

Abstract
Type III Collagen (Col3), a fibril-forming collagen, is a major extracellular matrix component in a variety of internal organs and skin. It is also expressed at high levels during embryonic skeletal development and is expressed by osteoblasts in mature bone. Loss of function mutations in the gene encoding Col3 (Col3a1) are associated with vascular Ehlers Danlos Syndrome (EDS). Although the most significant clinical consequences of this syndrome are associated with catastrophic failure and impaired healing of soft tissue structures, several studies have documented skeletal abnormalities in vascular EDS patients. However, there are no reports of the role of Col3 deficiency on the murine skeleton. We compared craniofacial and skeletal phenotypes in young (6-8 weeks) and middle-aged (>1 year) control (Col3+/+) and haploinsufficient (Col3+/−) mice, as well as young null (Col3−/−) mice using microcomputed tomography (μCT). Although Col3+/− mice did not have significant craniofacial abnormalities based upon cranial morphometrics, microCT analysis of distal femur trabecular bone demonstrated significant reductions in bone volume (BV), bone fraction volume (BV/TV), connectivity density (ConnD), structure model index (SMI) and trabecular thickness (TbTh) in young adult, female Col3+/− mice relative to wild-type littermates. The reduction in BV/TV persisted in female mice at one year of age. Next we evaluated the role of Col3 in vitro. Osteogenesis assays revealed that cultures of mesenchymal progenitors harvested from Col3−/− embryos display decreased alkaline phosphatase activity and reduced capacity to undergo mineralization. Consistent with this data, a reduction in osteogenic markers (type I collagen, osteocalcin and bone sialoprotein) correlates with reduced bone Col3 expression in Col+/− mice and with age in vivo. A small but significant reduction in osteoclast numbers was found in Col3+/− compared to Col3+/+ bones. Taken together, these findings indicate that Col3 plays a role in development of trabecular bone through its effects on osteoblast differentiation.
Keywords: Type III collagen, Extracellular matrix, Bone formation, Osteogenesis, Osteoporosis, Mineralization
Introduction
Osteoporosis, the most common bone disease in humans, is characterized by low bone mass, deterioration of bone architecture, compromised bone strength and an increase in the risk of fracture [1]. Aging is associated with significant bone loss in women and men. In fact, in developed countries, approximately 50% of female and 20% of male quinquagenarians will experience a fracture over the course of their remaining lives [2]. Medical and surgical therapies for osteoporosis, including those which treat sequelae such as fractures, have been estimated to increase healthcare costs by an estimated 19 billion dollars per year in the United States and are anticipated to escalate given the expanding elderly population [3]. Three major mechanisms thought to contribute to osteoporosis include insufficient deposition of peak bone mass by late adolescence/early adulthood, inadequate new bone formation during remodeling, and excessive bone resorption, particularly beyond the fifth decade of life [1]. Therefore, identification of critical factors which mediate bone deposition early in life is of significant clinical importance for life-long bone health.
Formation and maintenance of the skeleton requires progenitor cell recruitment, replication and differentiation, processes regulated in part by the extracellular matrix (ECM)[4, 5]. Approximately 90% of the matrix in bone is comprised of collagen, which serves as a tissue scaffold but also provides a substrate for cell anchorage and regulates bioavailability of growth factors and cytokines. Type III collagen (Col3), a homotrimeric fibril-forming collagen, is found mainly in extensible tissues such as blood vessels, skin, and lung [6-8] in heterotypic fibrils with type I collagen (Col1)[9, 10]. A small amount of Col3 is also present in Col1-containing fibrils in bone [11-13]. A critical role for Col3 in skeletal development is suggested by its appearance in mesenchymal condensations preceding cartilage and bone formation[14], its requirement for growth acceleration of osteoblasts [15] and its possible role in preserving osteogenic potential of mesenchymal stem cells [16].
Vascular Ehlers-Danlos Syndrome (EDS) is a dominantly inherited connective tissue disorder caused by mutations in the Col3 gene. Individuals with vascular EDS suffer significant morbidity and mortality due to rupture of large vessels. They also display skeletal manifestations, including a distinctive facial appearance, spinal deformity, scoliosis, and osteoporosis [17-19]. However, the mechanisms mediating the effects of Col3 on skeletal development are unknown.
Recognizing this potential role for Col3 in development and maintenance of the skeleton, our objective was to use mice deficient in Col3 [20] to investigate the important role of this protein in craniofacial and skeletal development. The data presented herein support our hypothesis that Col3 plays a positive role in development of trabecular bone through its effect on osteoblastogenesis.
Materials and Methods
Production and genotyping of Col3-deficient mice
Animal utilization and care were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Pennsylvania and followed guidelines set forth in the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All mice for this study were generated in a colony established at the University of Pennsylvania from breeder pairs of Col3A1 heterozygous (Col3+/−) mice originally purchased from Jackson Laboratories (Bar Harbor, ME). These mice had been generated by homologous recombination by replacement of the promoter region and first exon of the Col3 gene with a 1.8kb PGKneo cassette[20]. Animals were genotyped for Col3 by PCR analysis of DNA extracted from tail biopsies as described previously[21]. At the time of genotyping, mice were microchipped for identification (Allflex FDX-B transponders, Allflex USA, Inc., Dallas, TX). All mice were identified based upon the last four digits of the implanted microchip so that analysis was performed in a blinded fashion with respect to genotype of the individual, as previously described[21]. Mice were euthanized by CO2 asphyxiation followed by cervical dislocation, in accordance with current AVMA guidelines on euthanasia. Femora and tibiae were removed and the majority of soft tissues were removed from the bone specimens immediately after euthanasia. Whole bone specimens were wrapped in gauze and stored at 4°C in 4% paraformaldehyde prior to μCT imaging.
Bone mRNA Expression Analysis
Hindlimbs were removed from young (10.4 ± 0.7 week; mean ± SD) and aged (92.6 ± 1.5 week) female Col3+/+ and Col3+/− mice (N=3-4 for each group), and muscle was removed from the femora and tibiae. The bones were stored in RNAlater (Life Technologies, Carlsbad, CA) at −20°C. Before RNA extraction, bones were flash frozen in liquid nitrogen and ground with a mortar and pestle cooled with dry ice. Total RNA was isolated from the resultant powdered tissue using QiaShredder and RNeasy Mini kits (Qiagen, Venlo, Netherlands). RNA concentration was quantified, and 300ng was subjected to single-strand reverse transcription using the Superscript III First-Strand Synthesis System (Life Technologies). The resultant cDNA was utilized for real-time PCR with oligonucleotides that were specific for Col3 (5’-cacagcagtccaacgtagatgaat-3’, 5’-tgacatggttctggcttcca-3’), type I collagen (Col1; 5’-aatggtgctcctggtattgc-3’, 5’-ggcaccagtgtctcctttgt-3’), bone sialoprotein (BSP; 5’-aagaagaggaagaggaagaaaatgagaacga-3’, 5’-gcttcttctccgttgtctcc-3’), osteocalcin (OCN; 5’-cgctctgtctctctgacctc-3’, 5’-tcacaagcagggttaagctc-3’) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH; 5’-ctacactgaggaccaggttgtct-3’, 5’-ggtctgggatggaaattgtg-3’), using the 7500 Fast Real-Time PCR System, and Power SYBR Green (Life Technologies). Each sample was analyzed in duplicate, and the resulting data were averaged.
Micro-computed tomography
The quantitative analysis of trabecular bone at the distal femoral metaphysis was performed using a vivaCT 40 system (Scanco Medical http://www.scanco.ch/, Switzerland) at a voxel resolution of 10μm per slice and at least 100 slices were morphed. Each scan was performed at 55 keV and 145 μA with an integration time of 200 μs. Images were thresholded to distinguish bone from non-bone voxels using a custom edge-detection algorithm. The region of interest analyzed for each femur was 20 slices obtained 50μm (5 slices) from the primary spongiosum in the metasphyseal region. Morphometric parameters defining trabecular bone mass and microarchitecture, including bone volume/total volume (BV/TV), trabecular thickness (Tb.Th.) and separation (Tb.S), structural model index (S.M.I.) and connectivity density (Conn.D) were calculated.
Cranial morphometric analysis
Three-dimensional reconstructions of skull μCT images were used to obtain distances between previously established developmental craniofacial landmarks [22]. Briefly, the distance between 1) the nasion to the bregma, 2) the nasion to the intersection of parietal and interparietal bones, 3) the joining of the squamosal body to the zygomatic process of the squamosal and the intersection of the parietal, temporal and occipital bones, and 4) the distance between the intersection of the parietal, temporal and occipital bones on the right and left side of the skull were calculated in 6-week old wild-type and heterozygote mice.
Mesenchymal progenitor isolation and alkaline phosphatase and mineralization assays
Mesenchymal progenitors were isolated from e18.5 embryos as previously described [23, 24] to allow comparison of differentiation capacity of Col3 wild-type and null cells. Cells were expanded in culture to generate sufficient numbers. Subconfluent cultures were passaged and replated at a density of 1.8 × 104 cells/cm2 in DMEM supplemented with 10% fetal bovine serum (FBS; Atlanta Biologicals, Inc., Lawrenceville, GA), 100 U/ml penicillin, 100 μg/ml streptomycin (Sigma, St. Louis, MO, USA) and 100 μg/ml of L-ascorbic acid 2-phosphate (Asc; Sigma, St Louis, MO, USA). The cells were incubated in this growth media at 37°C in a humidified 5% CO2 atmosphere, as previously described [25, 26]. Early passage cells were utilized (<P10) for all experiments. Cells were isolated from five different embryos of each genotype for analysis.
Alkaline phosphatase (AP) activity assays were performed as previously described [26]. Briefly, cells were seeded in growth media at an initial plating density of 3.4 ×104 cells/cm2. After 24 hours, growth media was replaced with fresh media with or without 100 ng/ml of BMP6 (R&D Systems, Minneapolis, MN, USA) and 100 μg/ml of Asc. The media was changed every 2-3 days. Six days after induction, cultures were harvested and assayed for AP activity, which was normalized to the number of viable cells per well as determined by the use of the MTS assay as previously described[26].
For longer term osteogenic cultures, cells were plated at a slightly lower initial density (0.54 ×104 cells/cm2). Staining with Alizarin red S was performed on day 14 after osteogenic induction. For staining, cells were washed with PBS once, rinsed with 50% EtOH for 3 minutes, stained with 1% w/v Alizarin red S in 0.01% ammonium hydroxide for 10 minutes at room temperature, and washed with deionized water three times. Images of control and osteogenic cultures were obtained prior to semi-quantitative analysis of mineralization, as described previously [27]. Briefly, Alizarin red S was released from the cell matrix by incubating with 10% cetylpyridinium chloride in 10 mM sodium phosphate (pH 7.0) for 30 minutes (Sigma). The concentration of released Alizarin red S was determined by measuring the absorbance at 562 nm with Varioskan Flash (Thermo, West Palm Beach, FL).
Assessment of osteoclast numbers
Osteoclasts were identified by positive staining for tartrate resistant acid phosphatase (TRAP) activity using the Acid Phosphatase, Leukocyte (TRAP) kit (Sigma), following the manufacturer's instructions. Histologic sections were obtained from an unpublished simple closed transverse tibial fracture study in young adult female Col3+/+ (N=7) and +/− (N=6) mice. Bones had been fixed in 4% PFA and subsequently decalcified in 15% formic acid overnight, then processed for paraffin embedding and sectioned. Paraffin was removed with xylene, and sections were rehydrated in decreasing concentrations of ethanol. For each sample, osteoclasts were counted in one 200X field of the proximal tibia, not including the growth plate or associated with an intrameduallary pin tract, and the bone surface perimeter was traced and quantified using ImageJ.
Data analysis
Values are expressed as mean ± standard deviation (SD) in the text and figures, unless otherwise stated. Unpaired student's t-tests were used to determine the significance of differences between genotypes. Study groups were compared utilizing SigmaPlot software (Systat Inc, Chicago, IL). Nonparametric analyses utilized the Shapiro-Wilk test for normality and the Mann-Whitney Rank Sum test. P-values <0.05 were considered statistically significant.
Results
Consistent with previous reports, we observed rare (<1%) survival of Col3−/− mice beyond the early perinatal period. In contrast, Col3+/− mice survived and were weaned without incident and grew at a rate comparable to wild-type mice (mean ± SD weight of female Col3+/+ and +/− mice at 10 weeks: 20.87 ± 1.54 g and 19.78 ± 0.84 g, respectively). As previously reported for other tissues, Col3 expression in bones of young Col3+/− mice was approximately half of that found in wild-type mice (Fig. 1). However, a dramatic reduction in Col3 mRNA expression occurs in aged mice, consistent with the decreased Col3 described in other tissues during aging [28-32]. Mice of both genotypes were found to have Col3 mRNA expression at levels of <10% of that seen in young wild-type mice.
Figure 1. Col3 expression in Col3+/− bones is approximately one-half the amount seen in wild-type (Col3+/+) bones of young adult mice and Col3 expression declines with age.
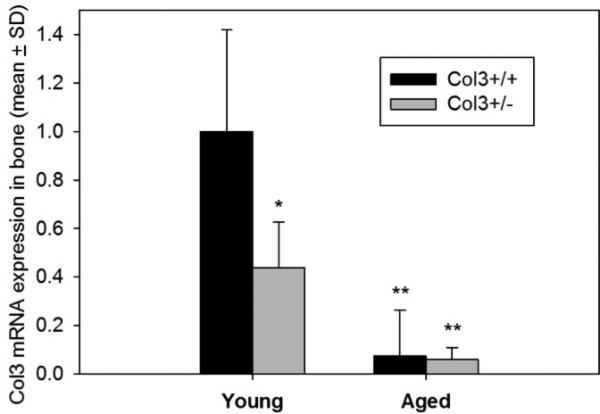
Quantitative real-time RT-PCR was performed on cDNA from young (8-12 week) and aged (>89 week) Col3+/+ and Col3+/− femora and tibiae. Col3 expression is presented relative to that in young Col3+/+ mice. N=4 for each group except young Col3+/+ (N=3). GAPDH expression was used as the endogenous control. Col3 expression values in young Col3+/− and aged Col3+/+ and +/− mice are significantly different from young Col3+/+ values at *p<0.05 and **p<0.01 via 1-way ANOVA followed by a Tukey post-hoc test.
Although Col3 heterozygous mice were initially reported to be phenotypically normal, more recent reports suggest that Col3 haploinsufficient mice develop pathology consistent with that seen in human patients with vascular EDS [33]. As facial dysmorphy is a common clinical feature of vascular EDS in human patients, we quantitatively assessed craniofacial phenotype in young adult Col3+/+ and Col3+/− littermates. This technique has previously been used to establish parallels in craniofacial maldevelopment between murine models and their respective human counterparts [22]. Four distinct linear measurements in the rostro-caudal and medio-lateral axes were obtained using the rendered μCT images from Col3+/+ and Col3+/− mice (Fig. 2A-C). Rostro-caudal length between either the nasion (#1 in Fig. 2A) or the bregma (#2) and the intersection of the parietal and interparietal bones (#3) were not significantly different between mice of either genotype. Additionally, no differences in length were found between the joining of the squamosal body to zygomatic process of squamosal (#4) and the intersection of parietal, temporal and occipital bones (#5). Furthermore, there was no difference in mediolateral length from the intersection of the parietal, temporal and occipital bones on the left side of the skull to this same point on the right (#5-6). Although no differences were noted in the cranial morphometrics analyzed in Col3 haploinsufficient mice and their wild-type counterparts, it was noted that the skull of a Col3−/− mouse, which survived to adulthood, appeared shortened in the craniocaudal direction (Fig. 2A). Also of particular interest was the finding of altered morphology and density of the mandibular condyle in the Col3−/− mouse (Fig. 2C), consistent with temporomandibular joint disease found in human patients with vascular EDS [17].
Figure 2. Effects of Col3 deficiency on craniofacial morphometry.
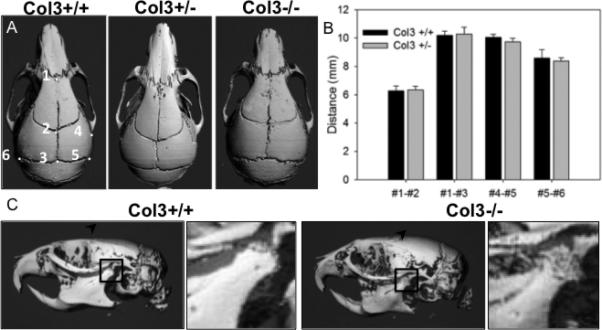
A, Dorsal views of three-dimensional reconstructions of skull μCT images of 6 week old Col3+/+ and Col3+/− mice were used to obtain distances between previously established craniofacial landmarks, labeled 1-6 in the panel on the left [21]. The skull of a Col3−/− mouse appears shortened in the craniocaudal direction. B, Quantitation of measurements comparing craniofacial morphometry of Col3+/+ and Col3+/− mice (N=5, each group). C, Lateral views of skulls of 6 week old Col3+/+ and Col3−/− mice. The Col3−/− mouse displays altered surface of the temporomandibular joint (inset showing magnified view of mandibular condyle within boxed region).
Col3-deficient mice have significantly reduced trabecular bone
As stated above, survival of Col3−/− mice to adulthood was extremely rare. In light of a previous report of juvenile osteoporosis in a young patient with vEDS[19], we investigated the effect of Col3 deficiency on bone mass in two available Col3−/− mice that survived until 6 weeks of age (one male, one female) and compared them to gender and age-matched littermates (Fig. 3). The μCT images show a dramatic decrease in trabecular bone mass in the distal femur of young adult Col3−/− mice compared to wild-type littermates. Heterozygous mice appeared to have an intermediate phenotype. In contrast to the noted changes in trabecular geometry, no significant differences in cortical bone geometry were noted between adult female Col3+/+ and Col3+/− mice (data not shown). In addition, femoral length was not significantly different between young adult female Col3+/+ and Col3+/− mice (14.1±0.94 vs 13.71±1.28 mm, respectively).
Figure 3. The effect of Col3 deficiency on trabecular bone density appears dose-dependent.
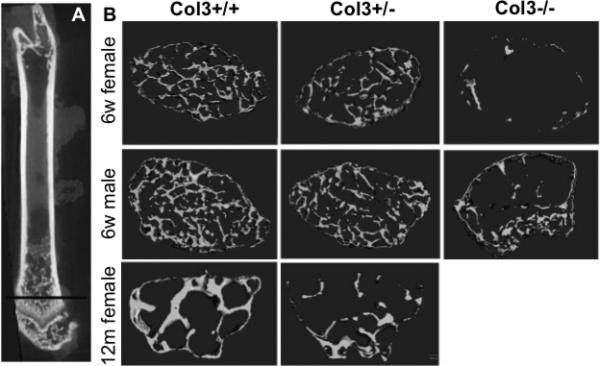
A, Longitudinal section through a femur demonstrating the plane of optical section used for sections in Panel B. Representative μCT scans of distal femurs of 6 week old Col3 +/+, Col3 +/− and Col3−/− mice of both genders and nulliparous 12 month old female Col3+/+ and +/− mice.
To further define the effects of Col3 deficiency on appendicular skeletal modeling and remodeling, we used microcomputed tomography (μCT) to assess distal trabecular bone of femurs isolated from both young and middle-aged (12 month old) nulliparous female Col3+/− mice. μCT morphometric analysis revealed a significant reduction in bone volume (BV) and bone fraction volume (BV/TV) in both young and aged Col3 +/− mice relative to controls (Fig.3B and Table 1).
Table 1. Trabecular bone structural parameters are altered by Col3 deficiency.
Microcomputed tomography was used to assess quantitative parameters of trabecular bone structure in young and aged female mice. Data are displayed as the mean ± SD. Significant differences between Col3+/+ and Col3+/− mice are indicated
| Female 6-8w | Female >ly | |||
|---|---|---|---|---|
| Col3+/+ (N=8) | Col3+./− (N=10) | Col 3+/+ (N=6) | Col3+/− (N=6) | |
| BV | 0.04±0.01 | 0.03±0.01* | 0.04±0.01 | 0.02±0.01* |
| TV | 0.33±0.02 | 0.31±0.01 | 0.34±0.05 | 0.32±0.05 |
| BV/TV | 0.12±0.02 | 0.08±0.02* | 0.12±0.05 | 0.07±0.02* |
| Conn. D. | 246.04±71.38 | 120.49±79.90* | 122.65±99.02 | 181.37±246.07 |
| SMI | 2.92± 0.09 | 3.27±0.20** | 2.13±0.46 | 2.74±0.54 |
| Tb.Th. | 0.033±0.0017 | 0.030±0.003* | 0.05±0.01 | 0.04±0.01 |
In addition, the connectivity density (Conn.D.) was decreased in Col3 heterozygous samples from young female mice, suggesting that trabeculae are less connected (Fig. 3 and Table 1). As expected, a reduction in Conn.D. was seen in older wild-type mice compared to younger mice, although no significant differences in Conn.D. were observed between older female mice of the two genotypes.
The structural model index (SMI) was also significantly increased in distal femurs of the young heterozygote female mice compared to that in wild-type mice (Table 1), suggesting the trabecular structure in Col3 haploinsufficient mice is more perforated or rod-like (Table 1). Although not statistically significant (p=0.06), a similar trend was seen in aging mice (>1 year). A small, but significant reduction in trabecular thickness (Tb.Th.) in young Col3+/− female mice compared to wild-type littermates was also found. An increase in Tb.Th. was noted in the older group of female mice compared to younger mice, although no significant effect of Col3 on Tb.Th. was seen in female mice greater than one year of age. Similar trends were noted for BV/TV, Conn.D., SMI and Tb.Th. in a small number of young male mice examined (N=4 for each genotype); however, these values were not statistically significant. As these data were not sufficiently powered, conclusions regarding the effect of gender in modulating Col3 effects in bone cannot be made. The decision to pursue larger numbers of female mice was based upon the established relationship between females and osteoporosis and the practical reason that female mice could be aged in grouped housing with greater ease (lack of fighting).
Osteoblastogenesis is reduced in mesenchymal progenitors from Col3-deficient mice
Previous studies have suggested that Col3 plays a positive role in regulating osteoblastogenesis[15] To explore potential mechanisms by which an absence of Col3 may regulate bone mass, we compared the effect of Col3 on osteogenesis and osteoclast activity. To first investigate the hypothesis that Col3 is critical for osteogenesis, we examined the ability of wild-type and Col3-deficient mesenchymal progenitors to undergo osteogenesis and produce a mineralized matrix in vitro. Mesenchymal progenitors from five Col3+/+ and Col3−/− embryos each were utilized for assessing alkaline phosphatase (AP) activity and mineralization in longer-term cultures (day 14), as described in Materials and Methods. As anticipated, cultures of wild-type mesenchymal progenitors exposed to appropriate inducers had high levels of alkaline phosphatase activity (Fig. 4A). There were not significant differences in MTS activity (used to normalize cell numbers between cultures) between cells of the two genotypes (data not shown). Although AP activity was significantly elevated in Col3−/− mesenchymal progenitor cultures under osteogenic conditions compared to non-inducing conditions, the response of these cells was dramatically diminished (<10%) compared to that seen in cultures of Col3+/+ cells (p<0.02). Similarly, while Col3+/+ mesenchymal progenitors underwent efficient mineralization when cultured in the presence of appropriate inducers, cultures of Col3−/− cells under osteogenic conditions exhibited relatively poor mineralization, as evidenced by Alizarin red staining (Fig. 4B). When Alizarin red was extracted and quantitated, there was a significant reduction (p<0.01) in mineralization of cultures of Col3−/− cells relative to those of Col3+/+ cells (Fig. 4C). The lack of Col3 in the matrix does not appear to prevent mineralization completely, however, as cultures of Col3−/− cells do show an increase in mineralization under osteogenic conditions, albeit a much smaller increase than wild-type cells. Next, we examined expression of the osteogenic markers type I collagen (Col1), bone sialoprotein (BSP), and osteocalcin (OCN) in our young and aged heterozygote mice (Fig. 4D). Reduction in all three markers is noted in Col3+/− mice relative to wild-type mice, with a further decline evident with age. Finally, to determine whether decreased trabecular bone density could be mediated by increased osteoclast activity in Col3 deficient mice, TRAP was performed on the distal proximal tibia of young adult female Col3+/+ and Col3+/− mice. Interestingly, these data revealed a small but significant decrease in osteoclast frequency in Col3+/− mice compared to Col3+/+ (Fig. 5; p<0.05).
Figure 4. Osteoblastogenesis is diminished in mesenchymal progenitors harvested from Col3−/− embryos relative to Col3+/+ embryos.
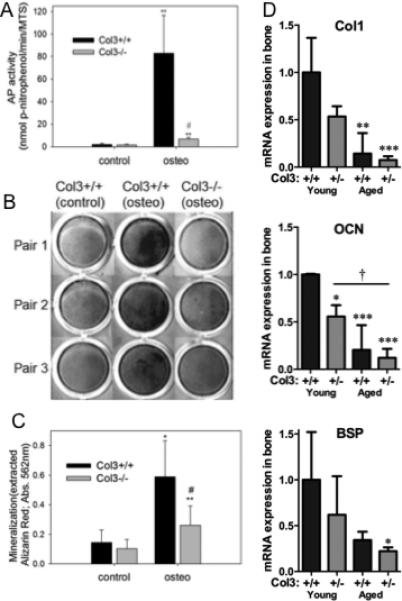
A. Following 6 days of osteogenic induction, AP activity was determined and normalized on the basis of cell number. Comparison of AP activity normalized to cell number revealed a significant decrease of AP in Col3-deficient mesenchymal progenitors relative to wild-type cells (**p<0.01, between control and osteogenic values; #p<0.01, between Col3+/+ and −/− under osteogenic conditions). B. Alizarin red S staining of mineralized matrix in representative cultures of mesenchymal progenitors from Col3+/+ and Col3−/− embryos under non-inducing and under osteogenic conditions. Col3+/+ cells cultured under osteogenic conditions show deposition of a richly mineralized matrix in contrast to cultures of donor cells from Col3−/− embryos which deposit minimal amount of Alizarin red S positive matrix. C. Quantitative analysis of Alizarin red S extracted from the matrix of control and osteogenic cultures of mesenchymal progenitors harvested from Col3+/+ and −/− embryos reveals a significant effect of Col3 on matrix mineralization (*p<0.05; **p<0.01 control vs. osteogenic conditions; #p<0.05 Col3+/+ vs Col3−/−). D. Quantitative real-time RT-PCR was performed on cDNA from young and aged Col3+/+ and Col3+/− femora and tibiae (samples identical to those used in Figure 1 to establish Col3 expression). Type I collagen (Col1), osteocalcin (OCN), and bone sialoprotein (BSP) expression is presented relative to that in young Col3+/+ mice (N=3-4 for each group). GAPDH expression was used as the endogenous control. For comparisons to young Col3+/+ mice,*p<0.05 and **p<0.01, and comparisons between other groups, †p<0.05, via 1-way ANOVA followed by a Tukey post-hoc test.
Figure 5. Diminished osteoclast density in Col3+/− bones relative to Col3 +/+ mice.
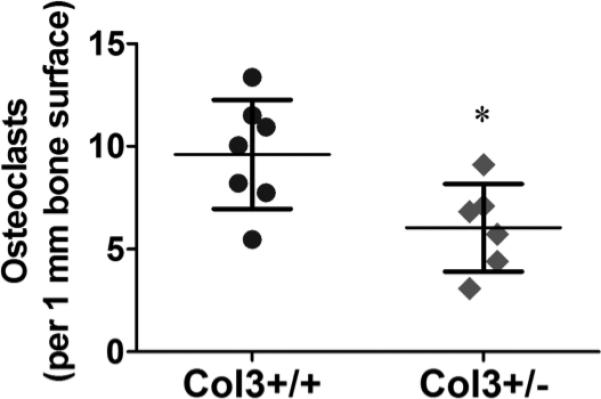
Osteoclast number was analyzed by positive TRAP staining in young female Col3+/+ (N=7) and +/− (N=6) proximal tibiae. Osteoclast number was normalized to the length of bone surface within the image field. Comparison of osteoclast density revealed a significant decrease in these cells in Col3-deficient bones relative to wild-type mice (*p<0.05, between Col3+/+ and +/− mice).
Discussion
The ECM plays an essential role in directing cellular activities during development, maintenance and repair of the skeletal system in part through its ability to act as a scaffold. In addition, the ECM also directly regulates cell behaviors such as migration, differentiation, proliferation, and survival through its ability to engage adhesion receptors, as well as its ability to regulate growth factor bioavailability and signaling [34]. More recently, a key role for mechanical control of cell fate through interaction with the ECM has emerged [35]. Col3 has been suggested to play important roles in development and in tissue repair and regeneration, due to its increased expression in embryonic relative to post-natal tissues, as well as its increased expression early in the repair process of a variety of tissues and organs [36-38].
Despite the fact that Col3 is among the most abundant collagens in tissues, relatively little is known about how this collagen directs key aspects of development and tissue maintenance and repair. A Col3-deficient mouse, which recapitulates many features of vascular EDS in humans, has been used by several investigators to examine the role of Col3 in these processes. Liu et al had previously established a critical role for Col3 in cardiovascular development through its ability to regulate type I collagen fibrillogenesis [20]. More recently, Col3 was shown to contribute to maintenance of vascular integrity, as the aortas of aged Col3 heterozygote mice develop a spectrum of lesions similar to those observed in human vascular EDS patients [33], in a process that appears to be regulated by increased tissue gelatinase activity [39]. Finally, Col3 has been shown to regulate proper lamination of the cerebral cortex by its ability to control neuronal migration in the developing brain through its interaction with the G-protein coupled receptor, GPR56 [40].
Col3 has been shown to be present in the mesenchymal condensations that precede chondrogenesis and osteogenesis [14] and in marrow stroma that serves as a reservoir of osteoprogenitor cells [41-43]; however, whether Col3 is present in mature bone is controversial. Early studies reported that Col3 disappears altogether from mature bone [44, 45]; however, recent studies with more sensitive techniques have reported that Col3 persists into maturity in trabecular bone [42] and as extensions of fibers connecting periosteum to bone [12, 46, 47]. Col3 expression in non-osseous tissues has been shown to decline with age; this decline has been suggested to correlate with decreased regenerative capacity [28-32]. These results are consistent with our expression data showing a significant reduction in bone Col3 with advanced age (Fig. 1). An approximate 50% reduction in Col3 expression in the bone of age-matched young heterozygote mice is consistent with previously reported Col3 expression in other tissues in these mice [20, 33, 48].
Although the major clinical signs that lead to diagnosis of vascular EDS include rupture of large vessels and the gastrointestinal or urogenital tracts, individuals with this condition have also been described to display numerous skeletal manifestions, including osteoporosis, as well as a distinctive facial appearance, spinal deformity, scoliosis and temporal mandibular disorder [17-19]. Distinctive facial features in vascular EDS patients include thin lips and philtrum, small chin, thin nose, and large eyes [49]. The skull of one Col3 homozygous null (Col3−/−) mouse was available that suggested shortening in the cranial-caudal direction (Fig. 2); however, the small number of null mice surviving to weaning precluded statistical analysis. Craniofacial morphometrics using four previously defined measurements for establishing parallels in craniofacial maldevelopment between murine models and their respective human counterparts failed to show any differences between Col3+/+ and Col3+/− littermates, suggesting either that the same features are not evident in this murine model or that the haploinsufficient state is not severe enough to cause morphometric abnormalities in these craniofacial bones. Of interest was the noted defect in the surface of the mandibular condyle in one Col 3−/− mouse (Fig. 2C), since temporomandibular joint (TMJ) disorder is commonly found in EDS patients, including those with the vascular form of the disease [17]. In addition to being a component of bone, Col3 is also a constituent of the collagenous matrix of the other structural components of the TMJ, including the fibrocartilage, supporting ligaments, disc and retrodiscal tissues [17].
Although craniofacial dysmorphism associated with Col3 deficiency was not established in Col3 +/− mice, the finding of diminished trabecular bone geometry in young mice (Fig. 3, Table 1) correlates with a previous report of juvenile osteoporosis in a 10-year-old vascular EDS patient [19]. Reduction in Col3 has previously been associated with an osteoporotic state [50], although whether Col3 deficiency contributed to or was the result of bone loss was not established. A significantly diminished bone volume fraction (BV/TV) in Col3+/− mice compared to wild-type littermates by 6-8 weeks of age suggests that Col3 plays an important role in establishing early accumulation of bone. Significant reduction in bone volume fraction was sustained in mice as they aged (over one year). A previous study examining bone density in Balb/c mice established that a significant decline in bone volume fraction is not seen until 20 months of age [51]. Because the older mice used in this study were of middle-age (~one year), conclusions cannot be drawn whether Col3 deficiency exacerbates age-associated osteoporosis. The distal femurs from only two Col−/− mice were available for analysis; however, the severity of trabecular bone loss evident on microCT suggests that the negative impact of Col3 loss on trabecular bone density is dose-dependent (Fig. 3).
In contrast to the effect of Col3 on trabecular bone density, there were no apparent differences in cortical bone geometry, at the level of the femoral mid-diaphysis, between Col3+/+ and Col3+/− mice (data not shown). Since cortical bone forms by a process resembling intramembranous bone formation, these results suggest that intramembranous bone is less severely affected than bone that is formed via endochondral ossification in Col3-deficient mice. This interpretation is also supported by the lack of evidence of significant craniofacial abnormalities in Col 3 +/− mice, since the majority of craniofacial bones develop via an intramembranous process. It should be noted that calvarial bone density was not assessed in our study.
Our results suggest that Col3 plays a role in trabecular bone formation and maintenance through its direct effect on osteogenesis, as supported by our in vitro and in vivo analysis of osteogenesis (Fig. 4). Furthermore, our data examining osteoclast numbers by TRAP staining did not reveal a proresorption phenotype associated with Col3 loss. In fact, the small but significant decrease in osteoclast numbers in Col3+/− is likely a result of diminished osteogenesis as these processes are well-established to be coupled[52]. Type III collagen has been shown to play a structural role in tissues [20], and the severity of the defects observed in skeletal development in the complete absence of Col3 could be due to altered collagen fibril architecture. However, two additional mechanisms by which Col3 may regulate cell fate and activity in tissue repair and maintenance have been proposed and are of relevance to bone, including regulation of bioavailability of members of the TGFβ family of growth factors, including bone morphogenetic proteins (BMPs) [21, 53, 54] or integrin expression [55]. Either or both of these mechanisms could alter the fate of mesenchymal progenitor cells, for example, by blocking their differentiation at an early stage or inducing an alternative differentiation path.
Acknowledgements
This work was supported by grants from the University of Pennsylvania Research Foundation and the Penn Center for Musculoskeletal Disorders (SLA and SWV) and the National Institutes of Health (K08AR053945 to SWV and RO1 AR044692 to SLA).
Footnotes
Disclosure of Potential Conflicts of Interest: The authors indicate no potential conflicts of interest.
References
- 1.Shen H, Recker RR, Deng HW. Molecular and genetic mechanisms of osteoporosis: implications for treatment. Curr Mol Med. 2003;3:737–757. doi: 10.2174/1566524033479375. [DOI] [PubMed] [Google Scholar]
- 2.Oot SG, editor. Bone Health and osteoporosis: a report of the Surgeon-General. U.S. Department of Health and Human Services; Rockville, MD: 2004. [Google Scholar]
- 3.Burge R, Dawson-Hughes B, Solomon DH, King A, Tosteson A. Incidence and economic burden of osteoporosis-related fractures in the United States, 2005-2025. J Bone Miner Res. 2007;22:465–475. doi: 10.1359/jbmr.061113. [DOI] [PubMed] [Google Scholar]
- 4.Franceschi RT. The developmental control of osteoblast-specific gene expression: role of specific transcription factors and the extracellular matrix environment. Crit Rev Oral Biol Med. 1999;10:40–57. doi: 10.1177/10454411990100010201. [DOI] [PubMed] [Google Scholar]
- 5.Ge C, Yang Q, Zhao G, Yu H, Kirkwood KL, Franceschi RT. Interactions between extracellular signal-regulated kinase 1/2 and p38 MAP kinase pathways in the control of RUNX2 phosphorylation and transcriptional activity. J Bone Miner Res. 2012;27:538–551. doi: 10.1002/jbmr.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Epstein EH., Jr (Alpha1(3))3 human skin collagen. Replease by pepsin digestion and preponderance in fetal life. J Biol Chem. 1974;249:3225–3331. [PubMed] [Google Scholar]
- 7.Smith LT, Holbrook KA, Byers PH. Structure of the dermal matrix during development and in the adult. J Invest Dermatol. 1982;79:93s–104s. doi: 10.1111/1523-1747.ep12545877. [DOI] [PubMed] [Google Scholar]
- 8.Tolstoshev P, Haber R, Trapnell BC, Crystal RG. Procollagen mRNA levels and activity and collagen synthesis during the fetal development of sheep lung, tendon and skin. J Biol Chem. 1981;256:9672–9679. [PubMed] [Google Scholar]
- 9.Birk DE, Mayne R. Localization of collagen types I, III, and V during tendon development. Eur J Cell Biol. 1997;72:352–361. [PubMed] [Google Scholar]
- 10.Henkel W, Glanville RW. Covalent crosslinking between molecules of type I and type III collagen. Eur J Biochem. 1982;122:205–231. doi: 10.1111/j.1432-1033.1982.tb05868.x. [DOI] [PubMed] [Google Scholar]
- 11.Keene DR, Sakai LY, Bachinger HP, Burgeson RE. Type III collagen can be present on banded collagen fibrils regardless of fibril diameter. J Cell Biol. 1987;105:2392–2402. doi: 10.1083/jcb.105.5.2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Keene DR, Sakai LY, Burgeson RE. Human bone contains type III collagen, type VI collagen and fibrillin. J Histochem Cytochem. 1991;38:59–69. doi: 10.1177/39.1.1983874. [DOI] [PubMed] [Google Scholar]
- 13.Reddi AH, Gay R, Gay S, Miller EJ. Transition in collagen types during matrix-induced cartilage, bone and bone marrow formation. Proc Natl Acad Sci USA. 1977;74:5589–5592. doi: 10.1073/pnas.74.12.5589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Silver MH, Foidart JM, Pratt RM. Distribution of fibronectin and collagen during mouse limb and palate development. Differentiation. 1981;18:141–149. doi: 10.1111/j.1432-0436.1981.tb01115.x. [DOI] [PubMed] [Google Scholar]
- 15.Maehata Y, Takamizawa S, Ozawa S, Izukuri K, Kato Y, Sato S, Lee MC, Kimura A, Hara RI. Type III collagen is essential for growth acceleration of human osteoblastic cells by ascorbic acid 2-phosphate, a long-acting vitamin C derivative. Matrix Biology. 2007;26:371–381. doi: 10.1016/j.matbio.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 16.Chen XD, Dusevich V, Feng JQ, Manolagas SC, Jilka R. Extracellular matrix made by bone marrow cells facilitates expansion of marrow-derived mesenchymal progenitor cells and prevents their differentiation into osteoblasts. J Bone Miner Res. 2007;22:1943–1956. doi: 10.1359/jbmr.070725. [DOI] [PubMed] [Google Scholar]
- 17.De Coster PJ, Martens LC, De Paepe A. Oral health in prevalent types of Ehlers-Danlos syndromes. J Oral Pathol Med. 2005;34:298–307. doi: 10.1111/j.1600-0714.2004.00300.x. [DOI] [PubMed] [Google Scholar]
- 18.Stanitski DF, Nadjarian R, Stanitski CL, Bawle E, Tsipouras P. Orthopaedic manifestations of Ehlers-Danlos syndrome. Clin Orthop Rel Res. 2000;376:213–221. doi: 10.1097/00003086-200007000-00029. [DOI] [PubMed] [Google Scholar]
- 19.Yen JL, Lin SP, Chen MR, Niu DM. Clinical features of Ehlers-Danlos syndrome. Formos Med Assoc. 2006;105:475–480. doi: 10.1016/S0929-6646(09)60187-X. [DOI] [PubMed] [Google Scholar]
- 20.Liu X, Wu H, Burne M, Krane S, Jaenisch R. Type III collagen is crucial for collagen I fibrillogenesis and for normal cardiovascular development. Proc Natl Acad Sci USA. 1997;94:1852–1856. doi: 10.1073/pnas.94.5.1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Volk SW, Wang Y, Mauldin EA, Liechty KW, Adams SL. Diminished type III collagen promotes myofibroblast differentiation and increases scar deposition in cutaneous wound healing. Cells Tissues Organs. 2011;194:25–37. doi: 10.1159/000322399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Richtsmeier JT, Baxter LL, Reeves RH. Parallels of craniofacial maldevelopment in Down Syndrome and Ts65Dn Mice. Developmental Dynamics. 2000;217:137–145. doi: 10.1002/(SICI)1097-0177(200002)217:2<137::AID-DVDY1>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 23.Garreta E, GenovÇ E, Borr¢s S, Semino CE. Osteogenic differentiation of mouse embryonic stem cells and mouse embryonic fibroblasts in a three dimensional self-assembling peptide scaffold. Tiss Eng. 2006;12:2215–2227. doi: 10.1089/ten.2006.12.2215. [DOI] [PubMed] [Google Scholar]
- 24.Legner CJ, Lepper C, van Wijnen AJ, Stein JL, Stein GS, Lian JB. Primary mouse embryonic fibroblasts: a model of mesenchymal cartilage formation. J Cell Physiol. 2004;200:327–333. doi: 10.1002/jcp.20118. [DOI] [PubMed] [Google Scholar]
- 25.Volk SW, Wang Y, Hankenson KD. Effects of donor characteristics and ex vivo expansion on canine mesenchymal stem cell properties: Implications for MSC-based therapies. Cell Transplant. 2012 doi: 10.3727/096368912X636821. Epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Volk SW, Diefenderfer DL, Christopher SA, Haskins ME, Leboy PS. Effects of osteogenic inducers on cultures of canine mesenchymal stem cells. Am J Vet Res. 2005;66:1729–1737. doi: 10.2460/ajvr.2005.66.1729. [DOI] [PubMed] [Google Scholar]
- 27.Spinella-Jaegle S, Roman-Roman S, Faucheu C, Dunn FW, Kawai S, GallÇa S, Stiot V, Blanchet AM, Courtois B, Baron R, Rawadi G. Opposite effects of bone morphogenetic protein-2 and transforming growth factor-[beta]1 on osteoblast differentiation. Bone. 2001;29:323–330. doi: 10.1016/s8756-3282(01)00580-4. [DOI] [PubMed] [Google Scholar]
- 28.Benatti BB, Silverio KG, Casati MZ, Sallum EA, Nociti FH., Jr. Influence of aging on biological properties of periodontal ligament cells. Connect Tissue Res. 2008;49:401–408. doi: 10.1080/03008200802171159. [DOI] [PubMed] [Google Scholar]
- 29.Furth JJ, Allen RG, Tresini M, Keogh B, Cristofalo VJ. Abundance of alpha 1(I) and alpha 1(III) procollagen and p21 mRNAs in fibroblasts cultured from fetal and postnatal dermis. Mech Ageing Dev. 1997;97:131–142. doi: 10.1016/s0047-6374(97)00051-1. [DOI] [PubMed] [Google Scholar]
- 30.Mays PK, Bishop JE, Laurent GJ. Age-related changes in the proportion of types I and III collagen. Mech Ageing Dev. 1988;45:203–212. doi: 10.1016/0047-6374(88)90002-4. [DOI] [PubMed] [Google Scholar]
- 31.Takeda K, Gosiewska A, Peterkofsky B. Similar, but not identical, modulation of expression of extracellular matrix components during in vitro and in vivo aging of human skin fibroblasts. J Cell Phys. 1992;153:450–459. doi: 10.1002/jcp.1041530303. [DOI] [PubMed] [Google Scholar]
- 32.Varani J, Dame MK, Rittie L, Fligiel SE, Kang S, Fisher GJ, Voorhees JJ. Decreased collagen production in chronologically aged skin: roles of age- dependent alteration in fibroblast function and defective mechanical stimulation. Am J Pathol. 2006;168:1861–1868. doi: 10.2353/ajpath.2006.051302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cooper TK, Zhong Q, Krawczyk M, Tae HJ, MÅller GA, Schubert R, Myers LA, Dietz HC, Talan MI, Briest W. The haploinsufficient Col3a1 mouse as a model for vascular Ehlers-Danlos Syndrome. Vet Pathol. 2010;47:1028–1039. doi: 10.1177/0300985810374842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schultz GS, Davidson JM, Kirsner RS, Bornstein P, Herman IM. Dynamic reciprocity in the wound microenvironment. Wound Rep Reg. 2011;19:134–148. doi: 10.1111/j.1524-475X.2011.00673.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leight JL, Wozniak MA, Chen S, Lynch ML, Chen CS. Matrix rigidity regulates a switch between TGF-beta 1 induced apoptosis and epithelial mesenchymal transition. Mol Biol Cell. 2012;23:677–689. doi: 10.1091/mbc.E11-06-0537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hurme T, Kalimo H, Sandberg M, Lehto JM, Vuorio E. Localization of type I and III collagen and fibronectin production in injured gastrocnemius muscle. Lab Invest. 1991;64:76–84. [PubMed] [Google Scholar]
- 37.Liu SH, Yang RS, Al-Haikh R, Lane JM. Collagen in tendon, ligament and bone healing. Clin Orthop Rel Res. 1995;318:265–278. [PubMed] [Google Scholar]
- 38.Merkel JR, DiPaolo BR, Hallock GG, Rice DC. Type I and type III collagen content of healing wounds in fetal and adult rats. Proc Soc Exp Biol Med. 1988;187:493–497. doi: 10.3181/00379727-187-42694. [DOI] [PubMed] [Google Scholar]
- 39.Briest W, Cooper TK, Tae HJ, Krawczyk M, McDonald DM, Talan MI. Doxycycline ameliorates the susceptibility to aortic lesions in a mouse model for the vascular type of Ehlers-Danlos Syndrome. J Pharmacol Exp Ther. 2011;337:621–627. doi: 10.1124/jpet.110.177782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Luo R, Jeong SJ, Jin Z, Strokes N, Li S, Piao X. G protein-coupled receptor 56 and collagen III, a receptor-ligand pair, regulates cortical development and lamination. Proc Natl Acad Sci USA. 2011;108:12925–12930. doi: 10.1073/pnas.1104821108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Apaja-Sarkkinen M, Autio-Harmainen H, Alavaikko M, Risteli J, Risteli L. Immunohistochemical study of basement membrane proteins and type III procollagen in myelofibrosis. Br J Haematol. 1986;63:571–580. doi: 10.1111/j.1365-2141.1986.tb07535.x. [DOI] [PubMed] [Google Scholar]
- 42.Becker J, Schuppan D, Benzian H, Bals T, Hahn EG, Cantaluppi C, Reichart P. Immunohistochemical distribution of collagens type IV, V, and VI and of pro-collagens types I and III in human alveolar bone and dentine. J Histochem Cytochem. 1986;34:1417–1429. doi: 10.1177/34.11.3772076. [DOI] [PubMed] [Google Scholar]
- 43.Bentley S, Alabaster O, Foidart JM. Collagen heterogeneity in normal human bone marrow. Br J Haematol. 1981;48:287–291. [PubMed] [Google Scholar]
- 44.Miller E. A review of biochemical studies on the genetically distinct collagens of the skeletal system. Clin Orthop Rel Res. 1973;92:260–280. doi: 10.1097/00003086-197305000-00024. [DOI] [PubMed] [Google Scholar]
- 45.Muller P, Raisch K, Matzen K, Gay S. Presence of type III collagen in bone from a patient with osteogenesis imperfecta. Eur J Pediatrics. 1977;125:29–37. doi: 10.1007/BF00470603. [DOI] [PubMed] [Google Scholar]
- 46.Carter D, Sloan P, Aaron JE. Immunolocalization of collagen types I and III, tenascin and fibronectin in intramembranous bone. J Histochem Cytochem. 1991;39:599–606. doi: 10.1177/39.5.1707904. [DOI] [PubMed] [Google Scholar]
- 47.Luther F, Saino H, Carter DH, Aaron JE. Evidence for an extensive collagen type III/VI proximal domain in the rat femur. Bone. 2003;32:652–659. doi: 10.1016/s8756-3282(03)00094-2. [DOI] [PubMed] [Google Scholar]
- 48.Stevenson K, Kucich U, Whitbeck C, Levin RM, Howard PS. Functional changes in bladder tissue from type III collagen-deficient mice. Mol Cell Biochem. 2006;283:107–114. doi: 10.1007/s11010-006-2388-1. [DOI] [PubMed] [Google Scholar]
- 49.Germain DP. Ehlers-Danlos syndrome type IV. Orphanet J Rare Dis. 2007;2:32–41. doi: 10.1186/1750-1172-2-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Balla B, K¢sa JP, Kiss J, Borsy A, Podani J, Tak†cs I, Laz†ry A, Nagy Z, B†csi K, Speer G, Orosz L, Lakatos P. Different gene expression patterns in the bone tissue of aging and postmenopausal osteoporotic and non-osteoporotic women. Calcif Tiss Intl. 2008;82:12–26. doi: 10.1007/s00223-007-9092-3. [DOI] [PubMed] [Google Scholar]
- 51.Willinghamm MD, Brodt MD, Lee KL, Stephens AL, Ye J, Silva MJ. Age- related changes in bone structure and strength in female and male BALB/c mice. Calcif Tiss Int. 2010;86:470–483. doi: 10.1007/s00223-010-9359-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Olsen BR, Reginato AM, Wang W. Bone Development. Annu Rev Cell Dev Biol. 2000;16:191–220. doi: 10.1146/annurev.cellbio.16.1.191. [DOI] [PubMed] [Google Scholar]
- 53.Oganesian A, Au S, Horst JA, Holzhausen LC, Macy AJ, Pace JM, Bornstein P. The NH2-terminal propeptide of type I procollagen acts intracellularly to modulate cell function. J Biol Chem. 2006;281:38507–38518. doi: 10.1074/jbc.M607536200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhu Y, Oganesian A, Keene DR, Sandell LJ. Type IIA procollagen containing the cysteine-rich amino propeptide is deposited in the extracellular matrix of prechondrogenic tissue and binds to TGF-beta1 and BMP2. J Cell Biol. 1999;144:1069–1080. doi: 10.1083/jcb.144.5.1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zoppi N, Gardella R, DePaepe A, Barlati S, Colombi M. Human fibroblasts with mutations in COL5A1 and COL3A1 genes do not organize collagens and fibronectin in the extracellular matrix, down-regulate α2β1 integrin, and recruit αvβ3 instead of α5β1 integrin. J Biol Chem. 2004;279:18157–18168. doi: 10.1074/jbc.M312609200. [DOI] [PubMed] [Google Scholar]