

Abstract
In this paper, we present a dynamic programming algorithm that runs in polynomial time and allows us to achieve the optimal, non-overlapping segmentation of a long RNA sequence into segments (chunks). The secondary structure of each chunk is predicted independently, then combined with the structures predicted for the other chunks, to generate a complete secondary structure prediction that is thus a combination of local energy minima. The proposed approach not only is more efficient and accurate than other traditionally used methods that are based on global energy minimizations, but it also allows scientists to overcome computing and storage constraints when trying to predict the secondary structure of long RNA sequences.
1. Introduction
It is known that noncoding RNA sequences are important for many biological processes, where they play largely regulatory functions rather than being used to encode proteins. The secondary structures of these noncoding RNAs have been shown to be very important to their functions, much like the structure of a protein is important for its cellular function [1]. The secondary structure of RNA is defined as the set of hydrogen bonds that form between the bases of a linear RNA sequence. The most common forms of RNA found in cells and viruses are single stranded. Since single-stranded RNA is considered to be less stable than its double-stranded counterpart, it tends to fold back on itself to form local regions of double-stranded RNA, resulting in formation of secondary structure elements such as stem-loops and pseuoknots. Stem-loop structures form when nucleotides within local areas of complementarity base pair with one another, forming a double-stranded stem topped by a single-stranded loop. Pseudoknots form when sequences in the single-stranded loop base pair with complementary sequences either up- or down-stream of the stem-loop.
Several available computer prediction programs can predict the secondary structure of an RNA sequence if given its primary sequence. Some of these programs use thermodynamic methods to find the structure with the lowest possible free energy (global energy minimum). These prediction programs employ known energy values for short RNA sequences (determined empirically in wet laboratories as a function of the temperature required to completely denature a given RNA sequence) [2] and use a dynamic programming algorithm to build the secondary structure prediction. Although these programs are accurate for sequences a few hundred bases in length, their accuracies diminish for longer sequences due to a lack of experimental energy results for long RNA sequences. Therefore we sought to discover an alternative method that does not include these values in the prediction algorithm. In addition, many of the existing algorithms do not predict pseudoknot structures. If one considers arbitrary pseudoknots to a given sequence, the prediction problem becomes NP-hard [3].
In our previous work, we used an alternative approach for predicting secondary structures in which we divided long sequences into overlapping chunks, predicted the structure of each chunk, and rebuilt the whole secondary structure from these component parts. Using this method, our predictions remain supported by the empirical thermodynamic evidence yet allow us to operate within existing limitations in computing power and memory. However, this raised the fundamental questions of how and where to subdivide the long sequence into chunks. We previously used a brute force approach in which we conducted a search of a small sub-space of overlapping chunks with fixed sizes by considering all the possible segmentations of the sub-space [4]. Although successful in terms of its accuracy, the approach was not optimal and was extremely time demanding. The investigation described in the present paper expands upon our previous results [4] and presents an optimal method using dynamic programming to segment a long sequence into non-overlapping sub-segments. Not only does the method produce accurate secondary structure predictions, it also conducts the search more efficiently. Computer experiments show that the combination of the secondary structures of the individual chunks result in an overall sequence secondary structure that possesses higher sensitivity and selectivity than the secondary structure obtained with traditional programs that consider the sequence as a whole for their global energy minimizations. It is more efficient because, although the possible combinations of the segmentations is O(2N), which is intractable for any RNA sequence of a practical length N, we can use dynamic programming techniques to search this space in O(N2) time.
The contributions of this paper are as follows:
We present an algorithm that explores the space of all the possible, non-overlapping nucleotide chunks into which a long RNA sequence can be divided and finds the set of chunks that rebuilds into a long secondary structure by maximizing a given scoring function.
We use our algorithm to find the optimal way to segment the sequence into non-overlapping chunks that maximize the similarity of the predicted structure to the structure observed in the laboratory and show that these rebuilt structures have better accuracy when compared to using a single whole sequence prediction.
We use our algorithm to predict secondary structures that could not otherwise be predicted due to lack of computing resources by limiting the size of the chunks while using energy as a scoring function and show that these predictions are comparable in accuracy to those using a single whole sequence prediction.
The remainder of this paper is organized as follows: in Section 2, we provide general background information and review related work. In Section 3, we describe the dynamic programming algorithm we used to find the optimal segmentation of a sequence for prediction. In Section 4, we compare the accuracy of the secondary structure prediction based on global minimizations versus the accuracy of the optimal segmented predictions provided by our method in two different scenarios, i.e., when the chunk length (or window size) is unlimited and when it is limited by computer resources (e.g., memory or computing power).
2. Related Work
In MFE (minimum free energy) approaches [5, 6, 7, 8], the entire nucleotide sequence is folded in such a way that the structure with the lowest free energy is returned as the predicted secondary structure. Unfortunately, this structure is not always the most similar to the observed structure in nature [2]. Indeed, in nature a structure with higher free energy may be favored, for example when a given structure must exist in equilibrium with an unfolded form to provide for a biological function. During replication of viral RNA, a secondary structure element such as a pseudoknot may be required for recognition of an RNA template by the RNA-dependent polymerase enzyme that copies it, yet it must unwind so that this enzyme can copy it. In order to find a more optimal structure, we explore all possible combinations of non-overlapping chunks for a given long RNA sequence and apply the same algorithm to calculate the MFE for each chunk. We then rebuild the overall secondary structure prediction from a combination of local minima rather than from the single global minimum. Since some of the MFE algorithms may require times up to O(N6) to predict pseudoknots [8], it becomes worthwhile to split up the RNA sequence into smaller manageable chunks and predict their structures in parallel. In this paper, we show that using several non-overlapping chunks can increase the accuracy over a single whole sequence prediction.
The problem of finding the best sequence segmentation to use for a prediction is similar to some problems in natural language processing, i.e., problems with word recognition in recorded audio, finding paragraph breaks in a body of text, or any other optimal segmentation of non-overlapping information given a metric for scoring each segment. In the 1960s, Vintsyuk first proposed the use of dynamic [9] programming methods for time-aligning a pair of speech utterances. Although the essence of the concepts of dynamic time warping, as well as rudimentary versions of the algorithms for connect-word recognition, were embodied in Vintsyuk's work, it was largely unknown in the West and did not come to light until the early 1980s – long after more formal methods were proposed and implemented by others. In our work, we used a similar approach to segment the input string (nucleotide sequence) and maximize the similarity between predicted structures from sub-segments and the structure found in nature.
3. Methodology for Searching the Space of Local Predictions
3.1. Algorithm Overview
Dynamic programming is an optimization technique that can be used when the optimal solution of the overall problem is composed of optimal solutions to sub-problems. In our case, we want to find the optimal non-overlapping segmentation of a long primary nucleotide sequence into chunks. In this case, the optimal segmentation is one that will provide us with the greatest similarity to the associated observed secondary structure once the secondary structure of each chunk has been predicted and combined into an overall secondary structure prediction for the long sequence. An initial approach to solve this problem is to enumerate all the possible segmentations and search for the segmentation that maximizes the similarity with the observed secondary structure among the 2N−1 alternatives, where N is the number of nucleotides in the RNA sequence. Since the search space grows exponentially, this approach is intractable for any practical value of N even on supercomputers. As an alternative approach, we can use dynamic programming to search the segmentation space in polynomial time, where optimal solutions for each chunk are part of the optimal solution for the whole sequence.
For the dynamic programming approach presented in this paper, given a nucleotide sequence x of length N with N ≫ 1 and j >= i, we first build an N by N predicted matrix that is filled as follows:
| (1) |
The function predict() takes the chunk starting at nucleotide i and terminating at nucleotide j and returns the predicted secondary structure of the chunk using the program prediction code. Any code for prediction can be used and, since the predictions are independent of one another, they can be performed in parallel and stored in a database. However, not all the chunks can be predicted for a very long sequence, due to resource constraints, such as limits in memory size and computing power. Each prediction code has a different Max_C length, which is the longest chunk length predictable. For example, the Pknots-RE [8] code can predict the structure of sequences up to 200 nucleotides in length, while Pknots-RG [7] can predict the structure of sequences up to 800 nucleotides long. Thus the predicted matrix is an upper triangular matrix. Furthermore, as Max_C ≪ N, this is actually an upper triangular band matrix, with band width Max_C.
The rebuilding process uses the upper right triangular band matrix, selects non-overlapping chunks and their predicted secondary structures from within the matrix, and combines them to build a secondary structure prediction for a nucleotide sequence longer than can be predicted otherwise. The selection of the chunks can be an a-posteriori or an a-priori selection.
A-priori selections are based only on the minimum energies of the chunks and can be used for blind predictions. In an a-priori selection using the prediction matrix, we build a score matrix and initialize its first row as follows:
| (2) |
where min_energy() is the lowest predicted energy of the chunk starting at nucleotide 1 and terminating at nucleotide j. Here we assume that j ≤ Max_C ≪ N. Using a recurrence relation, we complete the upper triangle of the score matrix as follows:
| (3) |
For each cell of the score matrix, score(i, j), we also store a pointer to the cell that gave the best score among the matrix cells score(k, i − 1)∀k with 1 ≤ k < i − 1.
A-posteriori selections use experimentally obtained secondary structures as references for the scoring and can be used for validation purposes only. In an a-posteriori selection using the prediction matrix, we build a score matrix and initialize its first row as follows:
| (4) |
where compare() compares the predicted secondary structure of the sub-segment starting at nucleotide 1 and terminating at nucleotide j to the corresponding experimentally observed secondary structure, observed(1,j). The comparison can be based on different criteria, e.g., sensitivity and selectivity. Using a recurrence relation, we complete the upper triangle of the score matrix, assuming that j ≤ Max_C ≪ N as follows:
| (5) |
Again, as for the a-priori approach described above, for each cell of the score matrix we store a pointer to the cell that gave the best score among the matrix cells score(k, i − 1)∀k with 1 ≤ k <= i − 1.
Once the score matrix is completed, we select the best value along the Nth column that corresponds to the minimum energy in an a-priori approach or to the maximum similarity score of the input sequence x in an a-posteriori approach:
| (6) |
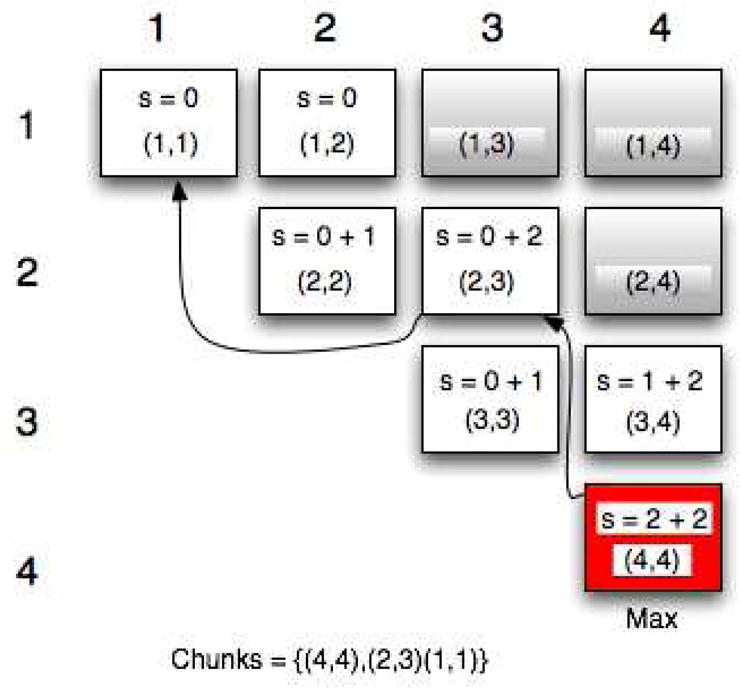
At this point we backtrack through the pointers and retrieve the segments that give the optimal similarity (best score). Figure 1 shows a mock example of a score matrix and the backtrack used to identify the optimal segmentation for a sequence of 4 nucleotides (N=4), with a code with maximum predictable length equal to 2 (Max_C=2). The values use are for demonstration purposes only and do not represent a real prediction scenario.
Figure 1. Example of score matrix with backtrack to rebuild the optimal segmentation with N = 4 and Max_C = 2.
3.2. Algorithm Complexity
The search for this optimal non-overlapping segmentation can be performed in time O(N2), where N is the length of the sequence. Both the prediction and score matrices have sizes of O(N2); thus completing them takes time O(N2). Furthermore, it is important to note that, although every cell depends on the maximum score of the i − 1st column, the maximum score for every row is the same. This is because row k represents all segments starting at nucleotide k and thus we need only to compute the maximum score of the k − 1 column, representing all the segments that end at nucleotide position k − 1. Once the maximal score for column k − 1 is computed, the value can be copied to every other cell in row k, keeping the running time at O(N2).
4. Evaluation Results
As part of the evaluation, we seek to understand whether predictions of a sequence set that consider each sequence as a whole is more or less accurate than predictions that consider each sequence as multiple, non-overlapping chunks. We also seek to understand whether the folding process favors local minimum energies rather than global minimum energies. This is relevant when the prediction of structures formed by very long sequences is not feasible. Therefore, when the global energy cannot be determined experimentally, the search for global energy must be replaced by the search across local minimum energies of shorter chunks.
4.1. Prediction Program, Scoring Metrics, and Data Set
Given the definition of our dynamic programming algorithm, any prediction code and score metric for RNA secondary structure prediction can be used. We choose to use a popular prediction tool such as Pknots-RG [7] because of its excellent performance in predicting the structure of sequences of up to 800 nucleotides and its ability to predict pseudoknot structures. We use this code to address questions of accuracy and sensitivity of our approach. Given both a predicted structure and an observed structure in parenthetical format, we measure the accuracy of the predicted structure in terms of sensitivity (i.e., ability to predict all true pairs) and selectivity (i.e., ability to only predict true pairs). Secondary structures of long RNA sequences, i.e., of the order of thousands of nucleotides, that have been experimentally validated are rare. Thus, for our analysis in this paper we used the longest nucleotide sequences from Group A in CONTRAfold [10], which have lengths ranging from 200 to 482 nucleotides.
4.2. Single-Segment Predictions vs. Predictions Using Chunks
To address whether predictions of a sequence set that consider each sequence as a whole are more or less accurate than predictions that consider each sequence as multiple, non-overlapping chunks, we select chunk sizes for our data set that are not limited by the prediction program or by computing resources. Therefore, our chunk sizes or Max_C range from 1 to N, where N is the length of each sequence in terms of nucleotides. We also select sequences whose secondary structures have been experimentally-determined and are thus available for building the score matrix in Equations 4 and 5, based on an a-posteriori approach. Finally, we use the average value of sensitivity and selectivity of predicted secondary structures versus experimental secondary structures as our metrics.
We first predict the secondary structure of each entire sequence using Pknots-RG. We then use our method and the same prediction code to identify the set of non-overlapping chunks with highest sensitivity and selectivity, as described in Section 3.1, Equations 4 and 5. The comparison of the two techniques, i.e., prediction based on the entire sequence and prediction based on non-overlapping chunks, can result in two possible outcomes. One possible outcome is that our approach based on chunks always converges towards the predictions based on the whole sequence. Alternatively, the best solutions are indeed combinations of chunks and in that case, the prediction can have higher, equal, or lower scores. Note that we do not assume any resource constraints and so we can always predict any chunk of any length. Also the scores are based on an a-posteriori approach and thus we are not driven by energy values in our selections. For each sequence, Table 1 presents the sequence name and length; the sensitivity and selectivity of the prediction considering the sequences as a single sequence (a single chunk); the sensitivity, selectivity, the number of optimal non-overlapping chunks used to rebuild the secondary structure, and the maximum chunk length (or maximum window size) used with our method.
Table 1. Comparison of sensitivity and selectivity for secondary structures predicted considering the sequence of nucleotides as a whole and as a set of non-overlapping chunks using an a-posteriori approach.
| Single Sequence | Multiple Chunks-unlim. window | |||||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Name | length(nt) | sen. | sel. | #segm. | sen. | sel. | #segm. | max window |
| RF00458_A | 202 | 0.39 | 0.58 | 1 | 0.64 | 0.78 | 4 | 146 |
| RF00193_A | 273 | 0.60 | 0.79 | 1 | 0.96 | 0.99 | 17 | 49 |
| RF00231_A | 275 | 0.41 | 0.71 | 1 | 0.71 | 0.97 | 9 | 68 |
| RF00503_A | 293 | 0.44 | 0.70 | 1 | 0.92 | 0.95 | 16 | 40 |
| RF00030_A | 297 | 0.48 | 0.68 | 1 | 0.59 | 0.67 | 13 | 59 |
| RF00216_A | 302 | 0.21 | 0.40 | 1 | 0.46 | 0.60 | 5 | 154 |
| RF00010_A | 312 | 0.63 | 0.77 | 1 | 0.63 | 0.77 | 3 | 306 |
| RF00009_A | 320 | 0.22 | 0.57 | 1 | 0.43 | 0.75 | 9 | 112 |
| RF00100_A | 330 | 0.23 | 0.40 | 1 | 0.70 | 0.81 | 8 | 107 |
| RF00036_A | 337 | 0.86 | 0.94 | 1 | 0.86 | 0.94 | 3 | 335 |
| RF00209_A | 379 | 0.46 | 0.75 | 1 | 0.76 | 0.90 | 10 | 218 |
| RF00024_A | 451 | 0.48 | 0.80 | 1 | 0.54 | 0.86 | 3 | 427 |
| RF00210_A | 462 | 0.56 | 0.80 | 1 | 0.74 | 0.91 | 7 | 295 |
| RF00177_A | 482 | 0.74 | 0.93 | 1 | 0.74 | 0.93 | 3 | 480 |
With unlimited chunk sizes and no resource limits, the chunk sets range from 3 to 17 sub-segments and their sizes are always smaller that the total sequence length. Only in 4 cases out of the 14 sequences considered (i.e, RF00010_A, RF00036_A, RF00024_A, and RF00177_A), did a single chunk cover the majority of the sequence. For these four cases Max_C = N. Our approach for these cases converges toward the whole-sequence prediction. In all the other cases, we observe equal or better sensitivity and selectivity when rebuilding the secondary structures from shorter non-overlapping sequence chunks. The better predictions can be either due to insufficiently accurate thermodynamic models for longer sequences (since the wet laboratory experiments are still missing or, in some cases, not feasible), or to the tendency for structures when folding to favor multiple localized minimum free energy structures rather than the global minimum free energy structure of the whole sequence, or it can be a combination of both.
4.3. Dealing with Resource Limits
To address the question of whether structures, when folding, tend to favor multiple localized minimum free energy structures rather than the global free energy structure, we explore all possible values for each sequence in our data set, of Max_C from 1 to N in Equations 2 and 3. This results in N score matrices, each exploring window sizes only up to the Max_C associated with the matrix. Figure 2 shows a simple example for a sequence with 4 nucleotides and four score matrices obtainable with this sequence (The scores are mock examples). For each score matrix we rebuild its lowest energy secondary structure given the limitation of the window size. Note that this time our scoring approach is an a-priori approach based on energy values only and not a comparison to the experimentally known structures as in the previous section.
Figure 2.
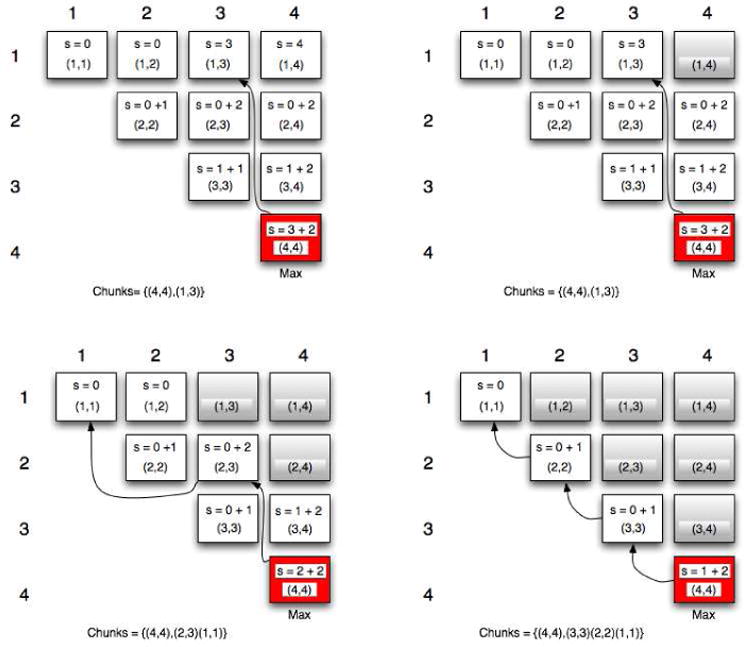
Examples of 4 score matrixes obtainable with a sequence of 4 nucleotides.
In Figure 3, we present two case studies from our data set to show how sensitivity and selectivity grow as a function of the window size. In both examples, we see that the whole sequence is not necessary to predict the best secondary structure. For Sequence RF00024_A with length 451 nucleotides and Sequence RF00210_A with length 462, with windows of 198 and 375 nucleotides respectively, we can already capture the best structures for the overall sequence. No further accuracy is gained by using longer chunks.
Figure 3.

Sensitivity and selectivity as a function of the chunk length for two sequences in our data set.
In Table 2, we compare sensitivity and selectivity of the prediction for all the 14 sequences in our data set using a global energy minimization (by feeding the whole sequence into the prediction code) with the sensitivity and selectivity of the prediction built from the best set of chunks obtained with our method. For the latter prediction, the table presents the number of chunks and the length of the longest chunk (Max_C). Using chunks that are limited in size (i.e., within the limitations of the computer resources) allows us to overcome computing and memory constraints. At the same time, we observe that for all 14 sequences we examined, the combination of non-overlapping chunks can predict secondary structures with either equal or better sensitivity and selectivity than those determined using the entire sequence. For only two sequences out of the 14 (RF00177_A and RF00036_A), the value of Max_C is almost as long as N, indicating the convergence of our method to the search for the global energy minimum.
Table 2. Comparison of sensitivity and selectivity for secondary structures predicted considering the sequence of nucleotides as a whole and as a set of non-overlapping chunks selected using an a-priori approach.
| Single Sequence | Multiple Chunks-lim. window | |||||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Name | length(nt) | sen. | sel. | #segm. | sen. | sel. | #segm. | max window |
| RF00458_A | 202 | 0.58 | 0.39 | 1 | 0.71 | 0.53 | 17 | 71 |
| RF00231_A | 275 | 0.70 | 0.41 | 1 | 0.96 | 0.66 | 19 | 73 |
| RF00193_A | 273 | 0.79 | 0.65 | 1 | 0.82 | 0.65 | 46 | 33 |
| RF00503_A | 293 | 0.70 | 0.44 | 1 | 0.94 | 0.77 | 49 | 43 |
| RF00030_A | 297 | 0.67 | 0.48 | 1 | 0.67 | 0.48 | 9 | 228 |
| RF00216_A | 302 | 0.39 | 0.20 | 1 | 0.60 | 0.41 | 37 | 49 |
| RF00010_A | 312 | 0.76 | 0.63 | 1 | 0.76 | 0.63 | 6 | 307 |
| RF00009_A | 320 | 0.56 | 0.21 | 1 | 0.68 | 0.30 | 55 | 23 |
| RF00100_A | 330 | 0.40 | 0.22 | 1 | 0.70 | 0.48 | 20 | 107 |
| RF00036_A | 337 | 0.93 | 0.85 | 1 | 0.93 | 0.85 | 3 | 335 |
| RF00209_A | 379 | 0.74 | 0.45 | 1 | 0.79 | 0.50 | 15 | 218 |
| RF00024_A | 451 | 0.79 | 0.48 | 1 | 0.85 | 0.52 | 30 | 198 |
| RF00210_A | 462 | 0.79 | 0.56 | 1 | 0.80 | 0.57 | 18 | 375 |
| RF00177_A | 482 | 0.92 | 0.74 | 1 | 0.92 | 0.74 | 3 | 480 |
These preliminary results produced by our approach clearly indicate the need for more accurate energy computations in existing MFE methods. In addition, our a-posteriori approach can be used as training data for intelligent prediction tools based on machine learning techniques such as Hidden Markov Models (HMM) and neural networks.
5. Conclusions and Future Work
In this paper, we show that it is possible to find an optimal way to segment a long sequence of nucleotides using a polynomial time dynamic programming algorithm and to rebuild accurate secondary structures from the collection of non-overlapping chunks given a scoring function that can be based on energy only. The results show that our approach can outperform MFE methods using dynamic programming to search for global energy minima 12 times out of 14 with the longest sequences in Group A in CONTRAfold. This suggests the need for more accurate energy computations in existing MFE methods for long nucleotide sequences.
Current work of the authors includes the design and training of HMM and neural network based tools to identify optimal segmentations when the experimental secondary structure is not available and the memory and computing resources are limited.
Acknowledgments
This research is supported in part by the Texas Higher Education Coordinating Board NHARP grant 003661-0013-2007, NSF grant DMS0800272, and NIH grants 5S06GM008012-39 and 2G12RR008124.
Contributor Information
Abel Licon, Email: licon@udel.edu.
Michela Taufer, Email: taufer@udel.edu.
Ming-Ying Leung, Email: leung@utep.edu.
Kyle L. Johnson, Email: kljohnson@utep.edu.
References
- 1.Meyer I. A Practical Guide to the Art of RNA Gene Prediction. Briefings in bioinformatics. 2007;8, no. 6:396. doi: 10.1093/bib/bbm011. [DOI] [PubMed] [Google Scholar]
- 2.Walter A, Turner D, Kim J, Lyttle M, Muller P, Mathews D, Zuker M. Coaxial Stacking of Helixes Enhances binding of Oligoribonucleotides and Improves Predictions of RNA folding. Proceedings of the National Academy of Sciences. 1994;91, no. 20:9218. doi: 10.1073/pnas.91.20.9218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lyngsø R, Pedersen C. RNA Pseudoknot Prediction in Energy-Based Models. Journal of Computational Biology. 2000;7, no. 3-4:409–427. doi: 10.1089/106652700750050862. [DOI] [PubMed] [Google Scholar]
- 4.Taufer M, Solorio T, Licon A, Mireles D, Leung M. On the Effectiveness of Rebuilding RNA Secondary Structures from Sequence Chunks. Proceedings of 7th IEEE Intl Workshop on High Performance Computational Biology (HiCOMB); 2008; pp. 1–8. [Google Scholar]
- 5.Dirks R, Pierce N. An Algorithm for Computing Nucleic Acid Base-Pairing Probabilities Including Pseudoknots. Journal of computational Chemistry. 2004;25, no. 10:1295–1304. doi: 10.1002/jcc.20057. [DOI] [PubMed] [Google Scholar]
- 6.Zuker M. Mfold Web Server for Nucleic Acid Folding and Hybridization Prediction. Nucleic acids research. 2003;31, no. 13:3406. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reeder J, Steffen P, Giegerich R. PknotsRG: RNA Pseudoknot Folding Including Near-Optimal Structures and Sliding Windows. Nucleic acids research. 2007 doi: 10.1093/nar/gkm258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rivas E, Eddy SR. A Dynamic Programming Algorithm for RNA Structure Prediction Including Pseudoknots. Journal of Molecular Biology. 1999 Feb;285, no. 5:2053–2068. doi: 10.1006/jmbi.1998.2436. [DOI] [PubMed] [Google Scholar]
- 9.Vintsyuk T. Speech Discrimination by Dynamic Programming. Cybernetics and Systems Analysis. 1968;4, no. 1:52–57. [Google Scholar]
- 10.Do C, Woods D, Batzoglou S. CONTRAfold: RNA Secondary Structure Prediction without Physics-Based Models. Bioinformatics. 2006;22, no. 14:e90. doi: 10.1093/bioinformatics/btl246. [DOI] [PubMed] [Google Scholar]