

Abstract
Dynamic reprogramming of the genome takes place during the gamete-to-embryo transition. This transition defines a period of continuous and global change but has been difficult to study because of extremely limited material and varying degrees of chromatin compaction. Improved methods of detecting chromatin and gene expression changes in the germ line and in the preimplantation embryo would greatly enhance the understanding of this crucial developmental transition. Here we describe a protocol developed and used by us that improves the sensitivity of existing fluorescence in situ hybridization (FISH) methods; the protocol described here has enabled us to visualize single-copy DNA targets and corresponding nascent RNA transcripts in preimplantation embryos and during spermatogenesis. Major improvements over alternative methods involve fixation and permeabilization steps. Chromatin epitopes can be visualized simultaneously by combining FISH with immunofluorescence; multicopy and repetitive element expression can also be reliably measured. This procedure (sample preparation and staining) requires 1–1.5 d to complete and will facilitate detailed examination of spatial relationships between chromatin epitopes, DNA and RNA during the dynamic transition from gamete to embryo.
Introduction
It has been proposed that the chromatin state of the male gamete can influence gene expression in early mouse development1,2. For example, imprinted X-chromosome inactivation (XCI) has been proposed to originate in the meiotic silencing of sex chromosomes3–7. In the male germ line, the haploid genome is continuously remodeled during spermiogenesis and, during the final stages of spermiogenesis, undergoes a dramatic genome-wide transformation in which core histones are replaced by protamines to enable compaction of sperm chromatin. The possibility of transgenerational carryover is supported by recent evidence, obtained by chromatin immunoprecipitation of mature spermatozoa, indicating that nucleosomes at imprinted and developmentally regulated genes are retained in the mature gamete8–10. Although clearly dynamic, much remains unknown about the manner in which chromatin is organized in the developing gamete and how it changes in the zygote. Development of sensitive cytological techniques to examine chromatin dynamics during the gamete-to-embryo transition would complement existing biochemical methods such as ChIP-seq8–10 and significantly enhance the understanding of spatial and temporal changes in chromatin structure. Although cytological methods such as RNA and DNA fluorescence in situ hybridization (FISH) and immunofluorescence are now routinely used in cell culture studies11–14, they have been applied, to a lesser extent, in the study of germ cells and early mouse embryos because of challenges presented by the following: extremely limited samples, high cytoplasm-to-nucleus ratios in preimplantation embryos and highly compacted chromatin in mature male germ cells.
We recently developed sensitive protocols to examine gene expression and chromatin states in the developing male gamete15 and early mouse embryo16. These protocols have enabled us to successfully carry out RNA and DNA FISH of single-copy targets in the two-cell embryo—a historically difficult developmental stage at which to perform cytological analysis. Indeed, the protocols can be applied to the detection of nascent RNA, single-copy DNA and protein localization in the nuclei of two-cell, four-cell and eight-cell embryos, as well as in blastocysts and male germ cells15,16. Using these protocols, we have found that the X chromosome is continuously remodeled during spermatogenesis, and that silencing of sex chromosome initiated by meiotic sex chromosome inactivation (MSCI) is maintained through the postmeiotic period15. In early mouse embryo, these methods have enabled us to conclude that the paternal X chromosome (XP) can be divided into two distinct chromatin domains, one comprising traditional coding genes (genic) and the other comprising intergenic repetitive elements, and that imprinted XCI occurs in two steps, with repeat silencing preceding genic inactivation16. We have therefore proposed that the imprint may be transmitted across generations by repetitive elements whose chromatin state is determined during male meiosis. Here we detail the technique used in these studies and discuss crucial steps in the protocols.
Optimization of protocol for male germ cells
Methods for immunostaining and FISH generally require, in order, the following steps: a permeabilization step to enable passage of antibodies or probes through cells; a fixation step, in which cellular material is fixed and preserved; a detection step, in which antibodies or nucleic acid probes are applied; and a final step, in which samples are washed and mounted for visualization by microscopy6,12–15,17–21. Although existing protocols share these general features, they can vary significantly in a number of crucial parameters. Prevailing methods for immunostaining cells in the male germ line call for hypotonic treatment of cells before fixation and application of probe and antibody22,23. These methods have worked well for some applications in which chromatin is condensed—in particular, for analysis of chromosome synapsis and recombination with axial markers, such as SCP3. The hypotonic treatment step swells nuclei and physically extends subnuclear structures, often facilitating access of antibodies to compacted chromatin. Such treatment is well suited for spreading out meiotic chromosomes so that axial elements can be captured in a single z-plane. Until recently, such protocols had not been applied extensively to germ cell analysis with FISH probes. When optimizing conditions for RNA and DNA FISH, we found that a significant downside to hypotonic swelling is a subtle but critical loss of three-dimensional (3D) chromatin architecture, which precluded visualization of finer subnuclear details. This point is exemplified by Cot-1 RNA FISH6,24 of postmeiotic sex chromatin (PMSC), a physically unique and epigenetically silent structure recently identified in mouse and marsupial male germ lines15,17,25,26.
Cot-1 RNA FISH is performed with probes derived from the Cot-1 genomic fraction—the fraction of the mammalian genome containing the most highly repetitive elements, such as long interspersed nuclear elements (LINEs), short interspersed nuclear elements (SINEs), long terminal repeats (LTRs) and other transposable elements—and provides a global view of transcriptional activity within a subnuclear region. When older slide preparation methods involving hypotonic treatment are used15,26,27, PMSC is found to be less distinct; chromatin structure is globally disrupted, dispersing Cot-1 signals (Fig. 1a,b)15. By contrast, the optimized RNA FISH protocol described in detail below yields Cot-1 RNA signals that are excluded from 4,6-diamidino-2-phenylindole (DAPI)-intense heterochromatic regions, including the PMSC structures associated with X- and Y chromosomes (Fig. 1c, d)15. Similarly, hypotonic treatment is detrimental to the detection of chromatin epitopes by immunostaining because of the physical distortion of chromatin structure, which results in lower signal-to-noise ratios15. Thus, for both RNA FISH and immunofluorescence to analyze chromatin structure, we recommend avoiding hypotonic treatment.
Figure 1.

Hypotonic treatment compromises nuclear architecture. (a) DAPI images of representative pictures of round spermatids after hypotonic treatment as described22. PMSC structure (arrow) is disrupted and not easily appreciated. (b) Cot-1 RNA FISH of round spermatids after hypotonic treatment. Note: Cot-1 RNA does not appear to be excluded from heterochromatic regions. (c) DAPI images of representative pictures of round spermatids fixed onto glass slides without hypotonic treatment using our described protocol. PMSC is clearly detected. Note that bright chromocenters (asterisks) and appended PMSC structures (arrows) are clearly visible by DAPI staining alone. (d) Cot-1 RNA FISH in the round spermatid using our protocol without hypotonic treatment. Cot-1 RNA (red signals) is depleted from PMSC. Arrows, PMSC.
The permeabilization and fixation steps are performed directly on seminiferous tubules and are followed by the mechanical dissociation of germ cells with forceps before Cytospinning onto slides. Our method can be applied to cells at any stage of spermiogenesis, including elongating and elongated spermatids, as well as round spermatids. For DNA FISH, especially on mature spermatozoa, we use an additional treatment with dithiothreitol (DTT) before fixation15. DTT disrupts sulfate bonds in protamines, which in theory helps to decondense the highly compacted sperm chromatin for probe annealing.
Our method yields preparations that retain the 3D nature of nuclei. By contrast, preparations including a hypotonic treatment step often yield more flattened nuclei. Thus, information regarding the 3D organization of subnuclear structures is often lost in such preparations, although the flattened and extended chromatin facilitates visualization of metaphase chromosomes and meiotic axial elements on a single plane. Traditional methods including a hypotonic treatment step may therefore be superior for viewing chromatin fibers and proteinaceous axial elements for 2D image acquisition; our protocol emphasizes preservation of RNA and 3D nuclear structures, and may therefore require image postprocessing that would include deconvolution to subtract out-of-focus light, z-sectioning and 2D or 3D reconstruction.
Optimization of protocol for preimplantation embryos
Similar considerations apply to preimplantation embryos. As is the case for experiments in male germ cells, previous methods for immunostaining and FISH of the early mouse embryo vary with respect to timing and order of each step (permeabilization, fixation and mounting of samples), as well as with respect to types and quantity of reagents used. Additional challenges include the limited availability of embryonic materials and the significant amount of cytoplasm, which makes probe penetration more difficult and results in higher background in the early embryo. DNA FISH has been especially problematic in this situation but is frequently necessary to verify that signals detected by RNA FISH are bona fide nascent transcripts from their corresponding genes16. As genic targets are typically single copy, the presence of any amount of background hybridization is often unacceptable. To overcome these problems and to enhance the signal-to-noise ratio, we have tested several published protocols6,19,28–30 and optimized crucial parameters such as order, concentration and duration of specific permeabilization and fixation steps.
Our efforts have led us to conclude that removal of cytoplasmic background is among the most critical factors for RNA and DNA FISH of preimplantation embryos, and a key determinant in reducing background is the order in which samples are fixed and permeabilized. Many methods involve fixing samples before permeabilizing18,19,31, typically resulting in higher cytoplasmic background. We have discovered that permeabilization before fixation or at the same time yields the best signal-to-noise ratios (Fig. 2). In our standardized protocol for the preimplantation embryo described below, we permeabilize with detergent and treat with fixative simultaneously; this approach has the added benefit of reducing the number of manipulations required—a significant consideration for extremely limited samples that may become desiccated with each medium change and/or become dislodged from the slide platform with every manipulation. This is an important improvement over our previous method6, which permeabilizes before fixing and therefore incurs additional mouth-pipetting steps by which embryos can be lost because of a more transparent appearance after the permeabilization step.
Figure 2.
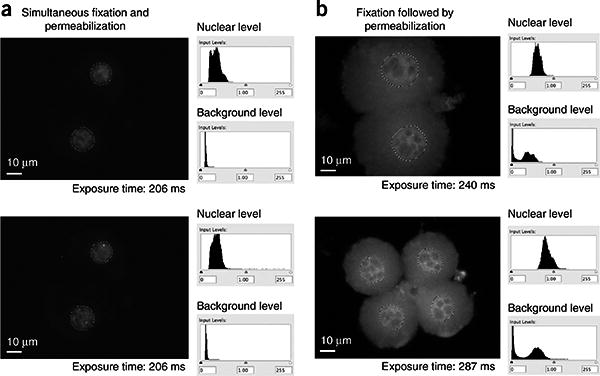
Order of fixation and permeabilization affects FISH analysis of preimplantation embryos. Shown are raw data from Cot-1 RNA FISH with indicated exposure times (without any contrast adjustments or deconvolution). Intensities of Cot-1 signals within the nucleus are quantified and compared with cytoplasmic background using ImageJ software. The nucleus is delimited by dotted lines. (a) Examples of images prepared using the protocol presented here. Note that there is very little cytoplasmic background and quantitation indicates a high signal-to-noise ratio. Two different z-sections of the same embryo are shown. (b) Examples of cells prepared using a protocol in which fixation precedes permeabilization19. The cytoplasmic background is relatively high.
Additional parameters critical for successful simultaneous permeabilization and fixation include the following:
Choice of detergent: Tergitol at a 0.05% (vol/vol) concentration works best, in our hands, when extraction of cytoplasmic components and fixation are to be performed simultaneously. However, Triton X-100 also works, and it can be substituted for Tergitol.
Choice of fixative: Methods using methanol/acetic acid fixation, especially in combination with hypotonic treatment, severely disrupt nuclear architecture and are not compatible with many RNA FISH experiments, especially if nascent Cot-1 RNA is to be visualized. Paraformaldehyde is our preferred fixative for both germ cells and preimplantation embryos.
Concentration of fixative: Best results are achieved using a 1.0% (wt/vol) paraformaldehyde formulation when simultaneous permeabilization with 0.05% (vol/vol) Tergitol is desired. This paraformaldehyde concentration differs from higher concentrations (e.g., 4.0%) typically used for cell culture preparations. In our hands, for preimplantation embryos, the 4.0% concentration impairs simultaneous permeabilization and inhibits probe and antibody penetration.
Our protocol can also be applied to embryos of the peri-implantation stage, specifically to examine the trophectodermal and trophoblastic lineages16. However, additional modifications may be necessary to visualize events in the inner cell mass because their protected location within the blastocyst makes probe penetration relatively challenging.
Experimental design
Probe consideration
DNA probes, labeled by random priming or nick translation, are used to perform RNA and DNA FISH. Random priming is used preferably for the Cot-1 probe (e.g., with the Prime-It Fluor kit (Stratagene) and Cy3-dUTP (GE Healthcare))6,15(Box 1). We use nick translation (e.g., Roche kit) for the preparation of all other DNA probes unless otherwise specified (see Box 2). Stratagene's Fluoro-12-dUTP seems to have the best signal-to-noise ratio among the green dyes tested. Cy3-dUTP (GE Healthcare) and Cy5-dUTP (GE Healthcare) provide good second and third colors for multiprobe combinations. For both random priming and nick translation, purification of labeled DNA and removal of unincorporated nucleotides (e.g., by the Illustra MicroSpin G-50 Columns) are helpful to reduce background signal for RNA and DNA FISH experiments.
Box 1. Probe Preparation By Random Priming (Cot-1 Probe) ● TIMING ∼2.5 H.
▲ CRITICAL STEP Random priming is used only for Cot-1 probe preparation. We use nick translation for preparation of all other DNA probes unless otherwise specified.
- For Cot-1 probe, add components tabulated below to a PCR tube. Mix and incubate for 5 min at 95 °C in a thermocycler.
Component Amount per reaction (μl) Final Template DNA (Cot-1 DNA: 1 mg ml−1) 1 1 μg Random-9mer primer (Prime-it Fluor kit) 10 Sterile water 27 Centrifuge briefly and place on ice.
- Add components tabulated below. Mix and incubate for 30 min at 37 °C in a thermocycler.
Component Amount per reaction (μl) Final concentration Mixture prepared from Step 2 38 5× nucleotide buffer (Prime-It Fluor kit) 9.2 Cy3-dUTP (1 mM) 0.8 16 μM Klenow (Prime-It Fluor kit) 2 Stop reaction by adding 2 μl of stop buffer (part of Prime-It Fluor kit).
Purify the stopped reaction by Illustra MicroSpin G-50 Columns. Before use, spin columns for 1 min at 900g using a microcentrifuge. Load 50 μl of reaction buffer. Spin for 2 min at 900g using a microcentrifuge). Collect the purified fraction in a 1.5-ml microcentrifuge tube.
Add 5 μl of herring sperm DNA (50 μg; 50-fold excess of Cot-1 DNA) and mix well.
Measure the total volume by pipette. Add 0.7× volume of 100% ethanol and 0.1× volume of 3 M sodium acetate (pH 5.2), mix well and centrifuge for 15 min at maximum speed (17,000g) at 4 °C.
Discard the liquid and add 200 μl of 70% (vol/vol) ethanol; mix well and centrifuge for 5 min at maximum speed at 4 °C.
Discard the liquid and dry the pellet for 10–30 min in room temperature.
Dissolve the precipitate in 20 μl of hybridization buffer. The precipitate is hard to dissolve. Pipette up and down thoroughly and use vortex mixer.
▪ PAUSE POINT Probes can be stored at −20 °C until use and are stable for more than 1 year.
Box 2. Probe Preparation By Nick Translation (Gene-Specific Probes For Rna and Dna Fish) ● TIMING ∼4 H.
- For probes, add components tabulated below to a PCR tube. Mix and incubate for 2 h at 15 °C in a thermocycler.
Component Amount per reaction (μl) Final Template DNA × 2.5 μg dATP (0.4 mM: Nick Translation kit) 7.4 60 μM dCTP (0.4 mM: Nick Translation kit) 7.4 60 μM dGTP (0.4 mM: Nick Translation kit) 7.4 60 μM labeled-dUTP (1 mM: see ▲ CRITICAL STEP below) 3 60 μM 10× Buffer (Nick Translation kit) 5 1× Enzyme mix (Nick Translation kit) 5 Sterile water added to 50 Stop reaction by heating to 65 °C for 10 min.
Purify the reaction using Illustra MicroSpin G-50 Columns. Before use, spin columns for 1 min at 900g with a microcentrifuge. Load 50 μl of reaction buffer. Spin for 2 min at 900g with a microcentrifuge. Collect the purified fraction in a 1.5-ml tube.
Add 12.5 μl of herring sperm DNA (125 μg; 50-fold excess of template DNA) and mix well.
Measure the total volume by pipette. Add 0.7× volume of 100% ethanol and 0.1× volume of 3 M sodium acetate (pH 5.2), mix well and centrifuge for 15 min at maximum speed (17,000g) at 4 °C.
Discard the liquid and add 200 μl of 70% (vol/vol) ethanol; mix well and centrifuge for 5 min at maximum speed at 4 °C.
Discard the liquid and dry the pellet for 10–30 min at room temperature.
Dissolve the precipitate in 50 μl of hybridization buffer. The precipitate is hard to dissolve. Pipette up and down thoroughly and use a vortex mixer.
▪ PAUSE POINT Probes can be stored at −20 °C until use and are stable for more than 1 year.
To detect nascent transcripts for specific genes by RNA FISH, we generally use genomic bacterial artificial chromosome (BAC) clones that cover ~150 to 200 kb, spanning the genes of interest as probe templates. BAC clones can be labeled without purifying inserts from vector DNA. However, a single BAC clone often contains multiple genes. Thus, BAC clones must be selected carefully to ensure that they include only the gene of interest. If such a BAC clone is not available, we recommend using smaller plas-mids that encompass all or only parts of the genic locus. We have successfully detected nascent transcripts using plasmids of 6 kb; for highly transcribed genes, smaller probes are possible (e.g., Tsix, Xist). For gene-specific DNA FISH, large BAC probes are ideal and can be visualized at single-copy targets with high efficiency using our protocol. Probes of only 2-kb length have also worked, but they offer much lower signal intensities. For both RNA and DNA FISH, when genomic probes containing repetitive elements are used, a preannealing step using unlabeled Cot-1 DNA is required to mask hybridization to repeats, enabling the specific detection of single-copy sequences.
Although we generally use directly labeled probes as described in this protocol, signal enhancement methods can be applied. Note that the biotin-avidin system cannot be used because endogenous biotin in preimplantation embryos would cause high background.
Confirmation of slide quality
To assess the quality of prepared slides, we highly recommend performing Cot-1 RNA FISH first. This is a crucial checkpoint to determine preservation of RNA structure and localization. As protein and DNA are generally more stable than RNA, confirmation of RNA targets implies preservation of chromatin epitopes and DNA as well. The Cot-1 probe is derived from the most highly repetitive sequences in the genome and is enriched for LINEs, SINEs, LTRs and other repeats. As Cot-1 RNA is abundant and expressed in all cell types, its localization generates a specific pan-nuclear stain and can be used in any sample to determine whether RNA is properly preserved on the slide. In a well-prepared sample, the Cot-1 signal should be visible on transcriptionally active euchromatic regions and excluded from silent heterochromatin and the nucleolus. As described in Box 1, Cot-1 probes can be prepared in large batches, stored in a −20 °C freezer for more than 1 year and used routinely to confirm the quality of newly prepared slides for general RNA, DNA and chromatin epitope staining.
Applications of the method and temporal considerations
Because of low background hybridization, these protocols can be applied to various analyses to detect nascent RNA, single-copy DNA, whole chromosomes and protein localization in germ cells and preimplantation embryos. Aside from improved signal- to-noise ratios in general, another notable advancement is the possibility of querying nucleic acids and proteins simultaneously with high sensitivity in these cell types. Some specific examples of what could be performed are (i) Cot-1 RNA FISH to examine global activity and RNA localization of repetitive elements in the Cot-1 fraction of the genome; (ii) RNA FISH of genic elements to examine nascent transcripts arising from traditional coding genes (e.g., Atrx, Hprt); (iii) DNA FISH using chromosome- specific paints to determine localization of specific chromosomes in the nucleus (e.g., Chromosome 1, X or Y); (iv) DNA FISH with single-copy probe to pinpoint the localization of a specific locus in the nucleus (e.g., X-inactivation center); and (v) immunofluo-rescence using antibodies against a chromatin-associated protein or chromatin modification to determine the epigenetic state of a subnuclear domain (e.g., H3-K27me3, HP1). Often, the question under investigation requires that RNA, DNA and/or protein be detected in the same nuclei. Our protocols can be adapted for simultaneous or serial detection of RNA and DNA (RNA-DNA FISH), for combined detection of RNA and protein epitopes (RNA-immunoFISH), for combined visualization of DNA and protein epitopes (DNA-immunoFISH) or for detection of all three (RNA/DNA-immunoFISH). Figure 3 shows possible combinations of these experiments.
Figure 3.

Possible combinations of RNA FISH, DNA FISH and immunofluorescence.
For RNA-immunoFISH combinations, it is highly recommended that the immunostaining precede RNA FISH. Exposure to formamide during FISH frequently compromises detection of epitopes by antibodies. We also found it helpful to reduce antibody incubation time during immunostaining, and to use RNAse inhibitors during the incubation step. When DNA FISH needs to be combined with either immunofluorescence or RNA FISH, we find it generally best to perform DNA FISH last, as indeed DNA seems more stable than RNA and protein under conditions used in these protocols. However, we note that it is very difficult to retain signals from earlier rounds (immunofluorescence or RNA FISH) after DNA FISH is performed. This is because of the harsh conditions under which DNA FISH must be performed (e.g., denaturation of samples in 70% (vol/vol) formamide and 80 °C). Thus, when serial RNA-DNA FISH or immunoFISH is undertaken, we recommend that images be acquired after each detection step, coordinates be marked computationally and images from subsequent detection steps be merged with the earlier acquisitions to visualize the combined stains. Signal intensities can then be quantified as described elsewhere16.
Materials
Reagents
Cot-1 DNA (Invitrogen, cat. no. 18440-016)
Prime-It Fluor kit, including Fluoro-12-dUTP (Stratagene, cat. no. 300380)
Herring sperm DNA (Invitrogen, cat. no. 15634-017)
Ethanol (100%; Pharmco-AAPER, cat. no. 111000200)
Ethanol (70%; vol/vol)
Sodium acetate (3 M, pH 5.2; Sigma-Aldrich, cat. no. S2889)
Hybridization buffer (see REAGENT SETUP)
Formamide (American Bioanalytical, cat. no. AB00600-00500)
Saline-sodium citrate buffer (SSC), 20× (see REAGENT SETUP)
Sodium citrate (Sigma-Aldrich, cat. no. C8532)
Bovine serum albumin (BSA, Lifeblood Medical, cat. no. 77110-100)
(50%, wt/vol) (see REAGENT SETUP)
Dextran sulfate sodium salt from Leuconostoc spp. (Sigma-Aldrich, cat. no. D6001)
Nick Translation Kit (Roche, cat. no. 10 976 776 001)
Cy3-dUTP (GE Healthcare, cat. no. PA53022)
Cy5-dUTP (GE Healthcare, cat. no. PA55022)
RPMI 1640 (Invitrogen, cat. no. A10491-01)
Cytoskeleton buffer (CSK) buffer (see REAGENT SETUP)
Acid Tyrode's solution (Sigma-Aldrich, cat. no. T1788). Split into 1.5-ml tubes and store at −20 °C. The solution is stable for more than 1 year. Thaw before use. Do not repeat freeze and thaw cycle.
NaCl (Sigma-Aldrich, cat. no. S3014)
Sucrose (Sigma-Aldrich, cat. no. S0389)
PIPES (Sigma-Aldrich, cat. no. P6757)
MgCl2 (Sigma-Aldrich, cat. no. M0250)
Triton X-100 (Sigma-Aldrich, cat. no. T8787)
Paraformaldehyde (Fluka, cat. no. 76240) ! CAUTION Hazardous; cross-linking agent; wear personal protective equipment and perform relevant steps under a fume hood when using paraformaldehyde.
NaOH (Sigma-Aldrich, cat. no. 221465) ! CAUTION NaOH is corrosive. Wear gloves and glasses to avoid contact with eyes and skin.
PBS (Sigma-Aldrich, cat. no. P4417)
Embryomax M2 medium (Chemicon, cat. no. MR-015-D)
BSA-PBS (6 mg ml−1) (see REAGENT SETUP) Keep the stock at −20 °C in a 1.5-ml tube. Thaw before use. Do not repeat freeze and thaw cycle.
Paraformaldehyde-PBS (1% (wt/vol), pH 7.4) (see REAGENT SETUP)
Paraformaldehyde-PBS (4% (wt/vol), pH 7.4) (see REAGENT SETUP)
Paraformaldehyde-PBS (1% (wt/vol), pH 7.4) with 0.05% (vol/vol) Tergitol (see REAGENT SETUP)
Tergitol (Sigma-Aldrich, cat. no. NP-40S: Tergitol contains 70% NP-40)
PBT (1% BSA, 0.1% TWEEN 20 in PBS) (see REAGENT SETUP)
TWEEN 20 (Sigma-Aldrich, cat. no. p5927)
RNA guard (Amersham, cat. no. 27-0816-01)
Vectashield with DAPI (DAPI-Vectashield; Vector laboratories, cat. no. H1200)
Ribonucleoside-vanadyl complex (New England Biolabs, cat. no. S1042S)
RNase solution (see REAGENT SETUP)
RNaseA (100 mg ml−1; Qiagen, cat. no. 19101)
HCl (0.2 N) with Triton X-100 (see REAGENT SETUP)
HCl (Sigma-Aldrich, cat. no. 258148) ! CAUTION HCl is corrosive. Wear gloves and glasses to avoid contact with eyes and skin.
Cy3-labeled X chromosome paint (Cambio, cat. no. 1200-XMCy3-02)
FITC-labeled Y chromosome paint (Cambio, cat. no. 1189-YMF-02)
Alexa Fluor 555 goat anti-mouse IgG (Invitrogen, cat. no. A21424)
RNA polymerase II C-terminal domain (Millipore, clone no. 8WG16)
DL-dithiothreitol (DTT; Sigma-Aldrich, cat. no. D0632)
Distilled water
Mice ! CAUTION Animal experiments must be carried out according to all relevant institutional and governmental guidelines.
Equipment
Illustra MicroSpin G-50 Columns (GE Healthcare, cat. no. 27-5330-01)
Microcentrifuge (Hettich, cat. no. Mikro120)
Tissue culture dish, small (Falcon, cat. no. 35 3001)
Forceps (VWR, cat. no. 300-050)
Tube (1.5 ml; ISC, cat. no. c-3217-1)
Cytospin 3 (Shandon; see EQUIPMENT SETUP)
Large coplin jar (Fisher, cat. no. 08-817)
Small coplin jar (Fisher, cat. no. 08-815)
Superfrost/Plus Microscope Slides (Fisher, cat. no. 12-550-15)
Dish (4 well; Nunc, cat. no. 179820)
MicroWell 60 high, 8 μm (Nunc, cat. no. 163118)
Mouth pipettes (see EQUIPMENT SETUP)
Calibrated micropipette (50 μl; Fisher, cat. no. 21-180-16)
Barrier tip (200 μl; Neptune, cat. no. BT200)
Silicone tubing; Nalgene 50 Platinum-Cured Silicone Tubing (I.D. = 3/16 inch, wall thickness = 3/32 inch, outer diameter = 3/8 inch. Fisher, cat. no. 14-176-332D)
Pasteur pipettes (9 inches long; CardinalHealth, cat. no. P5202-2) (see EQUIPMENT SETUP)
Sigmacote (Sigma-Aldrich, cat. no. SL-2) ! CAUTION Hazardous; wear personal protective equipment and perform relevant steps under a fume hood when using paraformaldehyde.
Tip-box chamber (see EQUIPMENT SETUP)
Parafilm (VWR, cat. no. 52858-000)
Kimwipes (Kimberly-Clark, cat. no. 34120)
Cover glass (Fisher, cat. no. 12-544-G: 22X60-1.5)
Razor blade (Fisher, cat. no. 12-640)
Rubber cement (Elmer's; cat. no. 231)
Air incubator, 37–42 °C (Incufridge, Fisher, cat. no. S90306)
Water bath, 37–45 °C (POLY PROBATH, The Lab Depot, cat. no. RS_PB-100)
Water bath, 80 °C (Thermo Precision, 280 series)
Vacuum evaporator (Labconco, cat. no. 78100-00)
Needles, 32-gauge
Thermocycler
Nikon Eclipse 90i microscope (Nikon)
Volocity 5 software (PerkinElmer)
Photoshop CS3 (Adobe)
ImageJ software (http://rsbweb.nih.gov/ij/)
Reagent Setup
Probe preparation
Probes are prepared by random priming (see Box 1) or by nick translation (Box 2).
Hybridization buffer
Mix components tabulated below to prepare 900 μl of hybridization buffer; this solution is used for probe stocks. This stock solution requires addition of 10% volume of RNase inhibitor solution (ribonucleoside-vanadyl complex) before hybridization; therefore, 900 μl of hybridization buffer corresponds to 1,000 μl of hybridization solution at final concentration.
| Component | Amount (μl) | Final concentration |
|---|---|---|
| Formamide | 500 | 50% (vol/vol) |
| 50% (wt/vol) Dextran | 200 | 10% (wt/vol) |
| 20× SSC | 100 | 2× |
| 1% (wt/vol) BSA | 100 | 0.1% (wt/vol) |
Add 1 in 10 volumes of 200 mM ribonucleoside-vanadyl complex or water before hybridization to match the final concentration. This solution remains stable at −20 °C for more than 1 year.
20× SSC buffer
Dissolve 175.3 g of NaCl and 88.2 g of sodium citrate in 800 ml of distilled H2O. Adjust the pH to 7.0 with a few drops of 1 M HCl.Adjust the volume to 1 liter with additional distilled H2O. Sterilize by auto-claving. It can be stored at room temperature (25 °C) for more than 1 year.
Dextran (50% (wt/vol))
Slowly add 50 g of dextran sulfate to water and stir at room temperature overnight. Final volume is adjusted with water to 100 ml and autoclaved. It can be stored at room temperature for more than 1 year. ▲ CRITICAL It may take hours to dissolve dextran sulfate. Dextran (50%, wt/vol) should be prepared in advance.
CSK buffer
CSK buffer comprises 100 mM NaCl, 300 mM sucrose, 10 mM PIPES and 3 mM MgCl2. Stir the solution overnight to dissolve PIPES completely. Adjust pH to 6.8 with a few drops of 1 M NaOH if necessary. Keep at 4 °C for no more than 6 months. ▲ CRITICAL It may take hours to dissolve PIPES. CSK buffer should be prepared in advance.
CSK buffer + 0.5% (vol/vol) Triton
Add 250 μl of Triton X-100 to 50 ml of CSK buffer. Stir the solution until Triton X-100 is dissolved. Keep at 4 °C for no more than 6 months.
BSA-PBS (6 mg ml−1)
Dissolve 300 mg of BSA in 50 ml of water. Aliquot to small tubes (500 μl each) and stock at −20 °C until use. The solution is stable for more than 1 year.
Paraformaldehyde-PBS (4% (wt/vol); pH 7.4)
Dissolve 4 g of paraformal-dehyde in approximately 80 ml of water. Add one drop of 4-N NaOH and stir the solution at 60 °C under a fume hood. It will take an hour to dissolve. Add 10 ml of 10× PBS, allow the solution to cool to room temperature and adjust pH to 7.4. Add water to adjust to a total volume of 100 ml. ▲ CRITICAL Always use freshly prepared solution.
Paraformaldehyde-PBS (pH 7.4)
Dilute 25 ml of 4% (wt/vol) paraformaldehyde-PBS, pH 7.4, with 75 ml of PBS, pH 7.4. ▲ CRITICAL Use freshly prepared solution.
Paraformaldehyde-PBS (pH 7.4) with 0.05% (vol/vol) Tergitol
Dissolve 25 μl of Tergitol in 50 ml of 1% (wt/vol) paraformaldehyde-PBS (pH 7.4). ▲ CRITICAL Keep in a coplin jar on ice and use freshly prepared solution.
PBT (1% (wt/vol) BSA, 0.1% (vol/vol) Tween-20 in PBS)
Dissolve 1 g of BSA and 100 μl of Tween 20 in 100 ml of PBS. Aliquot to small tubes (1 ml each) and stock at −20 °C until use. The solution is stable for more than 1 year.
Ribonucleoside-vanadyl complex (RNase inhibitor)
Treat ribonucleoside-vanadyl complex stock at 65 °C for 10 min according to the manufacturer's instructions. Split into small tubes (1 ml each) and stock at −20 °C. It is stable for more than 1 year.
RNase solution
Add 10 μl of Qiagen RNaseA (100 mg ml−1) to 500 μl of PBS before use.
HCl (0.2 N) with Tween-20
Prepare stock solution of 0.2 N HCl: dilute 8.3 ml of 37% (wt/wt) HCl with water to attain a volume of 500 ml. Add 0.2 ml of Triton X-100 in 40 ml of 0.2 N HCl before use.
Equipment Setup
Cytospin
Set up the Cytospin centrifuge according to the manufacturer's instructions. ▲ CRITICAL Be sure to use reusable chambers (disposable chambers are sold but do not work properly for these applications).
Pasteur pipettes for embryo manipulation
Prepare very fine glass pipettes by pulling the tip of Pasteur pipettes above a gas burner. Siliconize the very fine pipettes with Sigmacote under a fume hood. With a rubber nipple for Pasteur pipettes, repeat the up-and-down action with the Sigmacote solution inside the fine glass pipettes three times. Sigmacote solution is reusable. Leave and dry the pipettes under the fume hood overnight.
Mouth pipette setup
A rubber aspirator tube and mouthpiece are then fitted to the siliconized Pasteur Pipette for embryo manipulation (see Fig. 4). The aspirator tube and mouthpiece come with the calibrated micropipette (50 μl). We also use a 200-μl barrier tip and silicone tubing to connect the aspirator tube and Pasteur pipette described above.
Figure 4.

Preparation of male germ cell and preimplantation embryo samples. (a) Slide preparation of male germ cells. (b) The mouth pipette for embryo manipulation. (1) Mouthpiece. (2) Aspirator tube. (3) 200-μl barrier tip. (4) Silicone tubing. (5) Siliconized Pasteur pipette. (c) Key steps in the preparation of preimplantation embryos. See PROCEDURE for further explanation.
Tip-box chamber
This will be used to incubate slides in a moist environment and prevent dehydration. Use empty plastic P1000 pipette tip boxes. Place the slides in the top chamber, and water in the bottom chamber to a depth of 2–3 cm. Preheat the chamber in a humidified incubator before performing experiments.
Microscope setup
We use a Nikon Eclipse 90i microscope workstation with Volocity Software (Improvision). If DNA FISH is to be performed after immunostaining or RNA FISH, be sure to capture images and mark their coordinates (using automated functions in Volocity or equivalent) immediately after immunostaining and RNA FISH before initiating the DNA FISH protocol. Immuofluorescence, RNA FISH and DNA FISH signals are then merged with Adobe Photoshop software (CS3) after experiments are completed.
Procedure
Slide preparation
-
To prepare slides for analyzing spermatogenesis, follow option A. To analyze embryogenesis, follow option B.
-
Spermatogenesis in mice ● TIMING 1 h
-
Euthanize male mice according to methods that comply with the regulations of your institution.
▲ CRITICAL STEP To prevent degradation of RNA, carry out Step 1A(i–iv) quickly.
Remove testis using forceps and scissors. Using forceps, place testis in a small dish containing RPMI 1640 or PBS on ice.
Remove extra tissues around the testis using forceps, rinse seminiferous tubules with RPMI 1640 or PBS on ice and transfer testis to a new dish with RPMI 1640 or PBS on ice.
Open up the testis, remove the tunica albuginea and unravel tubules using forceps in RPMI 1640 or PBS on ice.
-
Transfer a bunch of tubules (we usually pick up 5–10 pieces of tubules about 5–10 mm in length) to a small dish with CSK buffer + 0.5% Triton on ice using forceps, and incubate for 6 min (Fig. 4a).
▲ CRITICAL STEP This permeabilization step is critical to reduce cytoplasmic background in FISH experiments. Incubation time should be optimized for each type of sample.
-
Transfer the tubules to a small dish with 4% (wt/vol) paraformaldehyde solution using forceps, and incubate for 10 min at room temperature Fig. 4a).
▲ CRITICAL STEP Prepare 12 Cytospin chambers during the incubations in Steps 1A(vi) and (vii).
Transfer the tubules to a small dish with PBS using forceps, and incubate for 5 min at room temperature.
Transfer the tubules to 20–30 μl of PBS to the back side of a glass slide (avoid the top side because of cationic charge), and tear tubules to pieces between the tips of two forceps. Clip tubules between the tips of forceps and pull forceps horizontally (Fig. 4a). We spend 10–20 s on this step.
Pipette the suspension up and down with a P20 pipette. The suspension should look slightly cloudy.
Remove chunks of tubular remnants using forceps. Transfer remaining suspension to a 1.5-ml tube and dilute with PBS to ~1.3 ml. Apply 100 μl to each of 12 Cytospin sample chambers.
Cytospin 2,000 r.p.m. for 10 min at room temperature and dry slides on bench for 5 min.
Wash slides in a large coplin jar with 50 ml of PBS for 5 min at room temperature.
-
Slides are now ready for immunostaining. Transfer slides to a large coplin jar with 50 ml of 70% (vol/vol) ethanol and leave slides for at least 2 min before initiation of RNA FISH.
▪ PAUSE POINT Slides are stable for a month in 70% (vol/vol) ethanol at 4 °C. For long-term storage, treat slides in 80% (vol/vol) ethanol for 2 min and in 100% ethanol for 2 min in coplin jars, respectively, and dry slides on bench completely to store in a Parafilm-sealed slide box at −80 °C. They are stable at −80 °C for several months.
-
-
Preimplantation embryos ● TIMING 30 min
Prepare embryos by natural mating or by superovulation of female mice32.
Euthanize female mice using methods that comply with institutional regulations.
-
Dissect with scissors and forceps to obtain oviducts (see details in ref. 32).
▲ CRITICAL STEP All steps from 1B(iii–xvi) need to be performed as quickly as possible to maintain embryo integrity.
Place oviduct in a four-well dish with 500 μl of M2 medium.
Flush oviduct with approximately 100 μl of M2 medium using a 32-gauge needle and 1-ml syringe under a stereomicroscope.
-
Collect embryos with mouth pipettes, transfer to a new well (all embryos in the same well) in the four-well dish and wash with M2 medium.
▲ CRITICAL STEP The number of embryos processed should be kept to a minimum. Step 1B(vii–xix) should be carried out with no more than 20 embryos at a time; extra embryos should be kept in M2 medium and Step 1B(vii–xix) should be repeated until all embryos are processed.
Transfer the embryos to an empty well in a MicroWell 60 with ∼10 μl of M2 medium (Fig. 4c).
-
Place 10 μl of acid Tyrode's solution to each of three vacant wells in the MicroWell 60 and 10 μl of 6 mg ml–1 BSA-PBS to each of three different vacant wells in the MicroWell 60 (Fig. 4c).
▲ CRITICAL STEP Dispense solutions right before use to avoid drying out the solutions.
By mouth pipette, transfer embryos to a well containing 10 μl of acid Tyrode's solution. Aspirate as little of the M2 solution as possible during transfer (Fig. 4c).
Within seconds, transfer embryos to the second well containing 10 μl of acid Tyrode's solution using the mouth pipette (Fig. 4c).
-
Again, within seconds of exposure to acid Tyrode's solution, transfer embryos to the third well containing 10 μl of acid Tyrode's solution. The zona pellucida should disappear at the time of third exposure (Fig. 4c).
▲ CRITICAL STEP Once the zona pellucida is removed, embryos become sticky and fragile. They can therefore become stuck inside glass pipettes. Exposure to acid Tyrode's solution must be kept to a minimum, only long enough to remove the zona pellucida.
Once the zona pellucida disappears, immediately transfer embryos to one of the wells containing 10 μl of 6 mg ml-1 BSA-PBS using the mouth pipette (Fig. 4c).
Within seconds after washing in BSA-PBS, transfer embryos to the second well containing 10 μl of 6 mg ml−1 BSA-PBS (Fig. 4c).
Wash briefly a third time in 6 mg ml−1 BSA-PBS (third well; Fig. 4c).
Collect all embryos in the last BSA-PBS well and then transfer onto a Superfrost/Plus Microscope Slide using the mouth pipette. Transfer as little BSA-PBS solution as possible (Fig. 4c).
-
Once the embryos are placed on the slide, aspirate as much of the BSA-PBS solution as possible without disturbing the embryos. This step facilitates air-drying of embryos (Fig. 4c).
▲ CRITICAL STEP It is important to remove all extra BSA-PBS quickly so that embryos can dry quickly. Rapid embryo drying is crucial to maintaining morphology and promoting adherence to the glass slide. When performed in this manner, permeabilization works well. If extra liquid is not aspirated, embryos dry slowly, do not adhere well to the glass slide, lose nuclear morphology and cannot be permeabilized readily.
-
After embryos are completely dry, place the slide in 50 ml of 1% (wt/vol) paraformaldehyde-PBS (pH 7.4) with 0.05% (vol/vol) Tergitol in a coplin jar on ice for 5 min (Fig. 4c).
▲ CRITICAL STEP This permeabilization step is critical to reduce cytoplasmic background in FISH experiments. Concentration of paraformaldehyde or detergent (Tergitol) should be optimized for each tissue type.
Transfer the slide to 50 ml of 1% (wt/vol) paraformaldehyde-PBS (pH 7.4) in a coplin jar on ice for 5 min (Fig. 4c).
-
Transfer the slide to 50 ml of 70% (vol/vol) ethanol in a coplin jar on ice (Fig. 4c).
▪ PAUSE POINT Keep the slide in 50 ml of 70% (vol/vol) ethanol in a coplin jar at 4 °C until use. Slides are stable for at least 2–3 weeks. At this point, the samples are ready for RNA or DNA FISH, for immunostaining or for any combination shown in Figure 3. As discussed above, it is helpful to check the quality of slides by performing Cot-1 RNA FISH on one slide.
-
-
Staining
To perform immunofluorescence, proceed as described in option A. To carry out Cot-1 RNA FISH, follow option B. Follow option C to carry out gene-specific RNA FISH and option D to perform DNA FISH.
-
Immunofluorescence ● TIMING 2.5–18 h
-
Rinse stored slide by placing it in a coplin jar with 1× PBS for several seconds.
▲ CRITICAL STEP When immunofluorescence is to be combined with RNA FISH, it is critical to add RNase inhibitor during blocking and incubation steps with primary and secondary antibodies (Steps 2A(ii), (iii) and (v)) and to keep manipulation time to a minimum in order to preserve RNA. RNase inhibitor is not necessary in the wash buffer.
-
Wipe excess liquid off the slide and place the slide in a tip-box chamber. Overlay the slide with 100 μl of blocking buffer (PBT with 0.4 U μl−1 RNA guard). Cover slides with a square of Parafilm cut approximately to shape. Incubate for 20 min at room temperature.
▲ CRITICAL STEP Do not ever let slides become dehydrated during the procedure. Keep the number of slides to a minimum to prevent dehydration.
-
Remove blocking buffer by tipping the slide onto a piece of Kimwipe. Add 100 μl of primary antibody solution in PBT with 0.4 U μl−1 RNA guard to the slide in the tip-box chamber, and cover slides with Parafilm cut approximately to shape. Incubate for 1 h at room temperature to overnight at room temperature or at 37 °C.
▲ CRITICAL STEP For male germ cells, Step 2A(iii) is performed overnight (14–15 h) at room temperature or at 37 °C. The overnight incubation improves penetration of antibodies, especially for heterochromatic regions. However, overnight incubation may interfere with subsequent RNA FISH, as RNA degradation is more likely to occur with prolonged incubation. Therefore, if RNA FISH is to be performed, antibody incubation should not proceed overnight. If RNA FISH is not to be performed after immunostaining, RNase inhibitor is not necessary.
Wash slide three times with 0.1% Tween-20 in PBS in serial coplin jars for 5 min at room temperature.
-
Remove excess liquid by tipping the slide onto a piece of Kimwipe. Add 100 μl of secondary antibody solution in PBT with 0.4 U μl−1 RNA guard to the slide in the tip-box chamber, and cover slides with Parafilm cut approximately to shape. Incubate for 0.5–1 h at room temperature.
▲ CRITICAL STEP If RNA FISH is not to be performed after immunostaining, RNase inhibitor is not necessary.
Wash slide three times with 0.1% Tween-20 in PBS in a coplin jar for 5 min at room temperature.
If RNA FISH is to be performed in combination, go to Step 2A(viii) immediately. If RNA FISH is not to be performed, remove excess liquid by tipping the slide onto a piece of Kimwipe, add 20 μl of DAPI-Vectashield to the slides and cover with a cover glass; slides are now ready for microscopy.
Before RNA FISH, fix immunofluorescence signals by transferring the slide to a coplin jar containing 4% (wt/vol) paraformaldehyde-PBS. Incubate for 10 min at room temperature.
-
Wash slides with PBS in a coplin jar for 5 min at room temperature. Proceed immediately to the RNA FISH method (Step 2B or C).
? Troubleshooting
-
-
Cot-1 RNA FISH ● TIMING 7–16 h
-
Prepare Cot-1 probe as described in Box 1. For Cot-1 RNA FISH, add components tabulated below to a PCR tube. Optionally, a gene-specific probe such as Xist probe can be prepared as described in Box 2 and added to Cot-1 RNA FISH. Mix and incubate for 10 min at 80 °C, followed by a preannealing step of 10–30 min at 42 °C using a PCR machine. Ribonucleoside-vanadyl complex (RNase inhibitor) is optional; it can be replaced with sterile water.
▲ CRITICAL STEP This protocol is optimized on the basis of a previously described method6. It can be applied to both germ cells and preimplantation embryos, not only for gene-specific analysis but also for assaying repetitive element expression16.
▲ CRITICAL STEP For Cot-1 RNA FISH, do not use competitors such as Cot-1 DNA and yeast tRNA, as they compete with the Cot-1 probe. Herring sperm DNA is recommended if necessary.
Dehydrate slides by serial treatment with 70%, 80% and 100% (vol/vol) ethanol in coplin jars for 2 min each; air-dry slides completely. Perform all steps at room temperature.
-
Preheat a tip-box chamber in a humidified 42 °C incubator in advance (the tip-box chamber should be filled with 2–3 cm of water in the lower chamber). Place slide on the top chamber. Briefly spin the preannealed probes and pipette carefully directly onto the sample. Do not create bubbles. Cover with a cover slip gently.
▲ CRITICAL STEP To place the cover slip without creating bubbles, tilt it slightly between the thumb and index fingers of both hands and gently lower onto the probe solution. It is crucial to avoid trapping bubbles under the cover slip because bubbles may impede hybridization.
Incubate at 42 °C for 6 h to overnight (14–15 h).
Prepare wash solutions (two coplin jars with 50% (vol/vol) formamide in 2× SSC, and two coplin jars with 2× SSC). Prewarm these solutions in a water bath at 42 °C for 30 min.
-
Remove cover slip using a razor blade from the edge of the cover glass. Open up vertically and do not scratch samples. Alternatively, submerge slide in 50% (vol/vol) formamide in 2× SSC in a coplin jar and allow the cover slip to slide off (this takes only 30 s or so). Wash in a coplin jar containing 50% (vol/vol) formamide in 2× SSC for 5 min at 45 °C. Repeat one more time.
▲ CRITICAL STEP Do not agitate the water bath when embryo slides are used, as agitation may cause embryos to detach from the slide.
Wash in 2× SSC for 5 min at 45 °C. Repeat one more time.
-
Mount the slides with DAPI-Vectashield solution. Blot extra liquid with a Kimwipe, add 20 μl of DAPI-Vectashield to the slides and cover with a cover glass. Apply very gentle pressure to the cover glass to remove extra mounting solution. Do not fix the cover glass with nail polish if additional experiments (e.g., DNA FISH) are to be performed.
? Troubleshooting
-
-
Gene-specific RNA FISH ● TIMING 16 h
-
Dry each competitor (10 μl of Cot-1 DNA (1 mg ml−1 stock) and 0.5 μl of yeast tRNA (20 mg ml−1 stock)) in a PCR tube using a vacuum evaporator. This step requires > 30 min.
▲ CRITICAL STEP This protocol is optimized according to a previously described method33. The following protocol is applicable to preimplantation embryos and can also be applied to male germ cells.
-
Probe preparation. For gene-specific probes, add components tabulated below to the PCR tube containing dried competitors. Mix and pipette up and down thoroughly. Incubate for 10 min at 80 °C to denature, and then preanneal with competitor for 10–30 min at 37 °C using a thermocycler. Ribonucleoside-vanadyl complex is necessary.
▲ CRITICAL STEP Up to three probes with different fluorophores can be combined. Preannealing with Cot-1 DNA is necessary because gene-specific probes often contain repetitive elements that need to be blocked before gene-specific hybridization.
▲ CRITICAL STEP Selection of probe DNA is critical. Longer DNA probes always have stronger signals. We usually use BAC clones that cover entire transcribed regions to detect specific gene activity. However, BAC sequences often contain multiple genes. See Experimental design for detailed explanation.
Dehydrate slides by serial treatment with 70%, 80% and 100% (vol/vol) ethanol in serial coplin jars for 2 min each and air-dry completely. Perform all steps at room temperature.
-
Preheat a tip-box chamber in a humidified 37 °C incubator in advance (the tip-box chamber should be filled with 2–3 cm of water in the lower chamber). Place slide on the top chamber. Briefly spin the preannealed probes and pipette carefully directly onto the sample. Do not create bubbles. Cover with a cover slip gently.
▲ CRITICAL STEP To place the cover slip without creating bubbles, tilt the slide glass slightly between the thumb and index fingers with both hands and gently lower onto the probe solution. It is crucial to avoid trapping bubbles under the cover slip.
Incubate at 37 °C overnight (14–15 h).
Prepare wash solutions (two coplin jars with 50% (vol/vol) formamide in 2× SSC, and two coplin jars with 2× SSC). Prewarm these solutions in a water bath at 37 °C for 30 min.
-
Remove the cover slip using a razor blade from the edge of the cover glass. Open up vertically and do not scratch samples. Alternatively, submerge slide in 50% (vol/vol) formamide in 2× SSC in a coplin jar and allow the cover slip to slide off (this takes only 30 s or so). Wash with a coplin jar containing 50% (vol/vol) formamide in 2× SSC for 5 min at 45 °C. Repeat one more time.
▲ CRITICAL STEP Do not agitate the water bath when embryo slides are being used, as agitation may cause embryos to detach from the slide.
Wash with 2× SSC for 5 min at 37 °C. Repeat one more time.
-
Mount slide with DAPI-Vectashield solution. Blot extra liquid with a Kimwipe, add 20 μl of DAPI Vectashield to the slides and cover with a cover glass. Apply very gentle pressure to the cover glass to remove extra mounting solution. Do not fix the cover glass with nail polish if additional experiments (e.g., DNA FISH) are to be performed.
? Troubleshooting
-
-
DNA FISH ● TIMING 18 h
-
Wash in a coplin jar with PBS for 5 min at room temperature. If you perform this step following immunofluorescence and/or RNA FISH, remove the cover glass as described above.
▲ CRITICAL STEP This protocol is optimized according to a previously described method34. If DNA FISH is to be performed after immunostaining or RNA FISH, be sure to capture images and mark their coordinates immediately after immunostaining and RNA FISH before initiating the DNA FISH protocol (see EQUIPMENT SETUP). Preparation for DNA FISH subjects the sample to very harsh conditions, often destroying immunofluorescence and RNA FISH signals.
-
Refix the sample by transferring slide to a coplin jar with 4% (wt/vol) paraformaldehyde containing 0.5% (vol/vol) Tergitol and 0.5% (vol/vol) Triton X-100. Fix for 10 min at room temperature.
▲ CRITICAL STEP This refixation step helps to retain nuclear morphology during harsh treatment steps. Simultaneous detergent treatment facilitates probe penetration for DNA FISH.
Wash slides in a coplin jar with PBS for 5 min at room temperature.
RNase treatment: Blot excess liquid off the slide and place in a tip-box chamber preheated to 37 °C. Add 100 microliters of RNase to each slide and cover with Parafilm cut approximately to shape. Incubate for 30 min at 37 °C.
During RNase treatment, you should dry the competitor DNA (30 μl of Cot-1 DNA: 1 μg ml−1 stock) in a PCR tube using a vacuum evaporator. The PCR tube containing dried competitor DNA is used to prepare the hybridization solution in Step 2D(viii). It will take >30 min.
Wash slides from Step 2D(iv) in a coplin jar with PBS for 5 min at room temperature.
Transfer the slides to a coplin jar with 0.2 N HCl with Triton X-100 for 10 min on ice.
-
During HCl treatment, start preparing probes; for gene-specific probe, add components tabulated below to the PCR tube containing dried competitor DNA (Step 2D(v)). Mix and pipette up and down thoroughly. Incubate for 10 min at 80 °C and preanneal for 10–30 min at 37 °C using a thermocycler.
▲ CRITICAL STEP Up to three colors are available. Three probes of different fluorophores can be combined in a single experiment. Chromosome paints can be used. For XY chromosome paint analysis, we recommend combining X-Cy3 and Y-FITC because X-Cy3 yields greater sensitivity and Y-FITI is less background than Y-Cy3 in general. The quality of chromosome paints varies between batches and needs to be verified.
Wash slides from Step 2D(vii) in a coplin jar with PBS for 5 min at room temperature.
-
Denature slide in 70% formamide, 2× SSC in a coplin jar that has been preheated to 80 °C. Incubate for 10 min in the 80 °C water bath.
▲ CRITICAL STEP It takes about an hour to warm up the water bath and coplin jar to 80 °C. Set up the reagent and equipment before the experiment.
Transfer slide immediately from Step 2D(x) to a coplin jar with 70% (vol/vol) ethanol in an ice box and incubate for 2 min.
Dehydrate slide by serial treatment with 80% (vol/vol) and 100% ethanol in serial coplin jars for 2 min each. Air-dry the slides on a paper towel. Perform all steps at room temperature.
-
Preheat a tip-box chamber in a humidified 37 °C incubator in advance (the tip-box chamber should be filled with 2–3 cm of water in the lower chamber). Place slide on the top chamber. Briefly spin the preannealed probes and pipette carefully directly onto the sample. Do not create bubbles. Gently cover with a cover slip, and then apply rubber cement around the cover slip.
▲ CRITICAL STEP To place the cover slip without bubbles, tilt the slide glass slightly between the thumb and index fingers with both hands and gently lower onto the probe solution. It is crucial to avoid trapping bubbles under the cover slip.
Incubate at 37 °C for 14–15 h.
Prepare wash solutions (two coplin jars with 50% (vol/vol) formamide in 2× SSC, and two coplin jars with 2× SSC). Prewarm these solutions in a water bath at 45 °C for 30 min.
-
Remove rubber cement with your fingers, and then remove the cover slip with a razor blade from the edge of the cover glass. Open up vertically and do not scratch samples. Alternatively, submerge slide in 50% (vol/vol) formamide in 2× SSC in a coplin jar and allow the cover slip to slide off (this takes only 30 s or so). Wash with a coplin jar containing 50% (vol/vol) formamide in 2× SSC for 5 min at 45 °C. Repeat one more time.
▲ CRITICAL STEP Do not agitate the water bath when embryo slides are being used, as agitation may cause embryos to detach from the slide.
Wash with 2× SSC for 5 min at 45 °C. Repeat one more time.
-
Mount slide with DAPI-Vectashield solution. Blot extra liquid with a Kimwipe, add 20 μl of DAPI-Vectashield to the slides, and then cover with a cover glass. Apply very gentle pressure to the cover glass to remove extra mounting solution.
? Troubleshooting
-
-
? Troubleshooting
Troubleshooting advice can be found in Table 1.
Table 1.
Troubleshooting table.
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 2A(ix) | High background/low signal specificity after immunofluorescence | Cytoplasm is incompletely extracted | Optimize the permeabilization step by longer incubation with the detergent or higher detergent concentration |
| 2B(viii) | No signal after Cot-1 RNA FISH | Poor-quality probe | Check the probe quality. Try another batch of probe |
| Low probe concentration | Optimize probe concentration | ||
| Probe has not penetrated the cell | Optimize the permeabilization step by longer incubation with the detergent or higher detergent concentration | ||
| Cot-1 DNA is used as a competitor in hybridization | Eliminate Cot-1 DNA from competitor | ||
| RNA is degraded | Try to keep the bench RNase free by using the detergent for RNase removal | ||
| Use freshly prepared slides | |||
| Be sure to use RNase inhibitor when the immunofluorescence assay is performed before RNA FISH | |||
| 2B(viii) | High background/low signal specificity after Cot-1 RNA FISH | Cytoplasm is incompletely extracted | Optimize the permeabilization step by longer incubation with the detergent or higher detergent concentration |
| Poor-quality probe | Check the probe quality. Try another batch of probe | ||
| 2C(ix) | No signal after gene-specific RNA FISH | Probe is inadequate for RNA FISH | Some genes are not amenable to RNA FISH analysis due to low expression. Try other genes |
| Hybridized region of the probe is too small | Try probes at least several kb in size | ||
| Poor-quality probe | Check the quality of probe. Try another batch of probe | ||
| Try DNA FISH to test probe quality | |||
| Try Cy3-labeled probe. Cy3 probe has the strongest sensitivity | |||
| Low probe concentration | Optimize probe concentration | ||
| Probe has not penetrated the cell | Optimize the permeabilization step by longer incubation with the detergent or higher detergent concentration | ||
| RNA is degraded | Try to keep the bench RNase free by using the detergent for RNase removal | ||
| Use freshly prepared slides | |||
| Be sure to use RNase inhibitor when the immunofluorescence assay is performed before RNA FISH | |||
| 2C(ix) | High background/low signal specificity after gene-specific RNA FISH | Cytoplasm is incompletely extracted | Optimize the permeabilization step by longer incubation with the detergent or with higher detergent concentration |
| Poor quality probe | Check the probe quality. Try another batch of probe | ||
| Try Cy3-labeled probe. Cy3 fluorophore is brightest | |||
| Low competitor DNA | Try higher concentration of competitor DNA to obtain specific signals | ||
| 2D(xviii) | No signal after gene-specific DNA FISH | Hybridized region of the probe is too small | Try probes with at least several kb |
| Poor-quality probe | Check the quality of probe. Try another batch of probe | ||
| Try Cy3-labeled probe. Cy3 probe has the strongest sensitivity | |||
| Low probe concentration | Optimize probe concentration | ||
| Probe has not penetrated the cell | Optimize the permeabilization step by longer incubation with the detergent or with higher detergent concentration | ||
| Make sure the RNase treatment and HCl-detergent steps work properly | |||
| 2D(xviii) | High background/low signal specificity after DNA FISH | Cytoplasm is incompletely extracted | Optimize the permeabilization step by longer incubation with the detergent or higher detergent concentration |
| Poor-quality probe | Check the probe quality. Try another batch of probe | ||
| Try Cy3-labeled probe. Cy3 fluorophore yields intense signals | |||
| Low competitor DNA | Try higher concentration of competitor DNA to obtain specific signals | ||
| 2D(xviii) | Nuclear structure is broken after DNA FISH | Pretreatment before DNA FISH is too harsh | Try milder conditions |
| Try fixation again with paraformaldehyde between immunofluorescence/RNA FISH and DNA FISH |
In RNA and DNA FISH experiments, each probe should be tested first using control cell lines, such as fibroblasts, before use on precious samples.
● TIMING
Step 1A, slide preparation—spermatogenesis in mice: 1 h
Step 1B, slide preparation—preimplantation embryos in mice: 30 min
Step 2A, immunofluorescence: 2.5–18 h
Step 2B, Cot-1 RNA FISH: 7–16 h
Step 2C, gene-specific RNA FISH: 16 h
Step 2D, DNA FISH: 18 h
Box 1: ∼2.5 h
Box 2: ∼4 h
Anticipated Results
Representative results for Cot-1 RNA FISH are shown in Figures 1 and 2. In general, the Cot-1 stain is an excellent technique to visualize broad regions of transcriptional activity and silencing, although we caution that it primarily detects expression of highly repetitive elements. The results do not necessarily imply expression of neighboring genic elements but do correlate very well with RNA Pol-II localization16. Highly expressed telomeric RNAs can also be visualized using this stain34. An example of RNA-immunoFISH in a two-cell embryo can be seen in Figure 5. This type of experiment enables investigators to localize a nascent transcript (e.g., Xist RNA) in relation to areas of Pol-II activity. In the example shown, it can be observed that Xist RNA—a long noncoding RNA that initiates XCI in eutherian female embryos—has only commenced transcription, but the X chromosome in its vicinity already shows signs of transcriptional inactivity and is also positioned next to the nucleolar precursor within DAPI-intense heterochromatic regions16 (Fig. 5). Single-copy targets such as unique sequence DNA or very low-copy targets such as nascent transcripts are visualized reliably, especially if probes are made using longer-length BAC sequences. In summary, the optimized protocols described here should enable the investigator to examine spatial relationships between RNA, DNA and chromatin landmarks of interest in germ cells and preimplantation embryos. Its careful application is expected to elucidate events surrounding the poorly understood transition from gamete to embryo.
Figure 5.
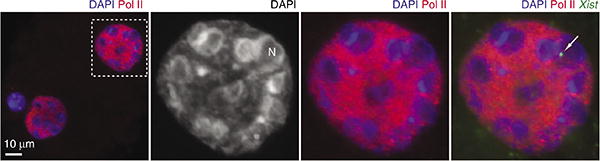
Combined RNA-immunoFISH experiment. Immunofluorescence was performed using a mouse monoclonal antibody against RNA polymerase II (Pol II) C-terminal domain and detected with secondary antibody conjugated with Alexa Fluor 555 anti-mouse IgG. Immunostaining is followed by RNA FISH with FITC-labeled Xist gene-specific probe. A representative blastomere (rectangle in the left panel) is magnified. Raw images of a single z-section are shown (without any contrast adjustments or deconvolution). In the corresponding original publication, we performed deconvolution analysis to subtract out-of-focus light in order to visualize true signals on a single z-plane. DNA was counterstained with DAPI.
Acknowledgments
We thank K. Hyunh, B. Payer and L.-F. Zhang for their assistance to optimize experimental conditions during the initial phases of these works. The studies have been supported by National Institutes of Health grant RO1-GM58839 to J.T.L. S.H.N. was supported by the research fellowships of the Japan Society for the Promotion of Science (JSPS) and the Charles King Trust. J.T.L. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Author Contributions S.H.N. and J.T.L. wrote the manuscript. S.H.N. designed the protocols, with guidance from lab protocols, and J.T.L. and S.H.N. conducted the experiments. J.T.L. supervised the projects.
Competing Financial Interests The authors declare no competing financial interests.
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions/.
References
- 1.Youngson NA, Whitelaw E. Transgenerational epigenetic effects. Annu Rev Genomics Hum Genet. 2008;9:233–257. doi: 10.1146/annurev.genom.9.081307.164445. [DOI] [PubMed] [Google Scholar]
- 2.Carrell DT, Hammoud SS. The human sperm epigenome and its potential role in embryonic development. Mol Hum Reprod. 2010;16:37–47. doi: 10.1093/molehr/gap090. [DOI] [PubMed] [Google Scholar]
- 3.Cooper DW. Directed genetic change model for X chromosome inactivation in eutherian mammals. Nature. 1971;230:292–294. doi: 10.1038/230292a0. [DOI] [PubMed] [Google Scholar]
- 4.Lyon MF. Imprinting and X chromosome inactivation. In: Ohlsson R, editor. Results and Problems in Cell Differentiation. Springer-Verlag; 1999. pp. 73–90. [DOI] [PubMed] [Google Scholar]
- 5.McCarrey JR. X-chromosome inactivation during spermatogenesis: the original dosage compensation mechanism in mammals? In: Xue G, et al., editors. Gene Families: Studies of DNA, RNA, Enzymes, and Proteins. World Scientific Publishing Co.; 2001. pp. 59–72. [Google Scholar]
- 6.Huynh KD, Lee JT. Inheritance of a pre-inactivated paternal X chromosome in early mouse embryos. Nature. 2003;426:857–862. doi: 10.1038/nature02222. [DOI] [PubMed] [Google Scholar]
- 7.Huynh KD, Lee JT. X-chromosome inactivation: a hypothesis linking ontogeny and phylogeny. Nat Rev Genet. 2005;6:410–418. doi: 10.1038/nrg1604. [DOI] [PubMed] [Google Scholar]
- 8.Hammoud SS, et al. Distinctive chromatin in human sperm packages genes for embryo development. Nature. 2009;460:473–478. doi: 10.1038/nature08162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arpanahi A, et al. Endonuclease-sensitive regions of human spermatozoal chromatin are highly enriched in promoter and CTCF binding sequences. Genome Res. 2009;19:1338–1349. doi: 10.1101/gr.094953.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brykczynska U, et al. Repressive and active histone methylation mark distinct promoters in human and mouse spermatozoa. Nat Struct Mol Biol. 2010;17:679–687. doi: 10.1038/nsmb.1821. [DOI] [PubMed] [Google Scholar]
- 11.Darzacq X, et al. Imaging transcription in living cells. Annu Rev Biophys. 2009;38:173–196. doi: 10.1146/annurev.biophys.050708.133728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clemson CM, et al. An architectural role for a nuclear noncoding RNA: NEAT1 RNA is essential for the structure of paraspeckles. Mol Cell. 2009;33:717–726. doi: 10.1016/j.molcel.2009.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cremer T, Cremer M. Chromosome territories. Cold Spring Harb Perspect Biol. 2010;2:a003889. doi: 10.1101/cshperspect.a003889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao R, Bodnar MS, Spector DL. Nuclear neighborhoods and gene expression. Curr Opin Genet Dev. 2009;19:172–179. doi: 10.1016/j.gde.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Namekawa SH, et al. Postmeiotic sex chromatin in the male germline of mice. Curr Biol. 2006;16:660–667. doi: 10.1016/j.cub.2006.01.066. [DOI] [PubMed] [Google Scholar]
- 16.Namekawa SH, Payer B, Huynh KD, Jaenisch R, Lee JT. Two-step imprinted X inactivation: repeat versus genic silencing in the mouse. Mol Cell Biol. 2010;30:3187–3205. doi: 10.1128/MCB.00227-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Turner JM, Mahadevaiah SK, Ellis PJ, Mitchell MJ, Burgoyne PS. Pachytene asynapsis drives meiotic sex chromosome inactivation and leads to substantial postmeiotic repression in spermatids. Dev Cell. 2006;10:521–529. doi: 10.1016/j.devcel.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 18.Okamoto I, et al. Evidence for de novo imprinted X-chromosome inactivation independent of meiotic inactivation in mice. Nature. 2005;438:369–373. doi: 10.1038/nature04155. [DOI] [PubMed] [Google Scholar]
- 19.Okamoto I, Otte AP, Allis CD, Reinberg D, Heard E. Epigenetic dynamics of imprinted X inactivation during early mouse development. Science. 2004;303:644–649. doi: 10.1126/science.1092727. [DOI] [PubMed] [Google Scholar]
- 20.Panning B. X inactivation in mouse ES cells: histone modifications and FISH. Methods Enzymol. 2004;376:419–428. doi: 10.1016/S0076-6879(03)76028-5. [DOI] [PubMed] [Google Scholar]
- 21.Erwin JA, Lee JT. Characterization of X-chromosome inactivation status in human pluripotent stem cells. Curr Protoc Stem Cell Biol. 2010;Chapter 1(Unit 1B):6. doi: 10.1002/9780470151808.sc01b06s12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peters AH, Plug AW, van Vugt MJ, de Boer P. A drying-down technique for the spreading of mammalian meiocytes from the male and female germline. Chromosome Res. 1997;5:66–68. doi: 10.1023/a:1018445520117. [DOI] [PubMed] [Google Scholar]
- 23.Barlow AL, Benson FE, West SC, Hulten MA. Distribution of the Rad51 recombinase in human and mouse spermatocytes. EMBO J. 1997;16:5207–5215. doi: 10.1093/emboj/16.17.5207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hall LL, et al. An ectopic human XIST gene can induce chromosome inactivation in postdifferentiation human HT-1080 cells. Proc Natl Acad Sci USA. 2002;99:8677–8682. doi: 10.1073/pnas.132468999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Namekawa SH, VandeBerg JL, McCarrey JR, Lee JT. Sex chromosome silencing in the marsupial male germ line. Proc Natl Acad Sci USA. 2007;104:9730–9735. doi: 10.1073/pnas.0700323104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Greaves IK, Rangasamy D, Devoy M, Marshall Graves JA, Tremethick DJ. The X and Y chromosomes assemble into H2A.Z, containing facultative heterochromatin, following meiosis. Mol Cell Biol. 2006;26:5394–5405. doi: 10.1128/MCB.00519-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khalil AM, Boyar FZ, Driscoll DJ. Dynamic histone modifications mark sex chromosome inactivation and reactivation during mammalian spermatogenesis. Proc Natl Acad Sci USA. 2004;101:16583–16587. doi: 10.1073/pnas.0406325101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Okamoto I, et al. Evaluating the role of inducible nitric oxide synthase using a novel and selective inducible nitric oxide synthase inhibitor in septic lung injury produced by cecal ligation and puncture. Am J Respir Crit Care Med. 2000;162(2 Pt 1):716–722. doi: 10.1164/ajrccm.162.2.9907039. [DOI] [PubMed] [Google Scholar]
- 29.Ohno M, Aoki N, Sasaki H. Allele-specific detection of nascent transcripts by fluorescence in situ hybridization reveals temporal and culture-induced changes in Igf2 imprinting during pre-implantation mouse development. Genes Cells. 2001;6:249–259. doi: 10.1046/j.1365-2443.2001.00417.x. [DOI] [PubMed] [Google Scholar]
- 30.Silva J, et al. Establishment of histone h3 methylation on the inactive X chromosome requires transient recruitment of Eed-Enx1 polycomb group complexes. Dev Cell. 2003;4:481–495. doi: 10.1016/s1534-5807(03)00068-6. [DOI] [PubMed] [Google Scholar]
- 31.Patrat C, et al. Dynamic changes in paternal X-chromosome activity during imprinted X-chromosome inactivation in mice. Proc Natl Acad Sci USA. 2009;106:5198–5203. doi: 10.1073/pnas.0810683106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nagy A, Gertsenstein M, Vintersen K, Behringer R. Manipulating the Mouse Embryo. Cold Spring Harbor Laboratory Press; 2003. [Google Scholar]
- 33.Beatty B, Mai S, Squire J. FISH A Practical Approach. Oxford University Press; 2002. [Google Scholar]
- 34.Zhang LF, Huynh KD, Lee JT. Perinucleolar targeting of the inactive X during S phase: evidence for a role in the maintenance of silencing. Cell. 2007;129:693–706. doi: 10.1016/j.cell.2007.03.036. [DOI] [PubMed] [Google Scholar]