci G, Manieli C & Godano U. 2010Xanthomatous hypophysitis mimicking a pituitary adenoma: case report and review of the literature. Journal of Oncology 2010. 10.1155/2010/195323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tashiro T, Sano T, Xu B, Wakatsuki S, Kagawa N, Nishioka H, Yamada S & Kovacs K. 2002Spectrum of different types of hypophysitis: a clinicopathologic study of hypophysitis in 31 cases. Endocrine Pathology 13183–195 10.1385/EP:13:3:183 [DOI] [PubMed] [Google Scholar]
- 3.Sugata S, Hirano H, Yatsushiro K, Yunoue S, Nakamura K & Arita K. 2009Xanthogranuloma in the suprasellar region. Neurologia Medico-Chirurgica 49124–127 10.2176/nmc.49.124 [DOI] [PubMed] [Google Scholar]
- 4.Leporati P, Landek-Salgado MA, Lupi I, Chiovato L & Caturegli P. 2011IgG4-related hypophysitis: a new addition to the hypophysitis spectrum. Journal of Clinical Endocrinology and Metabolism 961971–1980 10.1210/jc.2010-2970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mittal R, Kalra P, Dharmalingam M, Verma R, Kulkarni S & Shetty P. 2012Lymphocytic hypophysitis masquerading as pituitary adenoma. Indian Journal of Endocrinology and Metabolism 16S304–S306 10.4103/2230-8210.98035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paulus W, Honegger J, Keyvani K & Fahlbusch R. 1999Xanthogranuloma of the sellar region: a clinicopathological entity different from adamantinomatous craniopharyngioma. Acta Neuropathologica 97377–382 10.1007/s004010051001 [DOI] [PubMed] [Google Scholar]
- 7.Jung CS, Schänzer A, Hattingen E, Plate KH & Seifert V. 2006Xanthogranuloma of the sellar region. Acta Neurochirurgica 148473–477 10.1007/s00701-005-0700-6 [DOI] [PubMed] [Google Scholar]
- 8.Kamoshima Y, Sawamura Y, Motegi H, Kubota K & Houkin K. 2011Xanthogranuloma of the sellar region of children: series of five cases and literature review. Neurologia Medico-Chirurgica 51689–693 10.2176/nmc.51.689 [DOI] [PubMed] [Google Scholar]
- 9.Burt MG, Morey AL, Turner JJ, Pell M, Sheehy JP & Ho KK. 2003Xanthomatous pituitary lesions: a report of two cases and review of the literature. Pituitary 6161–168 10.1023/B:PITU.0000011177.43408.56 [DOI] [PubMed] [Google Scholar]
- 10.Arai A, Nishihara M, Sasayama T, Aihara H, Hosoda K, Itoh T, Sakagami Y, Kuwamura K & Kohmura E. 2010Xanthogranuloma of the sellar region–case report. Neurologia Medico-Chirurgica 50488–491 10.2176/nmc.50.488 [DOI] [PubMed] [Google Scholar]
- 11.Hama S, Arita K, Tominaga A, Yoshikawa M, Eguchi K, Sumida M, Inai K, Nishisaka T & Kurisu K. 1999Symptomatic Rathke's cleft cyst coexisting with central diabetes insipidus and hypophysitis: case report. Endocrine Journal 46187–192 10.1507/endocrj.46.187 [DOI] [PubMed] [Google Scholar]
- 12.Yokoyama S, Sano T, Tajitsu K & Kusumoto K. 2004Xanthogranulomatous hypophysitis mimicking a pituitary neoplasm. Endocrine Pathology 15351–357 10.1385/EP:15:4:351 [DOI] [PubMed] [Google Scholar]
- 13.Bao SS & Rapp R. 2014Xanthogranuloma as an unsuspected cause of Idiopathic central diabetes insipidus. Endocrine Practice 20e42–e46 10.4158/EP13116.CR [DOI] [PubMed] [Google Scholar]
- 14.Nishioka H, Shibuya M, Ohtsuka K, Ikeda Y & Haraoka J. 2010Endocrinological and MRI features of pituitary adenomas with marked xanthogranulomatous reaction. Neuroradiology 52997–1002 10.1007/s00234-010-0675-8 [DOI] [PubMed] [Google Scholar]
- 15.Amano K, Kubo O, Komori T, Tanaka M, Kawamata T, Hori T & Okada Y. 2013Clinicopathological features of sellar region xanthogranuloma: correlation with Rathke's cleft cyst. Brain Tumor Pathology 30233–241 10.1007/s10014-012-0130-0 [DOI] [PubMed] [Google Scholar]
- 16.Miyajima Y, Oka H, Utsuki S & Fujii K. 2011Rathke's cleft cyst with xanthogranulomatous change–case report. Neurologia Medico-Chirurgica 51740–742 10.2176/nmc.51.740 [DOI] [PubMed] [Google Scholar]
- 17.Zada G, Lin N, Ojerholm E, Ramkissoon S & Laws ER. 2010Craniopharyngioma and other cystic epithelial lesions of the sellar region: a review of clinical, imaging, and histopathological relationships. Neurosurgical Focus 28E4. 10.3171/2010.2.FOCUS09318 [DOI] [PubMed] [Google Scholar]
- 18.Singh K, Rajan KDA & Eberhart C. 2010Orbital necrobiotic xanthogranuloma associated with systemic IgG4 disease. Ocular Immunology and Inflammation 18373–378 10.3109/09273948.2010.490629 [DOI] [PubMed] [Google Scholar]
- 19.Verdijk RM, Heidari P, Verschooten R, van Daele PL, Simonsz HJ & Paridaens D. 2014Raised numbers of IgG4-positive plasma cells are a common histopathological finding in orbital xanthogranulomatous disease. Orbit 3317–22 10.3109/01676830.2013.842252 [DOI] [PubMed] [Google Scholar]
- 20.Müller HL, Gebhardt U, Faldum A, Warmuth-Metz M, Pietsch T, Pohl F, Calaminus G & Sörensen N. 2012Xanthogranuloma, Rathke's cyst, and childhood craniopharyngioma: results of prospective multinational studies of children and adolescents with rare sellar malformations. Journal of Clinical Endocrinology and Metabolism 973935–3943 10.1210/jc.2012-2069 [DOI] [PubMed] [Google Scholar]
- 21.Liu Z-H, Tzaan W-C, Wu Y-Y & Chen H-C. 2008Sellar xanthogranuloma manifesting as obstructive hydrocephalus. Journal of Clinical Neuroscience 15929–933 10.1016/j.jocn.2007.05.028 [DOI] [PubMed] [Google Scholar]