

Abstract
Imaging, cerebrospinal fluid (CSF) and blood-based biomarkers have the potential to improve the accuracy by which specific causes of dementia can be diagnosed in vivo, provide insights into the underlying pathophysiology, and may be used as inclusion criteria and outcome measures for clinical trials. While a number of imaging and CSF biomarkers are currently used for each of these purposes, this is an evolving field, with numerous potential biomarkers in varying stages of research and development. We review the currently available biomarkers for the three most common forms of neurodegenerative dementia, and give an overview of research techniques that may in due course make their way into the clinic.
Keywords: ALZHEIMER'S DISEASE, DEMENTIA, FRONTAL LOBE, BRAIN MAPPING, COGNITION
Introduction
A biomarker is a characteristic that can be objectively measured and evaluated as an indicator of normal biological or pathogenic processes or pharmacological responses to a therapeutic intervention.1 An ideal biomarker is reproducible, stable over time, widely available and reflects directly the relevant disease process.1 For the dementias, biomarkers may be used to distinguish different aspects of the underlying pathology; detect presymptomatic pathological changes; predict decline or conversion between clinical disease states; and/or monitor disease progression and response to treatment. In this review we provide an overview of currently available biomarkers, with a particular focus on the three commonest neurodegenerative dementias: Alzheimer's disease (AD), frontotemporal dementia (FTD) and dementia with Lewy bodies (DLB).
Biomarkers of dementia currently in clinical use
Biomarkers in dementias can be divided into imaging modalities, which are already widely used in clinical and research settings; and cerebrospinal fluid (CSF) measures whose clinical use varies widely between countries and centres. Currently, no blood-based or urine-based biomarkers are available for routine clinical use.
Imaging
Structural brain imaging
Structural brain imaging (CT or MRI) is recommended in all patients being investigated for dementia, according to UK, European and US guidelines.2 Moving away from excluding ‘surgical’ causes (eg, mass lesions) of cognitive impairment, structural imaging (and MRI in particular) can usefully assess vascular damage, white matter signal changes with a wide range of causes,3 and spongiform and gliotic changes as seen in prion disease. The pattern of regional brain loss (atrophy) reflecting neuronal loss has positive predictive value for different dementias2 and is incorporated into diagnostic criteria for several dementia syndromes (discussed below). Atrophy can be assessed using simple visual rating scales or more complex quantitative manual or automated techniques. Serial imaging—particularly with MRI, which provides superior grey/white contrast without radiation exposure—is widely used as safety and outcome measures for clinical trials, with rates of atrophy considered surrogate markers of neurodegeneration.
Functional imaging
Positron emission tomography (PET) using 18-F-flourodeoxyglucose (FDG) and single photon emission tomography (SPECT) using tracers such as 99mTc-hexamethylpropyleneamine (HMPAO), allows for visualisation and quantification of patterns of brain hypometabolism and hypoperfusion which show characteristic patterns that differ in different dementia syndromes.4 Dopamine transporter scanning can be used to determine central dopaminergic depletion, as seen in DLB, Parkinson's disease dementia, as well as a range of other movement disorders associated with dementia.5 The development of PET tracers that bind to and label-specific brain proteins, including fibrillar amyloid or Tau allows for aspects of the molecular pathology underlying certain dementias—and AD in particular—to be imaged in vivo.6
Functional MRI (fMRI) measures alterations in regional cerebral blood flow using a linked blood–oxygen-level-dependent (BOLD) signal change in the magnetic properties of cerebral venous blood. fMRI techniques can measure intrinsic fluctuations in BOLD signal in the waking brain at rest (‘resting state’ or rsfMRI) or BOLD changes in response to a particular stimulus or task in the scanner (‘activation’ fMRI). At present fMRI techniques require considerable expertise and a dedicated infrastructure to implement and analyse, which limits their widespread application as biomarkers.
Fluid biomarkers
Cerebrospinal fluid
In the context of dementia, CSF examination has traditionally been used to exclude infection, malignancy and neuroinflammation, as reflected by guidelines recommending CSF examination in individuals with cognitive impairment under the age of 55, individuals with rapid disease course, ‘unusual’ dementia syndromes or those who are immunosuppressed.7 In degenerative forms of dementia, the cell count is not usually raised, and there is no evidence for CNS-specific immune responses. Where abnormalities are present, these should prompt consideration of unusual forms of dementia, including infectious and inflammatory conditions.7 In cases of rapidly progressive dementia, the presence of a positive 14-3-3 protein, elevated S100B, elevated total-tau tau to phospho-tau ratio, and recently the use of real-time quaking-induced conversion (RT-QUIC) technology8 have positive predictive value for prion disease. CSF analysis using a variety of immunochemical techniques allows a range of neuronal-specific or neuronal-enriched proteins to be measured (see below). The use of neuronal-enriched CSF markers β-amyloid and tau in the routine evaluation of patients with dementia varies considerably between countries and between clinicians; these markers have been incorporated into new AD diagnostic criteria (see below, and table 2), and are increasingly included as inclusion/outcome measures for clinical trials. There is considerable variability in the methods used to collect and analyse CSF, leading to initiatives to harmonise preanalytical handling and standardise laboratory practices (see table 1). Developing normal reference ranges and reliable cut points for clinical use in the absence of postmortem confirmation of the underlying pathology is a significant challenge.9
Table 2.
Biomarkers currently used in diagnostic criteria
| Criteria | Comments |
|---|---|
| AD | |
| Biomarkers of amyloid pathology: Low CSF Aβ1-42 on CSF examination Positive amyloid PET scan | Either evidence of low CSF Aβ1-42 or positive amyloid PET scan required for diagnosis of amyloid brain deposition15 |
| Biomarkers of neuronal injury: Elevated CSF tau and phospho-tau; Hypometabolism on FDG-PET; Disproportionate atrophy of medial, basal and lateral temporal lobe, and medial parietal cortex on structural MRI | Either elevated CSF tau, FDG-PET changes or structural MRI changes required for a diagnosis of neuronal injury15 |
| McKhann criteria15 require evidence of amyloid pathology and neuronal injury to support a diagnosis of highly probable AD (biomarker evidence only recommended in individuals who do not meet the core clinical criteria for probable AD dementia).Dubois criteria (IWG2)26 require specific clinical features of AD (typical or atypical) plus evidence of in vivo AD pathology. Evidence of in vivo AD pathology: low CSF Aβ1-42 together with increased total-tau or phospho-tau or positive amyloid PET or proven mutation in PSEN1, PSEN2, or APP or other proven genes (including Down’s syndrome trisomy 21). | |
| Frontotemporal dementia | |
| bvFTD | |
| Frontal and/or anterior temporal lobe atrophy on MRI or CT | Either structural or PET imaging changes required for a diagnosis of probable bvFTD100 |
| Frontal and/or anterior temporal lobe hypoperfusion or hypometabolism on PET or SPECT | |
| Progressive non-fluent aphasia | |
| Predominant left posterior frontoinsular atrophy on MRI | Either structural or PET imaging changes required for an imaging supported diagnosis58 |
| Predominant left posterior frontoinsular hypoperfusion or hypometabolism on SPECT or PET | |
| Semantic dementia | |
| Predominant anterior temporal lobe atrophy | Either structural or PET imaging changes required for an imaging supported diagnosis58 |
| Predominant anterior temporal hypoperfusion or hypometabolism on SPECT or PET | |
| Dementia with Lewy bodies | |
| Relative preservation of medial temporal lobe structures on CT/MRI | Supportive feature (commonly present but not proven to have diagnostic specificity) |
| Generalised low uptake on SPECT/PET perfusion scan with reduced occipital activity | Supportive feature (commonly present but not proven to have diagnostic specificity) |
| Abnormal (low uptake) MIBG myocardial scintigraphy | Supportive feature (commonly present but not proven to have diagnostic specificity) |
| Abnormal uptake on PET/SPECT (eg, 123 I-FP CIT- DaTSCAN) | Supportive feature (used to differentiate DLB from AD and some forms of FTD) |
AD, Alzheimer's disease; APP, amyloid precursor protein; bvFDT, behavioural variant frontotemporal dementia; CSF, cerebrospinal fluid; FDG, 18-F-flourodeoxyglucose; MIBG, metaiodobenzylguanidine ; p-tau, tau phosphorylated at 181; PET, positron emission tomography; SPECT, single photon emission tomography; t-tau, total tau.
Table 1.
Optimal practice for CSF collection and processing
| Confounding variable | Ideal situation |
|---|---|
| Preanalytical factors | |
| Time of collection | 8:00–12 noon to avoid potential for diurnal variation in CSF biomarkers |
| LP needle | Needle gauge or design not known to influence measured biomarker concentration but gauge is related to risk of post-LP headache. Smallest size practical to use in diagnostic LP is 22G. Atraumatic needles are associated with reduced post-LP headache but increased failure rate |
| Use of lumbar catheters/manometers | Aβ1-42 may adhere—to be avoided, if possible |
| Collection vessel | Polypropylene tube recommended. Aβ1-42 and other proteins adhere to polystyrene and glass significantly reducing measured concentrations. Tube brand may also influence measured biomarker concentrations |
| Fasting | Not required |
| Blood contamination/blood–brain barrier dysfunction | Blood contamination of up to 5000 erythrocytes/µL cells does not alter measured biomarker concentration, but blood–brain barrier dysfunction equivalent to CSF/serum albumin ratio >11 results in reduced measured Aβ1-42 concentration and should be interpreted with care |
| Optimal volume | In addition to CSF collected for routine clinical examination (eg, cell count, oligoclonal bands, cytology, etc) 15 mL can safely be collected without increased risk of post LP headache |
| Centrifugation | Within 30 min of LP at 3000 rpm for 10 min to remove cells and other debris |
| Aliquot storage volume | Samples that are frozen prior to analysis should be stored in aliquots of a standardised volume which fits the tube size well. Specifically, one should strive for as high volume to surface ratio as possible (well-filled tubes). Volume to surface ratio and number of tube transfers influence measured Aβ1-42 concentration, probably due to protein adsorption |
| Freeze thawing | One or less freeze thaw cycles is recommended. Measured Aβ1-42 concentration drops by 20% after 3 freeze thaw cycles. Aβ1-42 and τ concentrations are stable at temperatures of −80°C |
| Choice of immunoassay/platform | Consistency required; variability in commercially available ELISA-based assays, calibration peptides and platforms mean interlaboratory and interassay consistency is poor |
CSF, cerebrospinal fluid; LB, lumbar puncture.
Blood and urine
While there are obvious advantages of blood-based or urine-based biomarkers for dementias, to date none have found utility in clinical practice. Brain derived proteins exist in much smaller concentrations in peripheral blood or urine than in CSF, due to the function of the blood–brain barrier and the large total volume of blood and urine in which they are diluted; further complications include significant binding of many proteins of interest and rapid clearance from the blood, which may make many conventional assays insufficiently sensitive. This may change with the development of more sensitive metabolomic and proteomic approaches being used for biomarker discovery.10
Online supplementary table shows the currently available biomarkers and those in current development.
Biomarkers of AD
AD is the commonest cause of dementia particularly in older individuals,11 and is characterised neuropathologically by amyloid plaques and tau containing neurofibrillary tangles. Other pathological changes include the presence of activated microglia around amyloid plaques12 and amyloid angiopathy and microhaemorrhages in some individuals with AD.13 Most cases of AD are sporadic, with autosomal dominant mutations in either the presenilin 1 (PSEN1), presenilin 2 (PSEN2) or amyloid precursor protein (APP) genes accounting for <1% of cases. In sporadic AD more than 20 genetic risk factors have been identified, implicating cholesterol transport, innate immunity and endosomal vesicle recycling in pathogenesis.14 Progressive impairment of episodic memory is the commonest clinical presentation of AD; rarer focal presentations include posterior cortical atrophy, logopenic aphasia (LPA) and a dysexecutive presentation.15 In clinical practice there is often significant overlap between these syndromes and a merging of symptoms as the disease progresses. The original clinical criteria for AD16 did not acknowledge this phenotypic diversity, and by requiring that an individual have dementia and multidomain cognitive impairment, precluded the early symptomatic and presymptomatic stages of AD. Newer criteria place an emphasis on using biomarkers to provide an earlier and more specific diagnosis17 18 (table 2). For research purposes only, this may extend to the prodromal phase of the disease, which may begin a decade or more before cognitive impairment occurs.17 19
Currently used biomarkers and their utility
Structural imaging
In AD, the typical imaging appearance is of global brain atrophy with early disproportionate symmetrical involvement of medial temporal lobe structures including the hippocampi.2 The presence of symmetrical medial temporal atrophy can differentiate AD from ageing with a sensitivity and specificity of around 80–85%20; and in a single centre pathologically proven study, established AD from DLB and vascular cognitive impairment with a sensitivity and specificity of 91% and 91%, respectively.21 The presence of medial temporal lobe atrophy per se is unable to differentiate AD from FTD in most instances,22 although patterns of (medial and lateral) temporal lobe atrophy may be very helpful in identifying specific FTD subgroups (see below). Medial temporal lobe atrophy can predict which individuals will develop clinical AD from a mild cognitive impairment (MCI) state with a sensitivity and specificity of 73% and 81%, respectively.20 22 Progressive atrophy of the parietal/occipital lobes is supportive of AD and in particular in distinguishing AD from FTD; incorporating visual ratings of posterior atrophy can improve the distinction of AD from other causes of dementia.2 Rates of whole brain and hippocampal atrophy, calculated from serial volumetric MRI are sensitive markers of progression of neurodegeneration and are increasingly used as outcome measures in trials of potentially disease modifying therapies in AD.22
Functional imaging
The typical AD pattern on FDG-PET or HMPAO-SPECT imaging is bilateral hypometabolism and hypoperfusion in the temporal and parietal cortices; the sensitivity and specificity in diagnosing AD versus other neurodegenerative diseases has been reported as 79% and 88%, respectively.23 These changes may predate brain atrophy or cognitive symptoms, and correlate with disease severity.24 Amyloid PET is a sensitive and specific means of imaging brain amyloid in individuals in vivo, and has been shown to correlate closely with autopsy measures of fibrillar amyloid load,25 and has considerable potential value in ruling in/out AD pathology as the cause of cognitive decline in a patient with cognitive impairment. Several amyloid PET agents are now licensed for clinical use, and amyloid imaging is now included in criteria for the diagnosis of AD.15 26 Importantly however as up to a third of elderly non-demented individuals may have a positive amyloid scan, the significance of which is as yet unclear, the clinical utility of amyloid PET imaging in older individuals is still to be determined and in particular it is not recommended in cognitively healthy individuals outside of research studies.17 27 Additionally, the presence of amyloid pathology does not always equate to a diagnosis of AD, as for instance a proportion of patients with DLB will also have amyloid deposition.28
Current guidelines (eg, by the UK Royal College of Radiologists and Physicians) advocate the use of amyloid PET in highly selected individuals with cognitive impairment after evaluation by a dementia expert where the presence or absence of amyloid pathology is expected to increase diagnostic certainty and influence management.29 At the time of writing, availability and cost of amyloid PET imaging has limited its use in clinical practice.
Cerebrospinal fluid
β-Amyloid
CSF levels of Aβ1-42, thought to be one of the key pathological forms of Aβ in brain tissue, are reduced in AD, with the degree of reduction correlating with brain amyloid plaque load.30 Reduction of CSF Aβ1-42 occurs years before symptom onset,31 and has good positive predictive value for conversion from MCI to clinical AD32; accordingly CSF Aβ1-42 is now included in new diagnostic criteria for MCI due to AD.15 In clinical practice, a normal CSF Aβ1-42 in a demented individual should prompt re-evaluation of a diagnosis of AD. Conversely however, a low CSF Aβ1-42, does not always reflect brain amyloid deposition, being seen in other conditions including multiple sclerosis, and more commonly due to CSF being collected, stored or processed incorrectly. Other forms of β-amyloid, notably Aβ1-40 can be measured in CSF and may better reflect both total brain Aβ burden than Aβ1-42.33 While some studies have suggested that addition of CSF Aβ1-40 may improve differential diagnosis in certain circumstances34 35 this has not yet entered routine clinical practice.
Tau and Phospho-tau 181
CSF levels of t-tau and tau phosphorylated at 181 (p-tau), are both increased in AD. Like Aβ1-42, t-tau and p-tau are usually measured using ELISA-based assays. There are also multiplexed assays for the three analytes. T-tau is increased after stroke, in inflammatory conditions and in other neurodegenerative diseases—most notably in Creutzfeldt-Jakob disease where levels are often orders of magnitude higher than in AD; p-tau elevation is thought to have high specificity for AD.36 Stability and reproducibility of t-tau and p-tau levels is good, and levels remain stable over periods of up to 6 months,37 suggesting that these biomarkers may be capable of detecting small biochemical changes induced by treatment. There are at present no data to support the use of tau/p-tau assays in peripheral blood, due to tau concentrations being below the lower limit of detection for most assays in the blood.
Combining CSF biomarkers
In 2006 a longitudinal study of 137 individuals followed for 4–6 years demonstrated that the combination of low CSF Aβ1-42, and elevated t-tau and p-tau could distinguish individuals with MCI/incipient AD from those without with 95% sensitivity and 87% specificity.38 These findings have since been replicated in several other studies.39 40 On a research basis, the combination of low Aβ42, elevated tau and p-tau has also been used to predict future cognitive decline in healthy older individuals.41 In clinical practice, the combination of low CSF Aβ1-42 and elevated tau (or p-tau) to Aβ1-42 ratio is often used to support the diagnosis of AD, with one recent study suggesting that tau:Aβ42 ratio is the most robust single biomarker combination.42
In practice, combining several of these different biomarkers, each of which provides different insights into the underlying disease process, may increase diagnostic certainty (see Case Study figure 1).
Figure 1.
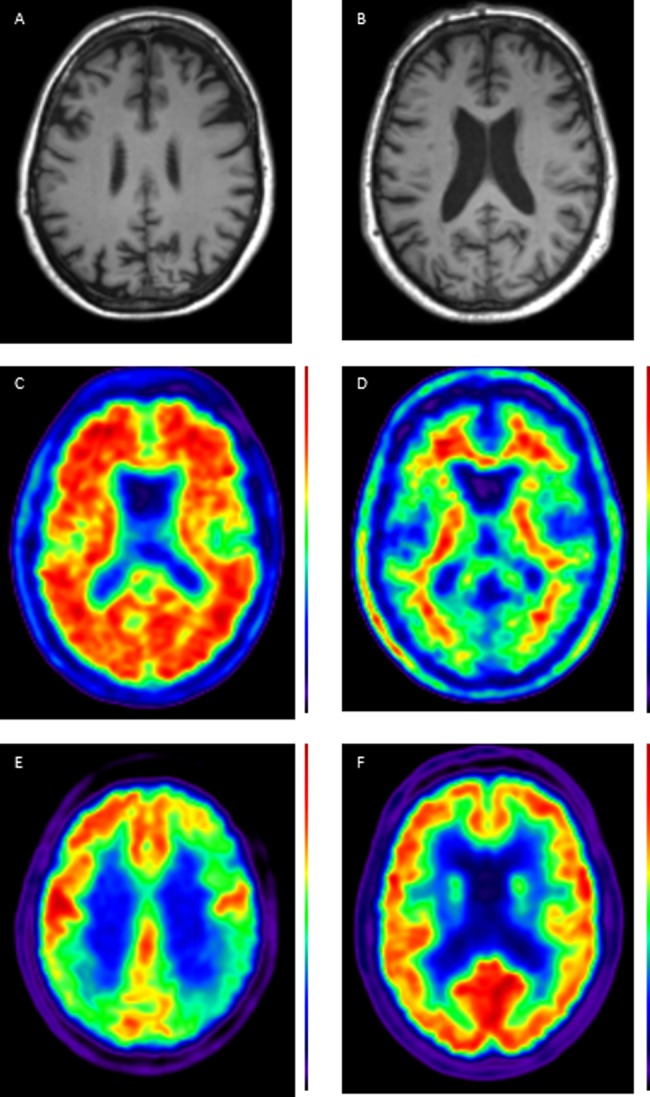
Case showing clinical use of biomarkers. A 56 year old patient presented with a 5–10-year history of ‘scattiness’. Three years ago she developed difficulties reading an analogue clock, her spelling had declined and she had difficulty reading, losing her place from line to line. She received a clinical diagnosis of posterior cortical atrophy. Subsequently episodic memory became impaired. At the time of scanning, the Mini-Mental State Examination score was 19/30. A T1 volumetric MRI of the brain demonstrated a posterior pattern of cortical atrophy (A) with preserved hippocampal volumes compared with a healthy control patient (B); A 18F-florbetpair amyloid positron emission tomography (PET) scans shows widespread cortical amyloid deposition (C) compared with a healthy control (D) fludeoxyglucose (18F) PET scan demonstrates a posterior dominant pattern of hypometabolism (E) SUVR 1.0–1.4, compared with an age matched healthy control (F) SUVR 1.0–1.5. Cerebrospinal fluid examination demonstrated an elevated t-tau: 1080 pg/mL (NR 146–595); Aβ1–42 360 pg/mL ((NR 627–1322) giving a tau/Aβ1–42 ratio of 3. This case illustrates how different biomarkers can provide complementary information including regional neuronal loss, more widespread metabolic dysfunction, as well as confirming the underlying pathology—in this case, Alzheimer's disease. (NB for clinical purposes, 18F-florbetapir images should be interpreted on a grey rather than colour scale.)
Emerging and future biomarkers
Advanced MRI
Current hypotheses predict that amyloid deposition, tau mediated neuronal dysfunction, neuroinflammation and synaptic loss precede the development of structural brain changes, that is, atrophy by several years, which in turn predates cognitive impairment. There is therefore considerable interest in determining imaging biomarkers to detect and quantify AD-related network disruption after the emergence of molecular pathology but before irreversible neuronal loss. Techniques including diffusion tensor imaging (DTI) and rsfMRI probing white matter tracts and functional integrity of neuronal networks, respectively, show promising results in group studies, but are only just being applied at the individual level. Using rsfMRI, presymptomatic individuals at increased risk of developing AD have shown altered resting connectivity in a distributed temporoparietofrontal network (the so-called default mode network) and altered task-related activations involving components of this network that may precede structural brain damage by up to many years.43 44 This suggests a potential future application in trial settings where rsfMRI might be used to assess the early impact of candidate interventions on brain function. Arterial spin labelling MRI has the potential to demonstrate cerebral blood flow pattern, which may provide a non-invasive means of obtaining similar information to FDG-PET.45
Tau PET imaging
Very recently, a number of tau PET tracers have been developed. At the time of writing, tau imaging has been performed only in small number of individuals with AD, and relatively little data is available.46 Once mature, tau PET imaging may prove valuable both for differential diagnosis, for prognostication, and as an outcome measure for clinical trials.
Fluid biomarkers
Biomarkers of amyloid processing
The dominant hypothesis for AD pathogenesis focuses on the production of toxic, amyloidogenic forms of Aβ.47The APP can be cleaved either sequentially by β-secretase and γ-secretase cleavage, producing amyloidogenic forms of Aβ, including Aβ1-42; or via other pathways to non-amyloidogenic forms. The β-site APP-cleaving enzyme 1 (BACE1), the major β-secretase in the brain, can be measured in the CSF. While some studies have determined higher levels of CSF BACE1 in patients with AD, results have generally been inconsistent.39
Secreted forms of APP reflecting the amyloidogenic and non-amyloidogenic pathways can also be measured in the CSF, but again have yet to show consistent results.39 There is also considerable interest in measuring CSF levels of soluble Aβ oligomers, which may be the most toxic Aβ species.39 Perhaps due to the fact that these moieties are predicted to have maximum effects very early in the disease, that they are thought to be present in very small quantities in CSF, and exist in multiple forms, reliable assays that show consistent differences between patients and controls have yet to be validated.
Biomarkers of neuroinflammation
A number of pathways related to neuroinflammation and more specifically microglial activation have been implicated in AD pathogenesis.48 The astrocytic and oligodendrocytic protein S100B is elevated in a range of conditions—most notably prion diseases, but possibly also in mild/moderate AD49 where it may correlate with rate of brain atrophy.50 The glycoprotein YKL-40 produced by astrocytes or activated microglia, plays a number of poorly defined roles in CNS inflammation, and is increased in CSF and serum in AD and FTD.51 F2-Isoprostanes, markers of membrane lipid peroxidation and inflammation, have been shown to predict conversion from MCI to AD.52 Other potential measures are shown in supplementary table.
Biomarkers for subcortical axonal degeneration and synaptic dysfunction
Elevated CSF neurofilament light protein (NFL) levels indicate involvement of predominantly large-calibre myelinated axons, and are elevated in a range of disorders including stroke, inflammatory disorders, vascular cognitive impairment and FTD (see below), but typically not ‘pure’ AD. A range of putative markers of synaptic function, and notably neurogranin show promise,53 but as yet their use in clinical practice is limited due to the difficulties of measuring their low concentration in CSF.
Blood-based and urine-based biomarkers
Despite much research, perhaps for the reasons discussed above, there are currently no established blood-based biomarkers for AD. Recent approaches using techniques where several biomarkers are analysed simultaneously have identified promising biomarkers,54 but the results have unfortunately been hard to replicate.55 Over 20 such studies, using different clinical cohorts and different methodology, now exist,55 but the majority of potential biomarkers, including a set of 10 lipids published in a recent study56 have only been detected in single studies. A small number of proteins have been identified as promising potential biomarkers across more than one study55; these will however need to be tested in larger cohorts with prospective follow-up to determine their clinical utility.
Biomarkers of FTD
The term FTD refers to a group of neurodegenerative disorders characterised by atrophy of the frontal and temporal lobes. Prevalence studies suggest that FTD is the second commonest cause of young onset dementia after AD.57 The two main clinical syndromes of FTD, are behavioural variant FTD (bvFTD), where there is deterioration in social function and personality; and primary progressive aphasia (PPA) where there is an insidious decline in language skills. PPA is further divided into several subtypes including semantic dementia (SD), progressive non-fluent aphasia (PNFA), logopenic aphasia (LPA) (typically an AD variant) and progressive apraxia of speech, based on the pattern of language breakdown.58 FTD and motor neuron disease show considerable overlap.59 Approximately a third to a half of patients with FTD—and particularly those with bvFTD—have a family history with an autosomal dominant pattern of inheritance. The three most common genes associated with FTD are hexanucelotide expansions in the C9orf72 gene (also linked to motor neuron disease phenotypes), mutations in the microtubule associated protein tau (MAPT) gene, and loss of function mutations in the progranulin (GRN) gene; a number of rarer genetic causes are also recognised.60
FTD occurring on a sporadic and an autosomal dominant basis is associated with a range of different underlying pathologies, based on the predominant protein accumulation. These include tau (4-repeat—progressive supranuclear palsy (PSP) or corticobasal degeneration (CBD) type, 3-repeat—Pick's disease type, and mixed 3-repeat and 4-repeat forms), Tar-DNA binding protein (TDP)-43 (types A–D) and fused in sarcoma protein (FUS). In bvFTD any of the pathological variants can be found, with tau and TDP-43 pathologies representing the majority in an approximately 50:50 split. SD and the behavioural/prosopagnosic presentations of SD (associated with more prominent right temporal lobe atrophy) are strongly associated with TDP type C pathology. The pathology of the other language forms is more complex, and includes AD (particularly in LPA), tauopathy (with PSP and CBD pathology commonly seen in patients with apraxia of speech), and TDP-43 proteinopathies. The co-occurrence of FTD and MND is strongly suggestive of underlying TDP-43 (typically type B) pathology.60
Currently used biomarkers and their utility
Structural brain imaging
While by no means 100% concordant, pattern of brain atrophy can provide important clues, not only to clinical phenotype but also for the underlying pathology and in some cases genetic basis of disease. Based on clinical phenotype, patients with bvFTD often show atrophy of mesial frontal, orbitofrontal and anterior insula cortices. As the disease progresses there is involvement of the other frontal neocortical grey matter regions, the striatum, hippocampi, posterior insulae and parietal lobes.61 In SD, patients show bilateral but typically highly asymmetrical (left)-sided atrophy of the anterior temporal lobes involving the polar and perirhinal cortices and anterior fusiform gyri. As the disease progresses this degeneration extends caudally into the posterior temporal lobes and rostrally into the posterior, inferior frontal lobes. The structural imaging findings in PNFA are very heterogeneous and scans are often remarkably normal in the earliest stages. Typically, however, there may be volume loss involving the anterior perisylvian, and inferior opercular and insular portions of the dominant hemisphere.62 As the disease progresses there is involvement of the dorsolateral prefrontal cortex, temporal cortex, orbital and anterior cingulate regions and parietal lobe.63 MRI-based quantification of atrophy rates of whole brain or lobar volumes are potentially useful objective biomarkers of progression in FTD64 .
When classified on the basis of pathology, 3-repeat tau Pick's disease is often associated with atrophy involving the prefrontal cortex with severe dorsolateral frontal atrophy, which can be markedly asymmetric.65 Patients with PSP and CBD can often present with an FTD-like syndrome, in which case certain features on structural imaging may be particularly helpful: patients with PSP classically have atrophy of the rostral midbrain, leading to the so-called Hummingbird sign on imaging66 this is however not always present in dementia dominant cases. In PSP and CBD atrophy may affect the posterior frontal lobes, while in CBD the atrophy will often involve the parietal lobes and can be very asymmetric. However, in pathologically ascertained series, the atrophy in ‘cognitive’ CBD is often surprisingly non-specific and symmetrical.65 67
Certain patterns of atrophy have been associated, although by no means invariably, with the different TDP-43 subtypes. Thus TDP-A is often associated with asymmetric atrophy involving the frontal, temporal and parietal lobes; TDP-B is associated with frontal atrophy, in keeping with the association of this pathology with the clinical syndrome of FTD/MND; and TDP-C with asymmetric anterior temporal lobe atrophy, consistent with the close clinicopathological correlation with SD.65 FUS pathology is associated with frontal atrophy often with striking atrophy of the caudate nuclei.65
From a genetic perspective, individuals with tau (MAPT) mutations, often have very symmetrical anterior and inferior medial temporal lobe atrophy, with involvement of the orbitofrontal cortices.68 In GRN mutations atrophy is typically highly asymmetric involving the temporal, inferior frontal and parietal lobes.68 No clear and consistent pattern of atrophy has yet emerged in C9orf72 mutation cases: atrophy is often symmetrical, but not always; and while often relatively mild, can on occasions be very widespread. It can involve the frontal and temporal lobes, and thalami, but may extend to the occipital lobe and cerebellum,69 where TDP-43 or p62-positive inclusions are frequently found at histology.
Functional imaging
FDG-PET typically shows frontal and temporal lobe hypometabolism in FTD. Demonstrating hypometabolism may be particularly valuable in cases with behavioural change with normal structural imaging, where a non-degenerative FTD-mimic is considered.4 Where the differential diagnosis is between AD and FTD, amyloid PET imaging may be very useful in ruling in/out the presence of amyloid pathology; this may prove particularly valuable in the progressive aphasias given their considerable clinical and pathological heterogeneity and also in corticobasal syndrome where it is difficult to distinguish CBD from AD as the underling cause.70
Cerebrospinal fluid
In the absence of specific CSF biomarkers for TDP-43, or to distinguish the different tauopathies, currently the most important clinical utility of CSF biomarkers in FTD is to distinguish underlying AD from other FTD pathologies. In particular, if CSF is appropriately handled and measured, reduction of Aβ1-42 level would not be expected in cases with tau or TDP-43 proteinopathies. Several studies71 have shown that CSF t-tau levels are lower in FTD than those seen in AD, but higher than that seen in controls,71 with t-tau levels correlating with neuropsychological, neuroimaging and prognosis in patients with FTD72; however in many cases with FTD, CSF t-tau levels can be normal. As discussed above, p-tau elevation is typically seen in AD rather than other neurodegenerative diseases, with one study of FTD finding that a reduced p-tau to t-tau ratio predicts TDP-43 pathology in FTD.73
Emerging and future biomarkers
Imaging
As with AD, DTI and rsfMRI have considerable potential to detect presymptomatic and disease-specific network breakdown in the various forms of FTD. Specific patterns of DTI breakdown have been associated with the clinical presentations in FTD74 and on a group level DTI may be helpful in discriminating between tau and TDP-43 proteinopathies, the latter showing greater white matter damage.75 There is considerable evidence again at least on a group level that breakdown in both functional connectivity in FTD involves different functional networks compared with AD, for example, targeting the so-called salience (environmentally directed) as opposed to default mode (internalised thought) network.76 Within the FTD spectrum, different diseases may have characteristic, molecularly determined network signatures or ‘nexopathies’77 It is however not clear if and how these techniques can be applied on an individual basis.
As with AD, ASL may provide a non-invasive alternative to FDG-PET, providing an MRI-based measure of cerebral perfusion. It is not yet clear to what extent available tau PET ligands bind different subtypes of tau pathology or how reliably they can distinguish tau, AD and TDP-43 pathologies, or whether this technique will prove a useful means of reliably distinguishing the various FTD pathologies in vivo. To date, no specific TDP-43-based ligands are available.
Fluid biomarkers
Tar-DNA binding protein-43
Increased TDP-43 levels have been found in CSF in FTD and MND cases.78 Kasai et al79 found increased levels in MND particularly early in disease progression, suggesting that CSF TDP-43 may be an early marker of TDP-43 proteinopathies. Patients with C9orf72 expanded repeats or GRN mutations, where TDP-43 pathology can reliably be predicted in vivo, have been shown to have increased plasma and CSF levels of phosphorylated TDP-43 compared with other patients with FTD and normal controls; this same study found plasma levels of total TDP-43 to be decreased in mutation carriers, possibly due to alterations in the ratio of phosphorylated to total TDP-43 in favour of the former.80
Progranulin
Serum progranulin levels show considerable promise as a biomarker of underlying progranulin mutations.81 Null mutations have been associated with a fourfold reduction in plasma progranulin levels compared to controls,82 with missense mutations resulting in a smaller reduction.83 Studies of large, mixed, FTD populations are required to determine the sensitivity and specificity of serum progranulin as a predictor of mutations and of the underlying pathology.
Neurofilament
Two recent studies84 85 have found that elevated CSF NFL levels correlate with FTD disease severity.84 The highest levels were in tau negative cases and SD,85 where TDP-43 is the predominant pathology. Different levels, and perhaps forms of neurofilament may therefore find utility in differentiating the underlying pathology in cases of FTD, in distinguishing AD from non-AD pathology, and for tracking disease in patients with confirmed disease; however, as NFL is elevated in several other conditions—and notably vascular disease—its utility in isolated unselected cases is less clear.
Biomarkers of neuroinflammation
As with AD, there is considerable interest in inflammation in FTD. A recent study86 showed increased serum TNF-α in two different conditions strongly associated with TDP-43 pathology, that is, SD and GRN mutation carriers. A number of inflammatory biomarkers have been reported to be elevated in FTD. One study has suggested that IL-23 may be specific for FTD associated with tau, while IL-17 may be specific for FTD associated with TDP-43 pathology; this however requires further confirmation.87
Biomarkers of DLB
DLB, the second most common cause of neurodegenerative dementia after AD, is characterised by a progressive deterioration in cognition, with core features including fluctuating cognition and variations in alertness; recurrent visual hallucinations; and features of motor parkinsonism.88 Neuropsychological testing typically reveals deficits in attention, executive function and visuospatial ability.88 The core pathology of DLB is the presence of Lewy bodies in brainstem, limbic regions or cortex comprising α-synuclein. DLB is widely thought to be on a continuum with Parkinson's disease dementia, and at autopsy is commonly associated with other pathologies including AD and vascular disease.89
Currently used biomarkers and their utility
Structural brain imaging
Typically, DLB is associated with preserved volume of the medial temporal lobes relative to that seen in AD.90 The extent to which hippocampal sparing as a marker of DLB as opposed to AD can be applied in individual cases is limited, with some studies finding that MRI is not useful in differentiating DLB from AD.91
Functional imaging
Current clinical diagnostic criteria include PET/SPECT (eg, 123 I-FP CIT—DaTSCAN) measures of dopaminergic loss in the basal ganglia as a suggestive feature of DLB.88 123 I-FP CIT SPECT imaging can be used to differentiate DLB from AD and some forms of FTD92 with reasonable diagnostic accuracy (sensitivity around 80% with 90% specificity), but is less useful in differentiating DLB from atypical parkinsonian conditions which can also show nigrostriatal abnormalities.93 FDG-PET occipital hypoperfusion may have utility in distinguishing DLB from AD,94 although changes can be seen in patients with posterior variants of AD, and in CBD. Several studies have found decreased metaiodobenzylguanidine myocardial scintigraphy reflecting impaired sympathetic nervous system function in DLB compared with AD.93
Emerging and future biomarkers
Imaging
Amyloid PET imaging is likely to have limited utility in distinguishing AD from DLB as both may have similar cortical amyloid load,95 with a recent review of 12 studies finding amyloid PET positivity in 57% of clinically diagnosed DLB cases.28 However, it may have a role in differentiating DLB from other parkinsonian conditions. There are as yet no α-synuclein PET ligands available.
CSF
Levels of CSF tau are very variable in DLB, typically being lower than in AD95 although in rapidly progressive cases they can be very elevated. Studies examining Aβ1-42 levels have found similar levels between DLB and AD.96 Some studies have reported reduced levels of CSF α-synuclein in DLB,97 while others have not.98 An oxidised form of Aβ1-40 has been proposed as a potential DLB marker.99 However, this oxidation may appear in CSF postsampling and the results need replication before any conclusions can be drawn on the diagnostic usefulness of this Aβ isoform.
Conclusion
In the correct clinical context, imaging and CSF biomarkers can provide in vivo evidence for the various pathological processes underpinning the different causes of dementia, which can in turn be used to improve diagnostic sensitivity and specificity, as inclusion and exclusion criteria and outcome measures in clinical trials, and to provide insights into pathogenesis. Biomarkers are now being incorporated into new diagnostic criteria (see table 2), and are being used—at least on a research basis—to allow for presymptomatic or paucity-symptomatic diagnosis. While the specific biomarkers vary across different dementias, there are common themes including their potential ability to allow presymptomatic diagnosis, track disease progression by the sequence of changes in biomarkers, and reflect underlying pathology. Many of the biomarkers have shown utility on a group level, but how they can be applied on an individual level and in clinical practice requires further investigation. This should not however diminish their importance especially in the setting of clinical trials, for patient stratification for entry and as outcome measures. No one biomarker is diagnostic of any one condition in its own right, each has limitations, and interpretation should always be done in the appropriate clinical context. Future studies will benefit from ever more sensitive and automated techniques, which will hopefully make blood-based and/or urine-based biomarkers a reality. Critical to the development and validation of any biomarker is standardisation of sample/scan collection, analysis and interpretation; and ultimately postmortem confirmation of diagnosis.
Supplementary Material
Footnotes
Correction notice: This paper has been made open access since it was first published online.
Acknowledgements: The authors are very grateful to Dr Manja Lehmann, PhD, for providing the images in figure 1, and they also gratefully acknowledge the support of the Leonard Wolfson Experimental Neurology Centre and Alzheimer's Research UK.
Contributors: RMA, RWP and JMS contributed to manuscript concept, writing and preparation; RWP, JDW, HZ, JTO’B, GMH and NCF contributed to manuscript writing and preparation.
Funding: This work was supported by the NIHR Queen Square Dementia BRU, by funding to Forefront, a collaborative research group dedicated to the study of frontotemporal dementia and motor neuron disease, from the National Health and Medical research Council of Australia (NHMRC) programme grant (#1037746) and the Australian Research Council Centre of Excellence in Cognition and its Disorders Memory Node (#CE110001021). The images in figure 1 were obtained in a study funded by AVID Radiopharmaceuticals (a wholly owned subsidiary of Eli Lilly) and the The National Brain Appeal—Frontotemporal Dementia Research Fund.
Competing interests: RMA holds a Royal Australasian College of Physicians and MND Research Australia scholarship, GMH is an NHMRC senior principal research fellow (#630434). JDW holds a Wellcome Trust Senior Clinical Fellowship (Grant No 091673/Z/10/Z).
Provenance and peer review: Commissioned; externally peer reviewed.
References
- 1.Biomarkers Definitions Working G. Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharmacol Ther 2001;69:89–95. [DOI] [PubMed] [Google Scholar]
- 2.Harper L, Barkhof F, Scheltens P, et al. An algorithmic approach to structural imaging in dementia. J Neurol Neurosurg Psychiatry 2014;85:692–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ahmed RM, Murphy E, Davagnanam I, et al. A practical approach to diagnosing adult onset leukodystrophies. J Neurol Neurosurg Psychiatry 2014;85:770–81. [DOI] [PubMed] [Google Scholar]
- 4.Kipps CM, Hodges JR, Fryer TD, et al. Combined magnetic resonance imaging and positron emission tomography brain imaging in behavioural variant frontotemporal degeneration: refining the clinical phenotype. Brain 2009;132:2566–78. [DOI] [PubMed] [Google Scholar]
- 5.Bajaj N, Hauser RA, Grachev ID. Clinical utility of dopamine transporter single photon emission CT (DaT-SPECT) with (123I) ioflupane in diagnosis of parkinsonian syndromes. J Neurol Neurosurg Psychiatry 2013;84:1288–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Svedberg MM, Rahman O, Hall H. Preclinical studies of potential amyloid binding PET/SPECT ligands in Alzheimer's disease. Nucl Med Biol 2012;39:484–501. [DOI] [PubMed] [Google Scholar]
- 7.Rossor MN, Fox NC, Mummery CJ, et al. The diagnosis of young-onset dementia. Lancet Neurol 2010;9:793–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Atarashi R, Sano K, Satoh K, et al. Real-time quaking-induced conversion: a highly sensitive assay for prion detection. Prion 2011;5:150–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bartlett JW, Frost C, Mattsson N, et al. Determining cut-points for Alzheimer's disease biomarkers: statistical issues, methods and challenges. Biomark Med 2012;6:391–400. [DOI] [PubMed] [Google Scholar]
- 10.Manwaring V, Heywood WE, Clayton R, et al. The identification of new biomarkers for identifying and monitoring kidney disease and their translation into a rapid mass spectrometry-based test: evidence of presymptomatic kidney disease in pediatric Fabry and type-I diabetic patients. J Proteome Res 2013;12:2013–21. [DOI] [PubMed] [Google Scholar]
- 11.Ott A, Breteler MM, van Harskamp F, et al. Prevalence of Alzheimer's disease and vascular dementia: association with education. The Rotterdam study. BMJ 1995;310:970–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Itagaki S, McGeer PL, Akiyama H, et al. Relationship of microglia and astrocytes to amyloid deposits of Alzheimer disease. J Neuroimmunol 1989;24:173–82. [DOI] [PubMed] [Google Scholar]
- 13.Charidimou A, Gang Q, Werring DJ. Sporadic cerebral amyloid angiopathy revisited: recent insights into pathophysiology and clinical spectrum. J Neurol Neurosurg Psychiatry 2012;83:124–37. [DOI] [PubMed] [Google Scholar]
- 14.Lambert JC, Ibrahim-Verbaas CA, Harold D, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet 2013;45:1452–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011;7:263–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McKhann G, Drachman D, Folstein M, et al. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology 1984;34:939–44. [DOI] [PubMed] [Google Scholar]
- 17.Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011;7:280–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Albert MS, DeKosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011;7:270–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jack CR, Jr, Knopman DS, Jagust WJ, et al. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol 2010;9:119–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Duara R, Loewenstein DA, Potter E, et al. Medial temporal lobe atrophy on MRI scans and the diagnosis of Alzheimer disease. Neurology 2008;71:1986–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Burton EJ, Barber R, Mukaetova-Ladinska EB, et al. Medial temporal lobe atrophy on MRI differentiates Alzheimer's disease from dementia with Lewy bodies and vascular cognitive impairment: a prospective study with pathological verification of diagnosis. Brain 2009;132:195–203. [DOI] [PubMed] [Google Scholar]
- 22.Frisoni GB, Fox NC, Jack CR, Jr, et al. The clinical use of structural MRI in Alzheimer disease. Nat Rev Neurol 2010;6:67–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hoffman JM, Welsh-Bohmer KA, Hanson M, et al. FDG PET imaging in patients with pathologically verified dementia. J Nucl Med 2000;41:1920–8. [PubMed] [Google Scholar]
- 24.Mosconi L, Berti V, Glodzik L, et al. Pre-clinical detection of Alzheimer's disease using FDG-PET, with or without amyloid imaging. J Alzheimers Dis 2010;20:843–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clark CM, Schneider JA, Bedell BJ, et al. Use of florbetapir-PET for imaging beta-amyloid pathology. JAMA 2011;305:275–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dubois B, Feldman HH, Jacova C, et al. Advancing research diagnostic criteria for Alzheimer's disease: the IWG-2 criteria. Lancet Neurol 2014;13:614–29. [DOI] [PubMed] [Google Scholar]
- 27.Chételat G, La Joie R, Villain N, et al. Amyloid imaging in cognitively normal individuals, at-risk populations and preclinical Alzheimer's disease. Neuroimage Clin 2013;2:356–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Donaghy P, Thomas AJ, O'Brien JT. Amyloid PET imaging in Lewy body disorders. Am J Geriatr Psychiatry 2013: pii: S1552-5260(14)00029-6 [Epub ahead of print, 3 Jul 2013]. [DOI] [PubMed] [Google Scholar]
- 29.Radiologists RCoPaRCo. Evidence- based indications for the use of PET-CT in the UK 2013 2013.
- 30.Seppala TT, Nerg O, Koivisto AM, et al. CSF biomarkers for Alzheimer disease correlate with cortical brain biopsy findings. Neurology 2012; 78:1568–75. [DOI] [PubMed] [Google Scholar]
- 31.Moghekar A, Li S, Lu Y, et al. CSF biomarker changes precede symptom onset of mild cognitive impairment. Neurology 2013;81:1753–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mattsson N, Zetterberg H, Hansson O, et al. CSF biomarkers and incipient Alzheimer disease in patients with mild cognitive impairment. JAMA 2009;302:385–93. [DOI] [PubMed] [Google Scholar]
- 33.Wiltfang J, Esselmann H, Bibl M, et al. Amyloid beta peptide ratio 42/40 but not A beta 42 correlates with phospho-tau in patients with low- and high-CSF A beta 40 load. J Neurochem 2007;101:1053–9. [DOI] [PubMed] [Google Scholar]
- 34.Verwey NA, Kester MI, van der Flier WM, et al. Additional value of CSF amyloid-beta 40 levels in the differentiation between FTLD and control subjects. J Alzheimers Dis 2010;20:445–52. [DOI] [PubMed] [Google Scholar]
- 35.Slaets S, Le Bastard N, Martin JJ, et al. Cerebrospinal fluid Abeta1-40 improves differential dementia diagnosis in patients with intermediate P-tau181P levels. J Alzheimers Dis 2013;36:759–67. [DOI] [PubMed] [Google Scholar]
- 36.Sjogren M, Davidsson P, Tullberg M, et al. Both total and phosphorylated tau are increased in Alzheimer's disease. J Neurol Neurosurg Psychiatry 2001;70:624–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blennow K, Zetterberg H, Minthon L, et al. Longitudinal stability of CSF biomarkers in Alzheimer's disease. Neurosci Lett 2007;419:18–22. [DOI] [PubMed] [Google Scholar]
- 38.Hansson O, Zetterberg H, Buchhave P, et al. Association between CSF biomarkers and incipient Alzheimer's disease in patients with mild cognitive impairment: a follow-up study. Lancet Neurol 2006;5:228–34. [DOI] [PubMed] [Google Scholar]
- 39.Rosén C, Hansson O, Blennow K, et al. Fluid biomarkers in Alzheimer's disease—current concepts. Mol Neurodegener 2013;8:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Paterson RW, Toombs J, Slattery CF, et al. Biomarker modelling of early molecular changes in Alzheimer's disease. Mol Diagn Ther 2014;18:213–27. [DOI] [PubMed] [Google Scholar]
- 41.Stomrud E, Hansson O, Blennow K, et al. Cerebrospinal fluid biomarkers predict decline in subjective cognitive function over 3 years in healthy elderly. Dement Geriatr Cogn Disord 2007;24:118–24. [DOI] [PubMed] [Google Scholar]
- 42.Duits FH, Teunissen CE, Bouwman FH, et al. The cerebrospinal fluid “Alzheimer profile”: easily said, but what does it mean? Alzheimers Dement 2014: pii: S1064-7481(13)00168-1 [Epub ahead of print, 7 Apl 2014]. [DOI] [PubMed] [Google Scholar]
- 43.Filippini N, MacIntosh BJ, Hough MG, et al. Distinct patterns of brain activity in young carriers of the APOE-epsilon4 allele. Proc Natl Acad Sci USA 2009;106:7209–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sala-Llonch R, Fortea J, Bartres-Faz D, et al. Evolving brain functional abnormalities in PSEN1 mutation carriers: a resting and visual encoding fMRI study. J Alzheimers Dis 2013;36:165–75. [DOI] [PubMed] [Google Scholar]
- 45.Alsop DC, Dai W, Grossman M, et al. Arterial spin labeling blood flow MRI: its role in the early characterization of Alzheimer's disease. J Alzheimers Dis 2010;20:871–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang W, Arteaga J, Cashion DK, et al. A highly selective and specific PET tracer for imaging of tau pathologies. J Alzheimers Dis 2012;31:601–12. [DOI] [PubMed] [Google Scholar]
- 47.Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science 1992;256:184–5. [DOI] [PubMed] [Google Scholar]
- 48.Schott JM, Revesz T. Inflammation in Alzheimer's disease: insights from immunotherapy. Brain 2013;136:2654–6. [DOI] [PubMed] [Google Scholar]
- 49.Peskind ER, Griffin WS, Akama KT, et al. Cerebrospinal fluid S100B is elevated in the earlier stages of Alzheimer's disease. Neurochem Int 2001;39:409–13. [DOI] [PubMed] [Google Scholar]
- 50.Petzold A, Jenkins R, Watt HC, et al. Cerebrospinal fluid S100B correlates with brain atrophy in Alzheimer's disease. Neurosci Lett 2003;336:167–70. [DOI] [PubMed] [Google Scholar]
- 51.Craig-Schapiro R, Perrin RJ, Roe CM, et al. YKL-40: a novel prognostic fluid biomarker for preclinical Alzheimer's disease. Biol Psychiatry 2010;68:903–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brys M, Pirraglia E, Rich K, et al. Prediction and longitudinal study of CSF biomarkers in mild cognitive impairment. Neurobiol Aging 2009;30:682–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Thorsell A, Bjerke M, Gobom J, et al. Neurogranin in cerebrospinal fluid as a marker of synaptic degeneration in Alzheimer's disease. Brain Res 2010;1362:13–22. [DOI] [PubMed] [Google Scholar]
- 54.Doecke JD, Laws SM, Faux NG, et al. Blood-based protein biomarkers for diagnosis of Alzheimer disease. Arch Neurol 2012;69:1318–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kiddle SJ, Sattlecker M, Proitsi P, et al. Candidate blood proteome markers of Alzheimer's disease onset and progression: a systematic review and replication study. J Alzheimers Dis 2014;38:515–31. [DOI] [PubMed] [Google Scholar]
- 56.Mapstone M, Cheema AK, Fiandaca MS, et al. Plasma phospholipids identify antecedent memory impairment in older adults. Nat Med 2014;20:415–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ratnavalli E, Brayne C, Dawson K, et al. The prevalence of frontotemporal dementia. Neurology 2002;58:1615–21. [DOI] [PubMed] [Google Scholar]
- 58.Gorno-Tempini ML, Hillis AE, Weintraub S, et al. Classification of primary progressive aphasia and its variants. Neurology 2011;76:1006–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Clark CM, Forman MS. Frontotemporal lobar degeneration with motor neuron disease: a clinical and pathological spectrum. Arch Neurol 2006;63:489–90. [DOI] [PubMed] [Google Scholar]
- 60.Seelaar H, Rohrer JD, Pijnenburg YA, et al. Clinical, genetic and pathological heterogeneity of frontotemporal dementia: a review. J Neurol Neurosurg Psychiatry 2011;82:476–86. [DOI] [PubMed] [Google Scholar]
- 61.Seeley WW, Crawford R, Rascovsky K, et al. Frontal paralimbic network atrophy in very mild behavioral variant frontotemporal dementia. Arch Neurol 2008;65:249–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gorno-Tempini ML, Dronkers NF, Rankin KP, et al. Cognition and anatomy in three variants of primary progressive aphasia. Ann Neurol 2004;55:335–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Grossman M. Primary progressive aphasia: clinicopathological correlations. Nat Rev Neurol 2010;6:88–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gordon E, Rohrer JD, Kim LG, et al. Measuring disease progression in frontotemporal lobar degeneration: a clinical and MRI study. Neurology 2010;74:666–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rohrer JD, Lashley T, Schott JM, et al. Clinical and neuroanatomical signatures of tissue pathology in frontotemporal lobar degeneration. Brain 2011;134:2565–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Massey LA, Micallef C, Paviour DC, et al. Conventional magnetic resonance imaging in confirmed progressive supranuclear palsy and multiple system atrophy. Mov Disord 2012;27:1754–62. [DOI] [PubMed] [Google Scholar]
- 67.Rankin KP, Mayo MC, Seeley WW, et al. Behavioral variant frontotemporal dementia with corticobasal degeneration pathology: phenotypic comparison to bvFTD with Pick's disease. J Mol Neurosci 2011;45:594–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Whitwell JL, Jack CR, Jr, Boeve BF, et al. Voxel-based morphometry patterns of atrophy in FTLD with mutations in MAPT or PGRN. Neurology 2009;72:813–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Whitwell JL, Weigand SD, Boeve BF, et al. Neuroimaging signatures of frontotemporal dementia genetics: C9ORF72, tau, progranulin and sporadics. Brain 2012;135:794–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rohrer JD, Schott JM. Primary progressive aphasia: defining genetic and pathological subtypes. Curr Alzheimer Res 2011;8:266–72. [DOI] [PubMed] [Google Scholar]
- 71.Riemenschneider M, Wagenpfeil S, Diehl J, et al. Tau and Abeta42 protein in CSF of patients with frontotemporal degeneration. Neurology 2002;58:1622–8. [DOI] [PubMed] [Google Scholar]
- 72.Borroni B, Cerini C, Archetti S, et al. Cerebrospinal fluid tau in frontotemporal lobar degeneration: clinical, neuroimaging, and prognostic correlates. J Alzheimers Dis 2011;23:505–12. [DOI] [PubMed] [Google Scholar]
- 73.Hu WT, Watts K, Grossman M, et al. Reduced CSF p-Tau181 to Tau ratio is a biomarker for FTLD-TDP. Neurology 2013;81:1945–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Whitwell JL, Josephs KA. Recent advances in the imaging of frontotemporal dementia. Curr Neurol Neurosci Rep 2012;12:715–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McMillan CT, Irwin DJ, Avants BB, et al. White matter imaging helps dissociate tau from TDP-43 in frontotemporal lobar degeneration. J Neurol Neurosurg Psychiatry 2013;84:949–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhou J, Greicius MD, Gennatas ED, et al. Divergent network connectivity changes in behavioural variant frontotemporal dementia and Alzheimer's disease. Brain 2010;133:1352–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Warren JD, Rohrer JD, Schott JM, et al. Molecular nexopathies: a new paradigm of neurodegenerative disease. Trends Neurosci 2013;36:561–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Steinacker P, Hendrich C, Sperfeld AD, et al. TDP-43 in cerebrospinal fluid of patients with frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Arch Neurol 2008;65:1481–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kasai T, Tokuda T, Ishigami N, et al. Increased TDP-43 protein in cerebrospinal fluid of patients with amyotrophic lateral sclerosis. Acta Neuropathol 2009;117:55–62. [DOI] [PubMed] [Google Scholar]
- 80.Suarez-Calvet M, Dols-Icardo O, Llado A, et al. Plasma phosphorylated TDP-43 levels are elevated in patients with frontotemporal dementia carrying a C9orf72 repeat expansion or a GRN mutation. J Neurol Neurosurg Psychiatry 2014;85:684–91. [DOI] [PubMed] [Google Scholar]
- 81.Schofield EC, Halliday GM, Kwok J, et al. Low serum progranulin predicts the presence of mutations: a prospective study. J Alzheimers Dis 2010;22:981–4. [DOI] [PubMed] [Google Scholar]
- 82.Ghidoni R, Benussi L, Glionna M, et al. Low plasma progranulin levels predict progranulin mutations in frontotemporal lobar degeneration. Neurology 2008;71:1235–9. [DOI] [PubMed] [Google Scholar]
- 83.Sleegers K, Brouwers N, Van Damme P, et al. Serum biomarker for progranulin-associated frontotemporal lobar degeneration. Ann Neurol 2009;65:603–9. [DOI] [PubMed] [Google Scholar]
- 84.Scherling CS, Hall T, Berisha F, et al. Cerebrospinal fluid neurofilament concentration reflects disease severity in frontotemporal degeneration. Ann Neurol 2014;75:116–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Landqvist Waldo M, Frizell Santillo A, Passant U, et al. Cerebrospinal fluid neurofilament light chain protein levels in subtypes of frontotemporal dementia. BMC Neurol 2013;13:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Miller ZA, Rankin KP, Graff-Radford NR, et al. TDP-43 frontotemporal lobar degeneration and autoimmune disease. J Neurol Neurosurg Psychiatry 2013;84:956–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hales CM, Hu WT. From frontotemporal lobar degeneration pathology to frontotemporal lobar degeneration biomarkers. Int Rev Psychiatry 2013;25:210–20. [DOI] [PubMed] [Google Scholar]
- 88.McKeith IG, Dickson DW, Lowe J, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology 2005;65:1863–72. [DOI] [PubMed] [Google Scholar]
- 89.Lippa CF, Duda JE, Grossman M, et al. DLB and PDD boundary issues: diagnosis, treatment, molecular pathology, and biomarkers. Neurology 2007;68:812–19. [DOI] [PubMed] [Google Scholar]
- 90.Burton EJ, Karas G, Paling SM, et al. Patterns of cerebral atrophy in dementia with Lewy bodies using voxel-based morphometry. Neuroimage 2002;17:618–30. [PubMed] [Google Scholar]
- 91.Barber R, Ballard C, McKeith IG, et al. MRI volumetric study of dementia with Lewy bodies: a comparison with AD and vascular dementia. Neurology 2000;54:1304–9. [DOI] [PubMed] [Google Scholar]
- 92.McKeith I, O'Brien J, Walker Z, et al. Sensitivity and specificity of dopamine transporter imaging with 123I-FP-CIT SPECT in dementia with Lewy bodies: a phase III, multicentre study. Lancet Neurol 2007;6:305–13. [DOI] [PubMed] [Google Scholar]
- 93.Sinha N, Firbank M, O'Brien JT. Biomarkers in dementia with Lewy bodies: a review. Int J Geriatr Psychiatry 2012;27:443–53. [DOI] [PubMed] [Google Scholar]
- 94.Gilman S, Koeppe RA, Little R, et al. Differentiation of Alzheimer's disease from dementia with Lewy bodies utilizing positron emission tomography with [18F]fluorodeoxyglucose and neuropsychological testing. Exp Neurol 2005;191(Suppl 1):S95–103. [DOI] [PubMed] [Google Scholar]
- 95.Gomperts SN, Rentz DM, Moran E, et al. Imaging amyloid deposition in Lewy body diseases. Neurology 2008;71:903–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cepek L, Steinacker P, Mollenhauer B, et al. Follow-up investigations of tau protein and S-100B levels in cerebrospinal fluid of patients with Creutzfeldt-Jakob disease. Dement Geriatr Cogn Disord 2005;19:376–82. [DOI] [PubMed] [Google Scholar]
- 97.Mollenhauer B, Cullen V, Kahn I, et al. Direct quantification of CSF alpha-synuclein by ELISA and first cross-sectional study in patients with neurodegeneration. Exp Neurol 2008;213:315–25. [DOI] [PubMed] [Google Scholar]
- 98.Spies PE, Melis RJ, Sjogren MJ, et al. Cerebrospinal fluid alpha-synuclein does not discriminate between dementia disorders. J Alzheimers Dis 2009;16:363–9. [DOI] [PubMed] [Google Scholar]
- 99.Bibl M, Mollenhauer B, Lewczuk P, et al. Validation of amyloid-beta peptides in CSF diagnosis of neurodegenerative dementias. Mol Psychiatry 2007;12:671–80. [DOI] [PubMed] [Google Scholar]
- 100.Rascovsky K, Hodges JR, Knopman D, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 2011;134:2456–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.