

Abstract
Background
Alkaline phosphatase (AP) catalyzes the hydrolytic cleavage of phosphate monoesters under alkaline conditions and plays important roles in microbial ecology and molecular biology applications. Here, we report on the first isolation and biochemical characterization of a thermolabile AP from a metagenome.
Results
The gene encoding a novel AP was isolated from a metagenomic library constructed with ocean-tidal flat sediments from the west coast of Korea. The metagenome-derived AP (mAP) gene composed of 1,824 nucleotides encodes a polypeptide with a calculated molecular mass of 64 kDa. The deduced amino acid sequence of mAP showed a high degree of similarity to other members of the AP family. Phylogenetic analysis revealed that the mAP is shown to be a member of a recently identified family of PhoX that is distinct from the well-studied classical PhoA family. When the open reading frame encoding mAP was cloned and expressed in recombinant Escherichia coli, the mature mAP was secreted to the periplasm and lacks an 81-amino-acid N-terminal Tat signal peptide. Mature mAP was purified to homogeneity as a monomeric enzyme with a molecular mass of 56 kDa. The purified mAP displayed typical features of a psychrophilic enzyme: high catalytic activity at low temperature and a remarkable thermal instability. The optimal temperature for the enzymatic activity of mAP was 37°C and complete thermal inactivation of the enzyme was observed at 65°C within 15 min. mAP was activated by Ca2+ and exhibited maximal activity at pH 9.0. Except for phytic acid and glucose 1-phosphate, mAP showed phosphatase activity against various phosphorylated substrates indicating that it had low substrate specificity. In addition, the mAP was able to remove terminal phosphates from cohesive and blunt ends of linearized plasmid DNA, exhibiting comparable efficiency to commercially available APs that have been used in molecular biology.
Conclusions
The presented mAP enzyme is the first thermolabile AP found in cold-adapted marine metagenomes and may be useful for efficient dephosphorylation of linearized DNA.
Keywords: Alkaline phosphatase, Metagenome, PhoX, Escherichia coli
Background
Alkaline phosphatase (AP; EC 3.1.3.1) is a ubiquitous enzyme widely distributed from microorganisms to humans [1] and functions to catalyze the hydrolytic cleavage of phosphate monoesters under alkaline conditions, releasing inorganic phosphate from many phosphate-containing compounds. This enzyme plays important roles in microbial ecology through its involvement in phosphate metabolism [2], signal transduction [3], and bacterial virulence [4]. Among bacterial APs, Escherichia coli phoA, which encodes an AP (termed ECAP), has been most extensively studied. ECAP is a homodimeric enzyme requiring two Zn2+ ions and one Mg2+ per monomer [5], and its active site has a conserved serine residue that is phosphorylated through catalysis [6]. PhoA-like APs are found in many other bacteria and have conserved domains with those in ECAP [5]. Recently, the novel AP genes phoX and phoK were identified in Vibrio cholerae [7] and Sphingomonas sp. strain BSAR-1 [8], respectively; these genes were shown to be activated by Ca2+ and share no homology with phoA.
AP is also a key enzyme in many molecular biology techniques, such as enzyme-linked immunosorbent assays [9], western blots, and immunodetection assays [10]. The favorable properties of AP for these applications are high catalytic activity and thermal instability, which allows simple inactivation by heat treatment [11]. Thus, several thermolabile APs have been isolated and characterized from psychrophilic microorganisms such as the Antarctic strain TAB5 [12], the marine bacterium Vibrio G15-21 [13], and the bacterium Shewanella sp. SIB1 [14]. Furthermore, the AP enzyme from the marine psychrophile Sphingomonas sp. strain BSAR-1 (SPAP) was found to be very efficient in bioprecipitation of uranium as uranyl phosphate under alkaline conditions [8].
Metagenomes have been successfully used for the discovery of novel biocatalysts and biomolecules for biotechnological and pharmaceutical applications [15,16]. To date, no thermolabile APs from metagenomes have been isolated or characterized. This might be due to the limited availability of sensitive and reliable screening systems, which enable the screening of large libraries of metagenome clones [17,18]. Recently, we developed a genetic circuit termed the Genetic Enzyme Screening System (GESS) that allows for the measurement of enzyme activity as in vivo fluorescence intensity and is highly useful for high-throughput metagenomic screening of enzymes [19]. The GESS is able to detect the activities of phenol-releasing enzymes, such as esterases/lipases, lyases, cellulases, and phosphatases, which have many applications in biotechnology [19].
Here, we report the first isolation and characterization of a novel thermolabile AP from metagenomes using the GESS. A metagenomic library was constructed with ocean-tidal flat sediments because they were collected from dynamic physicochemical environments with remarkable microbial diversity. We repeatedly screened approximately 106 metagenomic fosmid clones and identified one positive clone that showed significant AP activity. The metagenome-derived AP (mAP) was expressed as a soluble form with a high yield in the periplasmic space of E. coli, purified to homogeneity, characterized biochemically, and then applied to the dephosphorylation of linearized plasmid DNA.
Results
Rationale of the GESS method
We previously developed a genetic circuit, GESS, for functional screening of phenol-generating enzymes from genomes or metagenomes [19]. DmpR from Pseudomonas sp. strain CF600 acts as a transcriptional activator that can be stimulated upon binding to its effector. It regulates the expression of the divergently transcribed dmp operons, which are involved in phenol degradation, from the Po promoter in response to aromatic effector compounds (e.g., phenol and cresol) [20]. We took advantage of this regulatory system by placing the Po promoter upstream of enhanced green fluorescent protein (egfp) gene to put EGFP expression under the control of DmpR, generating a GESS for detection of phenol and its derivatives [19]. For functional screening of AP, the DmpR of GESS can be activated upon binding to p-nitrophenol (pNP), which is released from p-nitrophenyl phosphate (pNPP) hydrolysis by AP, and then turn on the egfp expression. Using this GESS, we conducted a systematic screening that allowed the high throughput in vivo detection of AP activity from metagenomes.
Isolation of an AP from a metagenome library
A metagenomic library was constructed from DNA isolated from ocean-tidal flat sediments and encompassed approximately 80,000 clones with an average insert size of 30 kb. This library was screened using pNPP as a substrate to detect AP activity. Using the strategy described in the Methods section, we discovered six clones exhibiting strong AP activity. Further screening resulted in the identification of a single positive fosmid clone, designated pFOS144C11, which carried a DNA insert of approximately 35 kb. After randomly shearing the DNA, 3 to 7 kb fragments were ligated into pSTV28 and a sublcone designated pSTV28-AP6 was identified based on fluorescence signals and AP activity. The sequence of this clone was determined (Figure 1). Among three intact ORFs in pSTV28-AP6, an ORF of 1,824 nucleotides encoding a putative AP of 607 amino acids, with a deduced molecular mass of 64 kDa, was found through a BLAST search. The deduced amino acid sequence of mAP gene was compared with other protein sequences of APs available from the NCBI database using the BLASTP program. mAP exhibited significant homology with the AP superfamily, and the level of sequence identity showed the highest similarity with nucleotide phosphodiesterases from psychrophilic Sphingopyxis sp. MC1 (85% amino acid sequence identity, GenBank accession no. WP 003042625) and Sphingopyxis alaskensis RB2256 (72% amino acid sequence identity, GenBank accession no. YP_615103). In addition, this mAP was also highly similar to APs from Sphingobium sp. SYK-6 (59% amino acid sequence identity, GenBank accession no. YP_004833865) and Sphingomonas sp. strain BSAR-1 (58% amino acid sequence identity, GenBank accession no. EF143994) [8].
Figure 1.
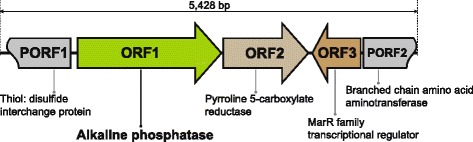
Identification of putative alkaline phosphatase from metagenome. Sequencing of subclone (pSTV28-AP6) expressing alkaline phosphatase activity resulted in the assembly of a 5,428-bp contig. Three intact ORFs (ORF1 - ORF3) and two partial ORFs (PORF1 and PORF2) with conserved domains were identified through BLAST search.
Sequence and phylogenetic analyses of mAP
A multiple-sequence alignment analysis of mAP was conducted using APs that showed high similarity with functionally characterized APs (Figure 2). Recently, the Thr89 of SPAP was found to be the catalytic residue [21]; this residue was conserved as Thr138 in mAP. In all other APs, the catalytic residue is a conserved serine (e.g., Ser102 for ECAP); thus, mAP was unique in that its catalytic residue was a threonine (Figure 2). The alignment also identified absolute conservation of six residues (D99, D397, H398, D352, H356, and W545 in mAP) of the active site between SPAP and mAP. The deduced amino acid sequence of mAP was then used for the construction of a phylogenetic tree with full-length amino acid sequences of APs from various organisms (Figure 3). Four distinct clusters were formed: (i) the PhoA cluster, including APs from E. coli, shrimp (SAP), and Antarctic bacterial TAB5 (AAP); (ii) the PhoX-I cluster containing most marine bacterial APs and the well-characterized Sinorhizobium meliloti AP (SMAP); (iii) the PhoD cluster; and (iv) the PhoX-II cluster, including functionally and structurally characterized SPAP, mAP, and mAP homologs from psychrophilic marine bacteria. The PhoX-I and PhoX-II are well-defined groups in PhoX family, mostly along phylogenetic lines [22]. PhoX-I includes a diverse group of α- and γ-Proteobacteria while PhoX-II includes the Gram-positive Actinobacteria, most the Cyanobacteria and many of the α-Proteobacteria encompassing Sphingopyxis, Sphingobium, and Sphingomonas.
Figure 2.

Multiple alignment of the deduced amino acid sequence of the mAP gene derived from the metagenome with other alkaline phosphatases (APs). APs are identified by their GenBank or PDB accession numbers: Sphingomonas sp. strain BSAR-1 AP (SPAP, ABL96598), Escherichia coli AP (ECAP, BAE76164), Vibrio sp. G15-21 AP (VAP, AAK94204), Antarctic bacterium TAB5 AP (AAP, CAB82508), shrimp AP (SAP, PDB 1SHQ), human placenta AP (HPAP, PDB 1ZEB), Shewanella sp. AP1 AP (SCAP, BAB85685), and Sinorhizobium meliloti 1021 AP (SMAP, NP 385195). Highly conserved amino acid residues are highlighted in black, and gaps are denoted by dots. Gray-shaded amino acids are conserved in at least six of the nine APs shown. The amino acid residues forming the metal binding site and catalytic residues of mAP are denoted by asterisks (★) and closed circle (●), respectively. Numbers along the sequences indicate the positions of the amino acid residues starting from the initial Met for each AP.
Figure 3.
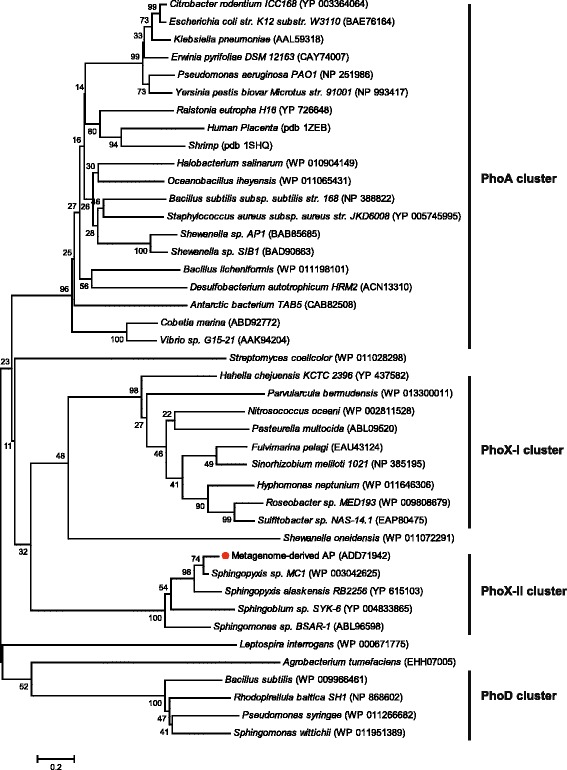
Phylogenetic analysis of mAP. A phylogenetic tree based on the similarities of full-length deduced amino acid sequences was constructed with MEGA 5.2 software using the neighbor-joining method. GenBank or PDB accession numbers are given after the species designation. Numbers at nodes are bootstrap values based on 1,000 samplings.
Expression and purification of mAP
To further characterize the mAP enzyme, the 1,824-bp mAP was cloned into the pET-21a plasmid and overexpressed in E. coli BL21(DE3). When mAP was expressed in E. coli cells at 37°C, no protein band with a calculated molecular mass of 64 kDa was detected on sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). However, when we expressed mAP at various temperatures (15°C, 20°C, or 30°C), mAP expression was increased as the temperature was decreased (Figure 4B). Interestingly, when whole cell extracts from cells expressing mAP were separated by SDS-PAGE, two bands were observed: one band having the calculated molecular mass of 64 kDa, and the other band having a lower molecular mass of approximately 56 kDa (Figure 4B). It is possible that the 56 kDa band represents the secreted and processed form from which the signal peptide cleaved. In order to test this possibility, the localization of mAP was examined when it was expressed at 15°C and 20°C. After cellular fractionation of periplasmic and cytoplasmic proteins using sucrose osmotic shock, SDS-PAGE was conducted. mAP with a molecular mass of 56 kDa was detected in the periplasmic fraction only at 15°C (Figure 4C), indicating that mAP was a cold-adapted AP. The His6-tagged mAP enzyme secreted into the periplasm of E. coli BL21(DE3) cells was purified using single-step Ni-NTA affinity chromatography (Figure 4D). The purified mAP enzyme exhibited a high specific activity of 1,528 U/mg when pNPP was used as the substrate at 37°C. Purified mAP was then used for subsequent biochemical characterization.
Figure 4.
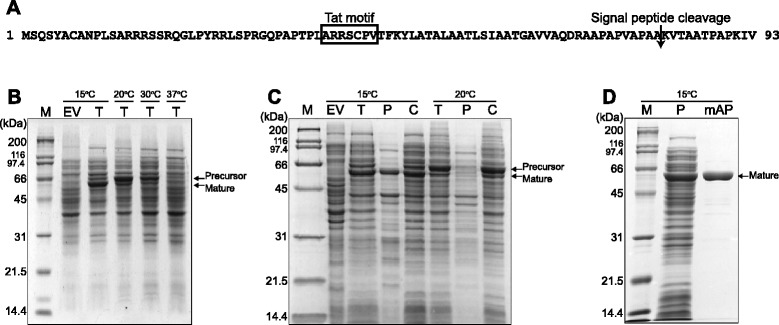
Signal peptide sequence, heterologous expression, and translocation of mAP. (A) N-terminal amino acid sequence of mAP showing the predicted Tat motif and signal peptide cleavage site at Ala81. (B) Expression of mAP in the E. coli BL21(DE3) transformant harboring pET-mAP at various temperatures from 15°C to 37°C. Lane M, protein standards; lane EV, total proteins expressed in BL21(DE3) cells with pET-21a at 15°C; lane T, IPTG-induced total proteins of BL21(DE3) with pET-mAP at various temperatures. (C) Secretion of mAP. mAP-expressing cells grown with 0.5 mM IPTG for 24 h were fractionated into total (lane T), periplasmic (lane P), and cytoplasmic/membrane (lane C) fractions and subjected to SDS-PAGE. Lanes M and EV are the same as in (B). (D) Purification of mAP. mAP expressed at 15°C was purified using the periplasmic fraction. Lane M, protein standards; lane P, IPTG-induced periplasmic fraction; lane mAP; purified mAP protein after dialysis and concentration. Arrows indicate precursor and mature mAP.
Type II secretion by a putative Tat signal peptide
Because there was a discrepancy between the calculated (64 kDa) and experimentally determined (56 kDa) molecular mass of mAP, a bioinformatic survey was carried out to predict the signal sequence of mAP. The SignalP 4.1 Server [23] did not identify any significant signal peptide cleavage sites in the deduced amino acid sequence of mAP. However, according to TatP 1.0 Server software [24], which can predict the presence and location of twin-arginine translocation (Tat) signal peptide cleavage sites in bacteria, the first 81 residues of mAP were predicted to be a Tat signal peptide. Indeed, N-terminal amino acid sequencing of the purified 64-kDa and 56-kDa mAPs confirmed the first 81 amino acids as a signal peptide (Figure 4A). The Tat signal peptide, which has the consensus motif SRRxFLK, followed by a membrane-spanning hydrophobic region [25], is a conserved feature among PhoX proteins [22]. Based on these results, we concluded that mature mAP had a length of 527 amino acids and a molecular mass of 56 kDa, in accordance with the expression results of mAP (Figure 4B and C). In order to determine the molecular mass of mAP in its native form, mAP was subjected to size exclusion chromatography. The molecular mass was determined by comparing its elution time with those of standard proteins. One major peak of mAP appeared when the elution time was 33.9 min. Regression analysis to compare the molecular mass of standard proteins and the elution time indicated that the molecular mass of active mAP was around 50 kDa (Figure 5), which suggested that mAP was monomeric in its native form.
Figure 5.
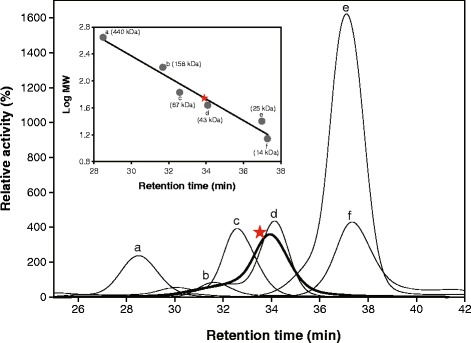
Molecular mass determination of mAP by size exclusion chromatography. mAP and six molecular mass standard proteins (peak a, ferritin; peak b, adolase; peak c, albumin; peak d, ovalbumin; peak e, chymotrypsinogen A; and peak f, RNase A) were each subjected to size exclusion chromatography on a Superose 6 HR column that had been pre-equilibrated with 25 mM Tris–HCl buffer containing 150 mM NaCl at pH 7.6. The molecular mass of mAP was estimated by comparing the retention time of mAP with the standard curve (plot of retention time versus logMW) obtained for the molecular mass standard proteins (inset).
Enzymatic properties of mAP
Next, we investigated the substrate preference of the purified mature mAP using various substrates. mAP demonstrated significant phosphatase activity with pNPP and with phosphorylated metabolites including nucleotides and carbohydrates (Table 1). Except for phytic acid and D-glucose 1-phosphate, mAP dephosphorylated all the tested substrates. While mAP showed broad substrate specificity, the highest activity was detected with ADP, followed by dATP, dGTP, and AMP. However, mAP was unable to hydrolyze bis-pNPP, indicating that it did not have phosphodiesterase activity. The previously reported SMAP also demonstrated significant phosphatase activity with most natural substrates, which cover all major classes of known phosphorylated metabolites [22].
Table 1.
Substrate specificity of purified mAP
| Substrates (1 mM) | P i released relative to that from p NPP* |
|---|---|
| pNPP | 100 |
| bis-pNPP | ND |
| AMP | 39 |
| ADP | 76 |
| ATP | 25 |
| dATP | 44 |
| dCTP | 38 |
| dGTP | 39 |
| dTTP | 37 |
| D-Glucose 1-phosphate | 0.2 |
| O-Phosphoethanolamine | 3 |
| Phosphoenolpyruvate | 18 |
| Phytic acid | ND |
| Paraoxaon | ND |
| Methyl parathion | ND |
*The Pi released from pNPP was set at 100%. ND: not detected. All results are the means of three experiments wherein variation was less than 15%.
The apparent optimal pH of mAP was 9.0 in 50 mM diethanolamine (DEA) buffer. The enzyme exhibited at least 70% of its optimal activity over a rather narrow pH range, from 8.5 to 9.5 (Figure 6A) and had significant activity over a pH range from 7.5 to 10.5. Furthermore, thermal activation studies demonstrated that the apparent optimum temperature was 37°C, and 25% of the maximum activity was found at 60°C (Figure 6B). Moreover, the enzyme exhibited at least 80% of its optimal activity over a temperature range from 30°C to 40°C, indicating that mAP was a marine-derived psychrophilic enzyme. Thermal stability was determined from enzyme incubation for 15 min over a range of temperatures (20 – 80°C). The psychrophilic mAP showed remarkable thermal instability, resulting in complete loss of its enzymatic activity, by incubation at 65°C for 15 min (Figure 6C).
Figure 6.
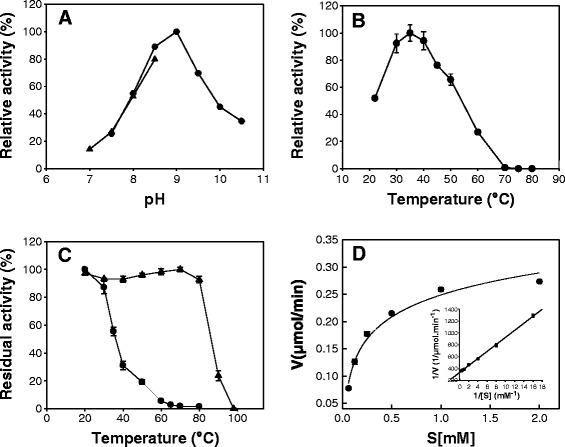
Effects of pH and temperature on mAP activity. AP activity was measured by a spectrophotometric method using p-nitrophenyl phosphate (pNPP). (A) Optimum pH. Enzymatic activity was assayed at 37°C in the presence of 0.5 mM pNPP as a substrate using 50 mM Tris–HCl buffer (pH 7.0–8.5) or 50 mM DEA buffer (pH 8.0 to 10.5). (B) Optimum temperature. Enzymatic activity was assayed at 20–80°C in the presence of 0.5 mM pNPP as a substrate using 50 mM DEA buffer (pH 9.0). (C) Thermal stability. Residual activity of mAP (●) was determined at 37°C after 15 min incubation at temperature ranges from 20°C to 100°C. Bacterial AP (▲) was also assayed under the same conditions. (D) Kinetic studies of mAP. Enzymatic activity was assayed under standard condition with 0.5 mM CaCl2 using a range of pNPP.
Divalent cations, such as Zn2+ and Mg2+, have been identified as essential cofactors for ECAP [26]. The activity of mAP was completely inhibited by 5 mM EDTA, indicating that it was a metalloenzyme (Table 2). To examine the metal specificity of mAP, the enzyme was dialyzed against 20 mM Tris buffer (pH 7.5) containing 5 mM EDTA. The resulting apoenzyme was assayed for phosphatase activity in the presence of various divalent cations (1 mM each; Table 2). Ca2+ was the best metal ion for reconstitution of enzyme activity, followed by Co2+ and Zn2+, suggesting that mAP was strongly dependent on multiple divalent cations. Mg2+ and Cu2+, however, had no significant effect on the restoration of enzymatic activity (Table 2).
Table 2.
Effects of metal ions on the activity of mAP
| Metal ions (1 mM) | % AP activity restored* |
|---|---|
| None | 0.11 |
| Ca2+ | 45.6 |
| Co2+ | 27.3 |
| Zn2+ | 12.8 |
| Ni2+ | 1.66 |
| Mn2+ | 1.44 |
| Mg2+ | 0.81 |
| Cu2+ | 0.24 |
*Results are expressed as the percentage of activity restored when compared with the enzyme that was not dialyzed against 5 mM EDTA (see Methods). All results are the means of three experiments wherein variation was less than 10%.
The kinetic parameters of mAP were estimated using the pNPP as substrate (Figure 6D). The Km of mAP is somewhat lower than the Km values reported for AAP, SAP, and calf intestinal AP (CIAP), but higher than that of SMAP which is a well-characterized PhoX. When compared with the Km value of ECAP, it is almost same (Table 3). In addition, mAP exhibited higher turnover number (kcat) and catalytic efficiency (kcat/Km) than those of ECAP, AAP, CIAP, and SMAP (Table 3).
Table 3.
Kinetic parameters of alkaline phosphatases
| Enzyme | Temperature (°C) | K m (mM) | k cat (s −1 ) | k cat /K m (s −1 M −1 ) | Reference |
|---|---|---|---|---|---|
| ECAP | 37 | 0.17 | 139 | 8.2 × 105 | [13] |
| AAP | 30 | 0.50 | 236 | 4.7 × 105 | [27] |
| SAP | 37 | 0.69 | 7,176 | 1.0 × 107 | [28] |
| CIAP | 37 | 0.40 | 43 | 1.0 × 105 | [29] |
| SMAP | 37 | 0.085 | 2.3 | 2.7 × 104 | [22] |
| mAP | 37 | 0.18 | 952 | 5.3 × 106 | This study |
ECAP, E. coli alkaline phosphatase; AAP, Antarctic alkaline phosphatase; SAP, shrimp alkaline phosphatase; CIAP, calf intestinal alkaline phosphatase; SMAP, Sinorhizobium meliloti alkaline phosphatase; mAP, metagenome-derived alkaline phosphatase.
DNA dephosphorylation by purified mAP
AP is widely used in molecular cloning to remove the 5′-terminal phosphate group from DNA fragments linearized by restriction enzymes, preventing religation of the linearized DNA. Thus, we tested the dephosphorylation efficiency (DE (%)) of mAP against cohesive (HindIII- or PstI-linearized pUC19) and blunt (SmaI-linearized pUC19) ends. The DE (%), which represents the ability of AP to prevent recircularization of linear DNA fragments, was estimated by comparing transformation rates after ligation of dephosphorylated and nondephosphorylated pUC19 plasmids [30]. mAP completely dephosphorylated DNA fragments with 3′-protruding cohesive ends generated by HindIII (Table 4). The DEs (%) of mAP were 91.1% and 99.6% for 5′-protruding cohesive ends generated by PstI and blunt ends generated by SmaI, respectively. The DE (%) of mAP was then compared with that of four different commercial APs (Table 4). The DEs (%) of mAP for both 3′-protruding cohesive ends and blunt ends were comparable with those of commercial APs, whereas the DE (%) of mAP for 5′-protruding cohesive ends was slightly lower than the values determined for SAP, CIAP, and APex heat-labile AP (HLAP) (Table 4).
Table 4.
Dephosphorylation activity of mAP and commercial APs
| Restriction enzyme | Dephosphorylation efficiency (%) | ||||
|---|---|---|---|---|---|
| mAP | AAP | SAP | CIAP | HLAP | |
| HindIII | 100 | 99.8 | 99.5 | 99.9 | 99.8 |
| PstI | 91.1 | 87.4 | 98.0 | 98.2 | 95.2 |
| SmaI | 99.6 | 99.4 | 98.9 | 100 | 99.7 |
mAP, metagenome-derived alkaline phosphatase; AAP, Antarctic alkaline phosphatase; SAP, shrimp alkaline phosphatase; CIAP, calf intestinal alkaline phosphatase; HLAP, APex heat-labile alkaline phosphatase. All results are the means of three experiments wherein variation was less than 10%.
Discussion
Using the GESS and metagenomic library of ocean-tidal flat sediments, we have identified a novel AP, called mAP, as a new member of the PhoX family of APs. To the best of our knowledge, this enzyme is the first thermolabile AP with functional activity isolated from a metagenome.
The mAP presented here clearly shows that it is distinct from the well-studied PhoA proteins;
-
(i)
The sequence identity between mAP and PhoA family (12% – 15%) was very low compared to the identity observed among PhoAs (30% – 50%). Based on a phylogenetic analysis, mAP evolutionarily belongs to the marine bacterial PhoX family, consistent with its isolation from flat tidal sediments. mAP and its homologs from the psychrophilic marine bacteria Sphingopyxis sp. MC1, Sphingopyxis alaskensis RB2256, Sphingobium sp. SYK-6, and Sphingomonas sp. strain BSAR-1 formed a phylogenetically distinct group that did not belong to the PhoA family.
-
(ii)
mAP activity is Ca2+-dependent whereas PhoA activity requires Mg2+ and Zn2+. ECAP is probably the most well-characterized PhoA to date. ECAP requires two Zn2+ ions to coordinate the phosphomonoester in the active center [5]. Interestingly, SPAP requires Ca2+ and Zn2+ for activation, while Mg2+ has no effect [21]. PhoX has a different catalytic center that requires Ca2+ instead of Zn2+ for enzyme activity [7]. Similarly, mAP also required Ca2+ for catalytic activity, indicating that its requirement for Ca2+ instead of Zn2+ has probably been a major factor in its selection over PhoA in flat tidal sediments, where Zn2+ often occurs at subnanomolar concentrations [31]. A recent study of the marine metagenomic Global Ocean Survey database showed that PhoX is more widely distributed among marine bacteria than PhoA [1].
-
(iii)
mAP is exported via the Tat pathway whereas PhoA translocation is dependent upon Sec pathway [5]. In contrast to the Sec pathway, the Tat secretion system is able to translocate fully folded proteins across bacterial membranes [32]. The Tat signal peptides are generally longer than Sec signal sequences [33]. mAP was found to have N-terminal 81 amino acids as a Tat signal peptide. The Tat signal peptide of mAP was longer than other previously identified PhoX family proteins that are ranging from 26 to 76 amino acids [22]. In addition, mAP contained a Tat motif (ARRSCPV, residues 39–45) that is slightly different from previously reported Tat signal peptides of PhoX family [34,35]. Interestingly, the Tat signal peptide of mAP was working at only low temperature (15°C) among all tested temperatures (15–37°C), suggesting that it has been evolved as a cold-adapted AP in ocean-tidal flat sediments with dynamic temperature changes. Although SPAP is also released to extracellular space, its signal peptide is not described yet.
-
(iv)
mAP exists as a monomeric enzyme while PhoA is active in the dimeric state. Among characterized PhoA family, ECAP and SPAP are present as a homodimer and multimeric protein, respectively.
Recently, the crystal structure of SPAP was reported and revealed that the key catalytic residue in SPAP is threonine, which is conserved in mAP, instead of serine found in all other APs [21]. In addition, the arginine conserved in key active site of PhoAs is deleted in SPAP. Based on these unique structural features, SPAP represents a new class of APs [21]. Although SPAP and mAP are structurally different from other APs, they share several enzymatic properties with typical PhoXs, such as high specific activity, low substrate specificity, and thermolability. In addition, both SPAP and mAP enzymes require Ca2+ for activation and mAP is secreted through the Tat pathway, which are similar to those of other PhoX proteins [1].
Psychrophilic enzymes are frequently associated with longer polypeptide chains compared to their mesophilic counterparts [36]; indeed, this was the case for mAP in the present study. Compared to the majority of the APs in the SwissProt Database, monomeric immature mAP was a relatively large protein (64 kDa) composed of 607 amino acids. Indeed, mAP exhibited remarkable thermal instability and was completely inactivated by incubation at 65°C for 15 min, in contrast to BAP, which retained 98% of its original activity at 70°C. In general, remarkable thermal instability, along with higher catalytic efficiencies at low and moderate temperatures, is properties of psychrophilic enzymes [37]. Therefore, an AP from psychrophiles exhibiting such properties could be of significant biotechnological interest.
APs are used in various molecular biology applications for the dephosphorylation of plasmid DNA prior to cloning in order to prevent recircularization, for the dephosphorylation of 5′-nucleic acid termini before 5′-end labeling by polynucleotide kinase, and for the removal of dNTPs and pyrophosphate from polymerase chain reaction assays. However, the AP has to be carefully removed after dephosphorylation to avoid interference with subsequent steps. Furthermore, ECAP and CIAP, which are preferred enzymes for these applications, are heat-stable and require the addition of detergents for inactivation. Thus, thermolabile APs are excellent alternatives as they are inactivated by moderate heat treatment and would therefore allow subsequent steps to be performed in the same test tube while minimizing nucleic acid losses. We examined the DE (%) using a linear cohesive or blunt-ended pUC19 plasmid and found that DEs (%) of mAP for cohesive and blunt ends were comparable with those of commercial APs. Therefore, because mAP exhibited excellent efficiency in linearized DNA dephosphorylation and was inactivated by simple heat treatment, further studies may be focused on elucidation of its potential use in molecular biology applications under optimal conditions.
Conclusions
This study provides a novel AP isolated from metagenomes of ocean-tidal flat sediments. The metagenome-derived AP is expected to show high potential for diverse molecular biology applications including the construction of recombinant plasmids. This was corroborated by its detailed biochemical characterization, which revealed high activity, catalytic efficiency, and thermal instability. In addition, the production of this enzyme with high yield in E. coli may be accelerating its applications.
Methods
Bacterial strains, plasmids, and chemicals
E. coli BL21(DE3) was used as a host strain for overexpression of mAP inserted into the pET-21a plasmid (Novagen, Darmstadt, Germany). DH5α cells and pUC19 plasmid (New England Biolabs, Ipswich, MA, USA) were used for plasmid preparation and dephosphorylation assays, respectively. Commercial bacterial AP (BAP, Invitrogen; Carlsbad, CA, USA), Antarctic AP (AAP; New England Biolabs), shrimp AP (SAP; Promega, Madison, WI, USA), calf intestinal AP (CIAP; Roche, Indianapolis, IN, USA), and APex heat-labile AP (HLAP; Epicentre, Madison, WI, USA) were purchased from each vendor. If not stated otherwise, E. coli cells were grown in Luria Bertani (LB) medium (5 g/L yeast extract, 10 g/L tryptone, and 5 g/L NaCl) supplemented with appropriate antibiotics. All chemical reagents used were of analytical-laboratory grade.
Isolation of DNA and fosmid library construction
Metagenomic DNA was isolated from the ocean-flat tidal sediments of the west coast of Korea (Taean, Korea) using a hydroshear machine (GeneMachines, Genomic Instrumentation Services, CA, USA). A metagenomic DNA fosmid library was constructed in E. coli EPI300 cells harboring fosmid pCC1FOS using a CopyControl Fosmid Library Production Kit (Epicentre) according to the manufacturer’s protocol. The size of the metagenomic library was estimated to be 600 Mbp, and the average insert size was 30 kb. For functional screening of AP, a pGESSv4 plasmid [19] was electrophoretically transformed into E. coli EPI300 cells carrying the metagenomic library. The resulting library cells were grown in LB broth containing ampicillin (50 μg/mL) and chloramphenicol (12.5 μg/mL) at 30°C for 12 h prior to fluorescence-activated cell sorting (FACS). False-positive cells were removed from total library cells using a FACSAria I (BD Bioscience, NJ, USA). A blue laser (488 nm) and a bandpass filter (530/30 nm) were used to analyze enhanced green fluorescent protein (EGFP) fluorescence. Forward and side scatter were used to exclude debris and dead cells. Nonfluorescent cells (106 cells) were collected and subsequently grown in LB broth containing 1 mM p-nitrophenyl phosphate (pNPP; Sigma Aldrich, St. Louis, MO, USA) at 30°C for 16 h. Next, approximately 200 fluorescent cells were sorted and grown on LB solid medium containing pNPP. After incubation at 30°C for 48 h, 47 colonies showed green fluorescence in a fluorescence image analysis. Among 47 colonies, six strongly fluorescent colonies were further examined for fluorescence intensity, representing the presence of the substrate, to confirm cellular phosphatase activity. Finally, a fosmid isolated from one colony displaying phosphatase activity was digested with BamHI and inserted into the pSTV28 plasmid (Takara Bio, Shiga, Japan) to yield a shotgun library in E. coli DH10B cells harboring pGESSv4. The shotgun library cells were grown on LB solid medium with pNPP, and six different colonies exhibiting fluorescence were analyzed by plasmid purification, followed by enzymatic digestion and DNA sequencing. The six purified plasmids were termed pSTV28-AP1–6.
Overexpression of mAP
The open reading frame (ORF) of putative mAP was amplified by polymerase chain reaction (PCR) with the pSTV28-AP6 plasmid as a template using the following primers: 5′-GGGAATTCCATATGTCACAATCCTATGCCTGTGC-3′ (italics: NdeI restriction site) and 5′-CCGCTCGAGTGAAATCCTCCCTCGATGCGGCAACTGCTCTCGG-3′ (italics: XhoI restriction site; bold: factor Xa cleavage site). The PCR product was then inserted into the pET-21a plasmid digested with NdeI and XhoI restriction enzymes, resulting in plasmid pET-mAP harboring the C-terminal His6 tag. For overproduction of mAP enzyme, E. coli BL21(DE3) cells transformed with pET-mAP were grown in LB medium supplemented with 50 μg/mL ampicillin at 37°C until reaching an optical density at 600 nm (OD600) of 0.4–0.5. Cells were then induced with 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG). The culture was transferred to 15°C and incubated for 40 h.
Cellular fractionation
For cytoplasmic and periplasmic protein preparations, we used a PeriPreps Periplasting kit (Epicentre), which uses osmotic shock to disrupt the outer cell membrane, followed by treatment with lysozyme to digest the cell wall. Briefly, the induced cells were harvested by centrifugation at 6,000 × g for 20 min at 4°C and resuspended in periplasting buffer (200 mM Tris–HCl [pH 7.5], 20% [w/v] sucrose, 1 mM EDTA, and 30 units/μL Ready-Lys lysozyme). After incubation on ice for 5 min, osmotic shock was accomplished by quickly adding an equal volume of cold water. This suspension was incubated on ice for 5 min and centrifuged at 12,000 × g for 2 min to remove the remaining cells. The supernatant was recovered as the soluble periplasmic protein fraction. The remaining insoluble fraction was resuspended in PeriPreps lysis buffer (10 mM Tris–HCl [pH 7.5], 50 mM KCl, 1 mM EDTA, and 0.1% [w/v] deoxycholate) with complete protease inhibitors cocktail (Roche) and incubated at room temperature for 5 min. Cell debris was removed by centrifugation, and the supernatant containing the cytoplasmic cellular protein fraction was transferred to a clean tube.
Purification of recombinant mAP
To purify the mAP enzyme, the periplasmic fraction was directly loaded on a HisTrap HP column (GE Healthcare Life Sciences) equilibrated with buffer A (25 mM Tris–HCl [pH 7.6], 1 mM phenylmethanesulfonyl fluoride (PMSF), 25 mM NaCl, 5 mM imidazole, and 1 mM CaCl2) using an automated chromatography system (ÄKTA FPLC, GE Healthcare Life Sciences, PA, USA). The enzyme was purified according to the manufacturer’s protocol for gradient elution of His-tagged protein. Protein fractions eluted with buffer B (25 mM Tris–HCl [pH 7.6], 25 mM NaCl, 500 mM imidazole, and 1 mM CaCl2) were desalted using Vivaspin centrifugal concentrators (Sartorius, Gottingen, Germany) with buffer C (25 mM Tris–HCl [pH 7.6], 1 mM CaCl2, and 10% glycerol). The purity of eluted protein was analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) on 12% gels, and the protein concentration was determined using a protein assay kit (Bio-Rad, Hercules, CA, USA) with bovine serum albumin as the standard.
AP activity assay
The standard assay for AP activity was carried out at 37°C for 5 min using 0.5 mM pNPP as the substrate in 50 mM DEA buffer (pH 9.0) containing 0.5 mM CaCl2, unless indicated otherwise. The release of p-nitrophenol (pNP) in the reaction mixture (1 mL) was monitored with a multilabel microplate reader (VICTOR X5, Perkin Elmer, MA, USA) at 405 nm over the linear period. One unit of enzyme activity is defined as the amount of enzyme required to release 1 μmol of pNP from pNPP in 1 min at 37°C. Each value is the mean of at least three assays.
Molecular mass determination by size-exclusion chromatography
Size exclusion chromatography was performed using a Superose 6 HR column (GE Healthcare Life Sciences) equilibrated with an equilibration buffer (25 mM Tris–HCl [pH 7.6], 150 mM NaCl, and 1 mM CaCl2). Separation was carried out isocratically at a flow rate of 0.5 mL/min at 25°C. To estimate the molecular mass, a gel filtration calibration kit (GE Healthcare Life Sciences) was used for molecular mass standards: ferritin, 440 kDa; aldolase, 158 kDa; albumin, 67 kDa; ovalbumin, 43 kDa; chymotrypsinogen A, 25 kDa; and RNase A, 14 kDa.
N-terminal sequencing
N-terminal sequencing was performed on purified precursor and mature mAP enzymes, which were electrophoresed on 12% SDS-PAGE gels and transferred to polyvinylamine difluoride (PVDF) membranes. The first 20 amino acids of the N-terminal sequences of both purified enzymes were determined by the Edman degradation method with an Applied Biosystems Precise Sequencer (Applied Biosystems, NY, USA) at the Korea Basic Science Institute (Daejeon, Korea).
Temperature and pH optima and thermal stability
The apparent optimum temperature was determined by running the standard assay at temperatures ranging from 20°C to 80°C in 50 mM DEA buffer (pH 9.0). The apparent optimum pH of the enzyme was determined by running the standard assay using 50 mM Tris–HCl buffer or 50 mM DEA buffer for the pH ranges 7.0–8.5 and 7.5–10.5, respectively. The thermostability of purified mAP enzyme or BAP (Invitrogen) was determined by incubation for 15 min at temperatures ranging from 20°C to 80°C and from 20°C to 100°C, respectively. Samples were taken at different time intervals, and the residual activity was determined by the standard assay.
Substrate specificity and divalent cation effects
To examine the substrate specificity of mAP, the following substrates were used at a final concentration of 0.5 mM: pNPP, D-glucose 1-phosphate, O-phosphoethanolamine, phosphoenolpyruvate, phytic acid, paraoxon, and methyl parathion in 50 mM DEA buffer (pH 9.0). In addition, adenosine monophosphate (AMP), adenosine diphosphate (ADP), adenosine triphosphate (ATP), deoxyadenosine triphosphate (dATP), deoxycytidine triphosphate (dCTP), deoxyguanosine triphosphate (dGTP), and deoxythymidine triphosphate (dTTP) were individually used as substrates in 50 mM Tris–HCl buffer (pH 8.5).
The effects of divalent cations were also examined for their influence on enzyme activity. After treating the purified mAP with 5 mM ethylenediaminetetraacetic acid (EDTA), several divalent cations (NiCl2•6H2O, CoCl2•6H2O, CaCl2, CdCl2, CuSO4•5H2O, MgCl2, or ZnSO4•5H2O) were individually added to the mAP enzyme solution. This enzyme solution was incubated for 15 min on ice, and the recovered activity was measured by the standard assay.
DNA dephosphorylation assay
The pUC19 plasmid was digested with HindIII, PstI, or SmaI to generate 3′-protruding cohesive, 5′-protruding cohesive, or blunt-ended linearized plasmids, respectively. Linearized plasmids (1.0 μg) were incubated with 1 U of each AP in dephosphorylation buffer for 2 h at 37°C, and the reaction was stopped by incubation at 65°C for 15 min. After linearization and dephosphorylation, DNA fragments were purified using a DNA purification kit (Qiagen). The dephosphorylation buffer for mAP was 50 mM Tris–HCl (pH 8.5) containing 1 mM CaCl2, while the buffers for commercial AAP, SAP, CIAP, and HLAP were provided by each manufacturer. A total of 0.1 μg of AP-treated linear plasmid DNA was ligated using 1 U of T4 DNA ligase (New England Biolabs) and incubated overnight at 16°C. Lastly, 10 ng of the ligation mixture was transformed into competent E. coli DH5α cells using the standard CaCl2 transformation protocol, and the cells were grown on LB solid medium supplemented with 50 μg/mL ampicillin. The control experiment was conducted using sterile water instead of APs in the dephosphorylation step. The percent dephosphorylation efficiency (DE (%)) was calculated from the following equation described in the previous study [30]:
where colony forming unit (CFU)/mL is the number of CFU per milliliter of transformed cells and CFU/mLAP and CFU/mLcontrol are estimated with dephosphorylated plasmids and nondephosphorylated plasmids, respectively.
Sequence analysis
Automated DNA sequencing of small inserted plasmids was carried out using an ABI377 instrument (Applied Biosystems, Foster City, CA, USA) and dye terminator chemistry according to the manufacturer’s protocol. We used 454 sequencing technology to establish large fosmid sequences. The amino acid sequence of the putative mAP was aligned with those of the homologous proteins using ClustalW (DNASTAR, WI, USA). Phylogenetic analysis was conducted using the MEGA 5.2 program [38].
Nucleotide sequence accession number
The complete nucleotide sequence of mAP was deposited in GenBank under accession number GQ250428.
Acknowledgments
This work was supported by the KRIBB Research Initiative Program, the Intelligent Synthetic Biology Center of Korea (2011-0031944) through the Global Frontier Research Program of the Ministry of Science, ICT and Future Planning (MSIP) and the New & Renewable Energy of the Korea Institute of Energy Technology Evaluation and Planning (KETEP: 20133030000300) grant funded by the Ministry of Trade, Industry and Energy (MTIE)).
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
DL and SL designed the experiments; SC, ER, SJK, SY, JHM and DL performed the experiments; DL and SL wrote the manuscript; SL supervised the work; and all authors contributed to the discussion of the research. All authors read and approved the final manuscript.
Contributor Information
Dae-Hee Lee, Email: dhlee@kribb.re.kr.
Su-Lim Choi, Email: sulimchoi@gmail.com.
Eugene Rha, Email: josephin@kribb.re.kr.
Soo Jin Kim, Email: sugar116@kribb.re.kr.
Soo-Jin Yeom, Email: sujin258@kribb.re.kr.
Jae-Hee Moon, Email: mrjy1469@kribb.re.kr.
Seung-Goo Lee, Email: sglee@kribb.re.kr.
References
- 1.Sebastian M, Ammerman JW. The alkaline phosphatase PhoX is more widely distributed in marine bacteria than the classical PhoA. ISME J. 2009;3:563–72. doi: 10.1038/ismej.2009.10. [DOI] [PubMed] [Google Scholar]
- 2.Torriani A. From cell membrane to nucleotides: the phosphate regulon in Escherichia coli. BioEssays. 1990;12:371–6. doi: 10.1002/bies.950120804. [DOI] [PubMed] [Google Scholar]
- 3.Hulett FM. The signal-transduction network for Pho regulation in Bacillus subtilis. Mol Microbiol. 1996;19:933–9. doi: 10.1046/j.1365-2958.1996.421953.x. [DOI] [PubMed] [Google Scholar]
- 4.Kadurugamuwa JL, Beveridge TJ. Natural release of virulence factors in membrane vesicles by Pseudomonas aeruginosa and the effect of aminoglycoside antibiotics on their release. J Antimicrob Chemother. 1997;40:615–21. doi: 10.1093/jac/40.5.615. [DOI] [PubMed] [Google Scholar]
- 5.Kim EE, Wyckoff HW. Reaction mechanism of alkaline phosphatase based on crystal structures: two-metal ion catalysis. J Mol Biol. 1991;218:449–64. doi: 10.1016/0022-2836(91)90724-K. [DOI] [PubMed] [Google Scholar]
- 6.Coleman JE. Structure and mechanism of alkaline phosphatase. Annu Rev Biophys Biomol Struct. 1992;21:441–83. doi: 10.1146/annurev.bb.21.060192.002301. [DOI] [PubMed] [Google Scholar]
- 7.Majumdar A, Ghatak A, Ghosh RK. Identification of the gene for the monomeric alkaline phosphatase of Vibrio cholerae serogroup O1 strain. Gene. 2005;344:251–8. doi: 10.1016/j.gene.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 8.Nilgiriwala KS, Alahari A, Rao AS, Apte SK. Cloning and overexpression of alkaline phosphatase PhoK from Sphingomonas sp. strain BSAR-1 for bioprecipitation of uranium from alkaline solutions. Appl Environ Microbiol. 2008;74:5516–23. doi: 10.1128/AEM.00107-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Suzuki C, Ueda H, Tsumoto K, Mahoney WC, Kumagai I, Nagamune T. Open sandwich ELISA with VH-/VL-alkaline phosphatase fusion proteins. J Immunol Methods. 1999;224:171–84. doi: 10.1016/S0022-1759(99)00020-4. [DOI] [PubMed] [Google Scholar]
- 10.Tomazic-Allen SJ. Recombinant bacterial alkaline phosphatase as an immunodiagnostic enzyme. Ann Biol Clin (Paris) 1991;49:287–90. [PubMed] [Google Scholar]
- 11.Kobori H, Sullivan CW, Shizuya H. Heat-labile alkaline phosphatase from Antarctic bacteria: rapid 5′ end-labeling of nucleic acids. Proc Natl Acad Sci U S A. 1984;81:6691–5. doi: 10.1073/pnas.81.21.6691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rina M, Pozidis C, Mavromatis K, Tzanodaskalaki M, Kokkinidis M, Bouriotis V. Alkaline phosphatase from the Antarctic strain TAB5. Eur J Biochem. 2000;267:1230–8. doi: 10.1046/j.1432-1327.2000.01127.x. [DOI] [PubMed] [Google Scholar]
- 13.Hauksson JB, Andrésson ÓS, Ásgeirsson B. Heat-labile bacterial alkaline phosphatase from a marine Vibrio sp. Enz Microb Technol. 2000;27:66–73. doi: 10.1016/S0141-0229(00)00152-6. [DOI] [PubMed] [Google Scholar]
- 14.Suzuki Y, Mizutani Y, Tsuji T, Ohtani N, Takano K, Haruki M, et al. Gene cloning, overproduction, and characterization of thermolabile alkaline phosphatase from a psychrotrophic bacterium. Biosci Biotechnol Biochem. 2005;69:364–73. doi: 10.1271/bbb.69.364. [DOI] [PubMed] [Google Scholar]
- 15.Schmeisser C, Steele H, Streit W. Metagenomics, biotechnology with non-culturable microbes. Appl Microbiol Biotechnol. 2007;75:955–62. doi: 10.1007/s00253-007-0945-5. [DOI] [PubMed] [Google Scholar]
- 16.Iqbal HA, Feng Z, Brady SF. Biocatalysts and small molecule products from metagenomic studies. Curr Opin Chem Biol. 2012;16:109–16. doi: 10.1016/j.cbpa.2012.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taupp M, Mewis K, Hallam SJ. The art and design of functional metagenomic screens. Curr Opin Biotechnol. 2011;22:465–72. doi: 10.1016/j.copbio.2011.02.010. [DOI] [PubMed] [Google Scholar]
- 18.Rabausch U, Juergensen J, Ilmberger N, Böhnke S, Fischer S, Schubach B, et al. Functional screening of metagenome and genome libraries for detection of novel flavonoid-modifying enzymes. Appl Environ Microbiol. 2013;79:4551–63. doi: 10.1128/AEM.01077-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Choi S-L, Rha E, Lee SJ, Kim H, Kwon K, Jeong Y-S, et al. Toward a generalized and high-throughput enzyme screening system based on artificial genetic circuits. ACS Syn Biol. 2014;3:163–71. doi: 10.1021/sb400112u. [DOI] [PubMed] [Google Scholar]
- 20.Shingler V, Bartilson M, Moore T. Cloning and nucleotide sequence of the gene encoding the positive regulator (DmpR) of the phenol catabolic pathway encoded by pVI150 and identification of DmpR as a member of the NtrC family of transcriptional activators. J Bacteriol. 1993;175:1596–604. doi: 10.1128/jb.175.6.1596-1604.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bihani SC, Das A, Nilgiriwala KS, Prashar V, Pirocchi M, Apte SK, et al. X-ray structure reveals a new class and provides insight into evolution of alkaline phosphatases. PLoS One. 2011;6:e22767. doi: 10.1371/journal.pone.0022767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zaheer R, Morton R, Proudfoot M, Yakunin A, Finan TM. Genetic and biochemical properties of an alkaline phosphatase PhoX family protein found in many bacteria. Environ Microbiol. 2009;11:1572–87. doi: 10.1111/j.1462-2920.2009.01885.x. [DOI] [PubMed] [Google Scholar]
- 23.Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8:785–6. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 24.Bendtsen J, Nielsen H, Widdick D, Palmer T, Brunak S. Prediction of twin-arginine signal peptides. BMC Bioinformatics. 2005;6:167. doi: 10.1186/1471-2105-6-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Berks BC, Palmer T, Sargent F. The Tat protein translocation pathway and its role in microbial physiology. Adv Microb Physiol. 2003;47:187–254. doi: 10.1016/S0065-2911(03)47004-5. [DOI] [PubMed] [Google Scholar]
- 26.Plocke DJ, Levinthal C, Vallee BL. Alkaline phosphatase of Escherichia coli: a zinc metalloenzyme. Biochemistry. 1962;1:373–8. doi: 10.1021/bi00909a001. [DOI] [PubMed] [Google Scholar]
- 27.Koutsioulis D, Wang E, Tzanodaskalaki M, Nikiforaki D, Deli A, Feller G, et al. Directed evolution on the cold adapted properties of TAB5 alkaline phosphatase. Protein Eng Des Sel. 2008;21:319–27. doi: 10.1093/protein/gzn009. [DOI] [PubMed] [Google Scholar]
- 28.Post MA. Alkaline phosphatase from psychrophile TAB5 and cold-adapted, northern shrimp (Pandalus borealis) are structurally similar yet functionally distinct. FASEB J. 2006;20:A479. [Google Scholar]
- 29.Chaudhuri G, Fau CS, Venu-Babu P, Fau V-BP, Ramasamy K, Fau RK, et al. Kinetic behaviour of calf intestinal alkaline phosphatase with pNPP. Indian J Biochem Biophys. 2013;50:64–71. [PubMed] [Google Scholar]
- 30.Zappa S, Rolland J-L, Flament D, Gueguen Y, Boudrant J, Dietrich J. Characterization of a highly thermostable alkaline phosphatase from the euryarchaeon Pyrococcus abyssi. Appl Environ Microbiol. 2001;67:4504–11. doi: 10.1128/AEM.67.10.4504-4511.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lohan MC, Statham PJ, Crawford DW. Total dissolved zinc in the upper water column of the subarctic North East Pacific. Deep Sea Res Part II Top Stud Oceanogr. 2002;49:5793–808. doi: 10.1016/S0967-0645(02)00215-1. [DOI] [Google Scholar]
- 32.Monds RD, Newell PD, Schwartzman JA, O’Toole GA. Conservation of the Pho regulon in Pseudomonas fluorescens p f0–1. Appl Environ Microbiol. 2006;72:1910–24. doi: 10.1128/AEM.72.3.1910-1924.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Berks BC. A common export pathway for proteins binding complex redox cofactors? Mol Microbiol. 1996;22:393–404. doi: 10.1046/j.1365-2958.1996.00114.x. [DOI] [PubMed] [Google Scholar]
- 34.van Mourik A, Bleumink-Pluym NMC, van Dijk L, van Putten JPM, Wösten MMSM. Functional analysis of a Campylobacter jejuni alkaline phosphatase secreted via the Tat export machinery. Microbiology. 2008;154:584–92. doi: 10.1099/mic.0.2007/012120-0. [DOI] [PubMed] [Google Scholar]
- 35.Wu J-R, Shien J-H, Shieh HK, Hu C-C, Gong S-R, Chen L-Y, et al. Cloning of the gene and characterization of the enzymatic properties of the monomeric alkaline phosphatase (PhoX) from Pasteurella multocida strain X-73. FEMS Microbiol Lett. 2007;267:113–20. doi: 10.1111/j.1574-6968.2006.00542.x. [DOI] [PubMed] [Google Scholar]
- 36.Feller G, Gerday C. Psychrophilic enzymes: molecular basis of cold adaptation. Cell Mol Life Sci. 1997;53:830–41. doi: 10.1007/s000180050103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gerday C, Aittaleb M, Arpigny JL, Baise E, Chessa J-P, Garsoux G, et al. Psychrophilic enzymes: a thermodynamic challenge. Biochim Biophys Acta. 1997;1342:119–31. doi: 10.1016/S0167-4838(97)00093-9. [DOI] [PubMed] [Google Scholar]
- 38.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–9. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]