

Abstract
Ombitasvir (ABT-267) is a hepatitis C virus (HCV) NS5A inhibitor with picomolar potency, pan-genotypic activity, and 50% effective concentrations (EC50s) of 0.82 to 19.3 pM against HCV genotypes 1 to 5 and 366 pM against genotype 6a. Ombitasvir retained these levels of potency against a panel of 69 genotype 1 to 6 chimeric replicons containing the NS5A gene derived from HCV-infected patients, despite the existence of natural sequence diversity within NS5A. In vitro resistance selection identified variants that conferred resistance to ombitasvir in the HCV NS5A gene at amino acid positions 28, 30, 31, 58, and 93 in genotypes 1 to 6. Ombitasvir was evaluated in vivo in a 3-day monotherapy study in 12 HCV genotype 1-infected patients at 5, 25, 50, or 200 mg dosed once daily. All patients in the study were HCV genotype 1a infected and were without preexisting resistant variants at baseline as determined by clonal sequencing. Decreases in HCV RNA up to 3.1 log10 IU/ml were observed. Resistance-associated variants at position 28, 30, or 93 in NS5A were detected in patient samples 48 hours after the first dose. Clonal sequencing analysis indicated that wild-type virus was largely suppressed by ombitasvir during 3-day monotherapy, and at doses higher than 5 mg, resistant variant M28V was also suppressed. Ombitasvir was well tolerated at all doses, and there were no serious or severe adverse events. These data support clinical development of ombitasvir in combination with inhibitors targeting HCV NS3/4A protease (ABT-450 with ritonavir) and HCV NS5B polymerase (ABT-333, dasabuvir) for the treatment of chronic HCV genotype 1 infection. (Study M12-116 is registered at ClinicalTrials.gov under registration no. NCT01181427.)
INTRODUCTION
Hepatitis C virus (HCV) is an enveloped, single-stranded, positive-sense RNA virus in the Flaviviridae family that infects approximately 170 to 200 million people worldwide (1, 2). Seven distinct HCV genotypes and 67 subtypes with significant variability in their geographic distribution have been characterized (3). HCV genotype 1, predominant in North America, Europe, and Japan, accounts for 60% of the global infections (4–6). Genotype 2 infections are most prevalent in North America, Europe, and Japan, while genotype 3, 6, and 7 infections are predominant within various parts of Southeast Asia (3, 7–9). In Egypt, HCV infections are almost exclusively genotype 4, while genotype 5 is common in South Africa (10, 11). The levels of nucleotide sequence diversity between genotypes and between subtypes are 30 to 35% and 20 to 25%, respectively (12). The viral dynamics are rapid for HCV, with 1012 virions being produced daily with a half-life of 45 min (13). Moreover, the RNA-dependent RNA polymerase of HCV is intrinsically error prone, and its lack of a proofreading function allows for introduction of approximately one nucleotide change per genome per replication cycle, which under drug pressure results in the expansion of preexisting drug resistant variants (13). These factors have created challenges in developing pan-genotypic HCV inhibitors with high genetic barriers to the development of resistance.
HCV replication can be inhibited at various points in the replication cycle by targeting viral or host cell functions (14, 15). For the treatment of HCV genotype 1, three HCV NS3/4A protease inhibitors (telaprevir, boceprevir, and simeprevir) and one nucleoside NS5B polymerase inhibitor (sofosbuvir), each in combination with pegylated interferon (pegIFN) and ribavirin (RBV), have received marketing approval in the United States and Europe. The sustained virologic response (SVR) rate increased from 40 to 52% with pegIFN and RBV regimens to 67 to 75% when telaprevir and boceprevir were used in combination with pegIFN and RBV (16, 17). The NS3/4A protease inhibitor simeprevir in combination with pegIFN and RBV improved the SVR rate to 80%, but in genotype 1a-infected patients with a Q80K polymorphism in the HCV NS3 protein, the SVR rate was reduced to 58% (18, 19). Sofosbuvir in combination with pegIFN and RBV yielded an SVR rate of 89% in genotype 1-infected patients; however, there were differences in SVR rate among genotype 1a (92%) and genotype 1b (82%) infected subjects (20). All direct-acting antiviral (DAA) regimens currently approved for treatment of HCV genotype 1- or genotype 4-infected patients must be coadministered with pegIFN and RBV, drugs that are associated with considerable, often treatment-limiting toxicities. Although there is a greater need for interferon-free regimens for the treatment of genotype 1 infection, the epidemiology of the numerous HCV genotypes and subtypes highlights the importance of developing pan-genotypic DAAs.
HCV NS5A has no known enzymatic function; however, it appears to play a critical role in the HCV replication cycle, both directly in viral RNA production and indirectly by modulating the host cell environment to favor viral replication (21–23). Studies have also suggested that NS5A plays a critical role in the assembly of viral particles into fully formed, infectious virions (24). The development of the in vitro HCV replicon system has aided in the discovery and optimization of NS5A inhibitors for the treatment of HCV (25). Several NS5A inhibitors, including daclatasvir, ledipasvir, samatasvir, GS5816, GSK-2336805, PPI461, PPI668, ACH-2928, ACH-3102, and MK-8742, are currently in various stages of clinical development (25). Our efforts to identify inhibitors of HCV replication led to identification of naphthyridine compounds that lacked activity against the HCV NS3/4A protease and NS5B polymerase enzymes (26, 27). Based on the selection pattern of resistance variants, the naphthyridine inhibitors were shown to target HCV NS5A in the HCV cell-based replicon system. A subsequent proof-of-concept study with a naphthyridine analog in a chimpanzee resulted in a robust HCV RNA viral load decline, and resistance analysis confirmed NS5A as the drug target (27). Structure-activity relationship studies and optimization of N-phenylpyrrolidine-based inhibitors led to the discovery of ABT-267, now known as ombitasvir (Fig. 1) (28).
FIG 1.
Structure of ombitasvir.
Ombitasvir is being developed for use in combination with ABT-450, a macrocyclic noncovalent peptidomimetic inhibitor of HCV NS3/4A protease (described in an accompanying article [29]), and the nonnucleoside NS5B polymerase inhibitor dasabuvir (ABT-333), with or without RBV, for the treatment of HCV (30–34). The pan-genotypic activity and the in vitro resistance profile of ombitasvir are described in this report. In addition, we report the viral load declines observed in patients infected with genotype 1 HCV who were treated for 3 days with ombitasvir (5, 25, 50, or 200 mg once daily), along with the resistance-associated variants that were present in virus isolated from these patients during the 3-day dosing period.
MATERIALS AND METHODS
Compound.
Ombitasvir, dimethyl([(2S,5S)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl]bis{benzene-4,1-diylcarbamoyl(2S)pyrrolidine-2,1-diyl[(2S)-3-methyl-1-oxobutane-1,2-diyl]})biscarbamate hydrate, was synthesized at AbbVie (28).
Replicon cell lines and measurement of in vitro activity.
The genotype 1a replicon construct contained the 5′ nontranslated region (NTR) from HCV strain 1a-H77 (GenBank accession number NC_004102) followed by a firefly luciferase reporter gene and the neomycin phosphotransferase (Neo) gene, which together comprised the first cistron of the bicistronic replicon construct. This was followed by the encephalomyocarditis virus (EMCV) internal ribosome entry site (IRES) and the second cistron containing the 1a-H77 NS3-NS5B-coding region with adaptive mutations encoding E1202G, K1691R, K2040R and S2204I and finally by the 1a-H77 3′ NTR. The genotype 1b-Con1 replicon construct contained the 5′ NTR from HCV strain 1b-Con1 (GenBank accession number AJ238799) followed by a firefly luciferase reporter gene and the Neo gene, which together comprised the first cistron of the bicistronic replicon construct. This was followed by the EMCV IRES and the second cistron containing the 1b-Con1 NS3-NS5B-coding region with adaptive mutations encoding K1609E, K1846T, and Y3005C and finally by the 1b-Con1 3′ NTR. In addition, the 1b-Con1 replicon construct contained a poliovirus IRES between the HCV 5′ NTR and the firefly luciferase gene. In order to assess the ability of compounds to inhibit NS5A from non-genotype 1 HCV, a 1b-Con1 replicon shuttle vector containing the Neo gene and luciferase reporter was constructed, which contained restriction site NotI, 90 nucleotides upstream of NS5A in the 3′ end of NS4B, and a BlpI site just after the NS5A amino acid 214 codon. Six chimeric subgenomic replicon cell lines were generated for evaluation of the activity of compounds. The NS5A regions for generation of chimeric replicons were amplified from the sera of patients infected with HCV genotype 2a, 2b, 3a, 4a, 5a, or 6a. Viral RNA was isolated from 140 to 280 μl of sera from HCV-infected patients using the QIAamp viral RNA isolation kit (Qiagen, Valencia, CA), according to the supplier's instructions. Reverse transcription-PCR (RT-PCR) using SuperScript III and Platinum Taq DNA polymerase (Invitrogen, Carlsbad, CA) and gene-specific primers was conducted on the RNA to generate a DNA fragment carrying the NS5A gene with NotI- and BlpI-compatible ends. This fragment was ligated into the replicon shuttle vector plasmid and transformed into competent Escherichia coli cells. After overnight growth in liquid culture, the plasmid DNA from the entire population was isolated, purified, and then linearized by restriction enzyme digestion. The TranscriptAid T7 high-yield transcription kit (Fermentas, Glen Burnie, MD) was used to transcribe HCV subgenomic RNA. This RNA was introduced into the human hepatoma cell line Huh-7 by electroporation to create a stable replicon cell line. All replicon cell lines were maintained in Dulbecco's modified Eagle medium (DMEM) containing 100 IU/ml penicillin, 100 μg/ml streptomycin, and 200 μg/ml G418 (all from Invitrogen, Carlsbad, CA) with 10% (vol/vol) fetal bovine serum (FBS) (Atlanta Biologicals, Flowery Branch, GA). The inhibitory effect of ombitasvir on HCV replication was determined in DMEM containing 5% FBS with or without 40% human plasma (Bioreclamation, Westbury, NY) by measuring the activity of the luciferase reporter gene. The cells were incubated for 3 days in the presence of ombitasvir and were subsequently lysed and processed according to the manufacturer's instructions (Promega, Madison, WI). The luciferase activity in the cells was measured using a Victor II luminometer (Perkin-Elmer, Waltham, MA). The 50% effective concentration (EC50) was calculated using nonlinear regression curve fitting to the 4-parameter logistic equation and GraphPad Prism 4 software. The cytotoxicity of ombitasvir was determined by the 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (5 mg/ml) (Sigma-Aldrich, St. Louis, MO) colorimetric assay (35). The 50% cytotoxicity concentration (CC50) was calculated from the optical density data using nonlinear regression curve fitting to the 4-parameter logistic equation and GraphPad Prism 4 software. Activity of ombitasvir against genotype 2a JFH-1 (GenBank accession number AB047639) was evaluated by Southern Research Institute, using a subgenomic replicon that did not contain a luciferase reporter, using a quantitative RT-PCR (qRT-PCR) assay (36, 37).
Phenotype against NS5A gene from clinical isolates.
A genotype 1b-Con1 HCV replicon-based shuttle vector cassette with the luciferase reporter but without the Neo gene was constructed for assessing the phenotypes of NS5A genes derived from individuals infected with HCV genotypes 1 to 6. A NotI restriction site was cloned into the 1b-Con1 subgenomic replicon vector 90 nucleotides upstream of NS5A in the 3′ end of NS4B, and a ClaI site was cloned after the NS5A amino acid 413 codon. The NS5A region from genotype 1-infected patients was inserted between the NotI and ClaI restriction sites. The 1b-Con1 shuttle vector with NotI and BlpI restriction sites (described in the previous section) was used to evaluate the ability of ombitasvir to inhibit the NS5A region encompassing amino acids 1 to 214 from non-genotype 1 HCV. The NS5A gene from clinical samples was amplified and ligated into the shuttle vector. In a transient assay, the HCV subgenomic replicon RNA containing the NS5A gene from each clinical sample was transfected via electroporation into a Huh-7 derived cell line (38). The cells were incubated in the presence of ombitasvir for 4 days. The luciferase activity in the cells was measured, and the EC50 was calculated as described above.
In vitro resistance analysis.
In order to characterize replicon variants with reduced susceptibility to ombitasvir, resistance selection was conducted in the chimeric genotype 1 to 6 HCV replicon-containing stable cell lines described above. Replicon cells (5 × 104 to 1 × 106) were plated in 150-mm cell culture plates and grown in the presence of G418 (400 μg/ml) and ombitasvir at a concentration that was 10-fold, 50-fold, 100-fold, or 1,000-fold above the EC50 for the respective cell line. After 3 weeks of treatment, the majority of replicon cells were cleared of replicon RNA and were therefore unable to survive in the G418-containing medium. The cells containing resistant replicon variants survived and formed colonies, and each of these colonies was picked and further expanded. In order to characterize the genotype of the resistant replicon variants, the expanded colonies were lysed in CellsDirect resuspension and lysis buffer (Invitrogen, Carlsbad, CA) to yield total RNA. The NS5A-coding region was amplified by RT-PCR, and the amplified samples were sequenced using the ABI Prism dye terminator cycle sequencing ready-reaction kit and were analyzed on an Applied Biosystems 3100 genetic analyzer.
Antiviral activity against a panel of resistant variants.
The methods used for measurement of the effects of individual amino acid variants on the activity of an inhibitor in HCV replicon cell culture assays were described previously (39). Briefly, the resistance-associated variants in NS5A were each introduced into the genotype 1a-H77 or 1b-Con1 or one of the chimeric genotype 2 to 6 replicons using the Change-IT multiple-mutation site-directed mutagenesis kit (Affymetrix, Santa Clara, CA). After the presence of the variant was confirmed by sequence analysis, the plasmid was linearized and the TranscriptAid T7 high-yield transcription kit (Fermentas, Glen Burnie, MD) was used to transcribe the HCV subgenomic RNA from the plasmid. In a transient assay, the replicon RNA containing the variant was transfected via electroporation into a Huh-7 cell line (38). The EC50s were calculated as described in the previous sections.
Clinical study design.
Study M12-116 (ClinicalTrials.gov registration no. NCT01181427) was the first study to evaluate the pharmacokinetics, safety, tolerability, antiviral activity, and resistance of ombitasvir in HCV-infected treatment-naive adults. All of the patients provided written informed consent. The study was performed in accordance with Good Clinical Practice guidelines and the principles of the Declaration of Helsinki, and the study protocol was approved by the relevant institutional review boards and regulatory agencies. Inclusion criteria included chronic HCV genotype 1 infection for at least 6 months prior to study enrollment, plasma HCV RNA level of >100,000 IU/ml at screening, and a liver biopsy within the past 3 years with histology consistent with HCV-related inflammation and fibrosis but no evidence of cirrhosis. Exclusion criteria included positive antibodies for hepatitis A or B virus or human immunodeficiency virus type 1 (HIV-1) or a history of clinically significant comorbidities. The primary endpoint was the maximum change from baseline in HCV RNA. The patients in the ombitasvir dose groups were enrolled sequentially, and within each group, patients were randomized (2:1) to either ombitasvir or placebo and treated under nonfasting conditions for 3 days while confined to the study site. The 200-mg dose group received a different formulation with higher bioavailability. Patients who received at least one dose of ombitasvir or placebo were provided the option to receive treatment with pegIFN/RBV for approximately 48 weeks once treatment with ombitasvir was completed. HCV RNA was measured using the Roche COBAS TaqMan HCV Test v2.0 real-time reverse transcriptase PCR assay (with a lower limit of quantification of 25 IU/ml and a lower limit of detection of 10 IU/ml). The virologic response was assessed as HCV RNA decrease from baseline in log10 IU/ml (40).
Resistance analysis of patient samples.
Resistance analysis was conducted on samples with ≥1,000 IU/ml HCV RNA at baseline (day 1), prior to dosing on day 3, and 72 h after dosing on day 3 (day 6). Viral RNA was isolated from plasma samples from HCV-infected patients by an automated method using the Abbott m2000 instrument (Abbott Molecular, Des Plaines, IL). RT-PCR using the Superscript III one-step RT-PCR system with Platinum Taq High Fidelity (Invitrogen, Carlsbad, CA) was performed using sense and antisense primers located outside the NS5A gene. Nested PCR was performed with Platinum Pfx DNA polymerase (Invitrogen, Carlsbad, CA) using sense and antisense primers specific for the NS5A gene. The NS5A PCR fragment was inserted into pJET1.2 vector (Fermentas, Glen Burnie, MD), plasmid DNA was isolated from individual colonies, and the NS5A region from the plasmid DNA was sequenced. Seventy-six clones, on average, were sequenced from each sample. Phenotypic analysis of clinical samples was conducted as described in the previous sections.
RESULTS
Activity in HCV replicons.
The activity of ombitasvir was previously presented by DeGoey et al. (28). Ombitasvir had EC50s of 14.1 and 5.0 pM against genotype 1a-H77 and 1b-Con1 subgenomic replicons, respectively. The antiviral activity of ombitasvir was attenuated 11- to 13-fold in the presence of 40% human plasma through sequestration of compound due to plasma protein binding. The CC50 of ombitasvir was greater than 32 μM, resulting in an in vitro therapeutic index that exceeded two millionfold. Ombitasvir demonstrated broad genotypic activity, with EC50s of 0.82 to 19.3 pM against genotype 2a, 2b, 3a, 4a, and 5a replicons and 366 pM against a genotype 6a replicon (Table 1).
TABLE 1.
Ombitasvir EC50s against stable subgenomic replicon cell lines
| HCV replicon subtype | Mean EC50 (pM) ± SD with human plasma ata: |
|
|---|---|---|
| 0% | 40% | |
| 1a-H77 | 14 ± 6.8 | 186 ± 63 |
| 1b-Con1 | 5.0 ± 1.9 | 56 ± 15 |
| 2a | 12 ± 2.7 | NDb |
| 2a JFH-1c | 0.82 ± 0.18 | ND |
| 2b | 4.3 ± 1.2 | ND |
| 3a | 19 ± 5.8 | ND |
| 4a | 1.7 ± 0.88 | ND |
| 5a | 3.2 ± 1.6 | ND |
| 6a | 366 ± 125 | ND |
Both the 0% and 40% human plasma assays also contain 5% fetal bovine serum.
ND, not determined.
Studies conducted at Southern Research Institute using qRT-PCR assay.
Selection of variants resistant to ombitasvir in genotype 1a, 1b, 2a, 2b, 3a, 4a, 5a, and 6a replicons.
The amino acid variants selected by ombitasvir across genotypes 1 to 6 are listed in Tables 2 and 3. The resistance profile of ombitasvir in genotype 1 was previously presented briefly by DeGoey et al. (28). In genotype 1a, the predominant variants selected by ombitasvir at 10-, 100-, or 100-fold over the EC50 were M28T, M28V, Q30R, Y93C, and Y93H. In the genotype 1a-H77 background, M28V conferred 58-fold resistance, while the M28T, Q30R, Y93C, and Y93H variants each conferred greater than 800-fold levels of resistance. The predominant variant selected by ombitasvir in genotype 1b was Y93H, which conferred 77-fold resistance to ombitasvir. In contrast to the observations for genotype 1a, single substitutions at amino acid position 28, 30, and 31 in genotype 1b conferred <10-fold resistance. At 100-fold or 1,000-fold over EC50 of ombitasvir, a number of clones contained double amino acid substitutions, primarily Y93H along with an additional substitution in the N-terminal region of NS5A, and the double variants conferred more than 400-fold resistance to ombitasvir.
TABLE 2.
Selection of resistant variants in NS5A by ombitasvir in genotype 1 replicon cell lines and resistance in transient assays
| Genotype | Variant | Prevalence in replicon assaysa |
Mean EC50 (pM) ± SDb | Fold resistance | Replication efficiency (%) | ||
|---|---|---|---|---|---|---|---|
| 10× | 100× | 1,000× | |||||
| 1a | Wild type | 2.7 ± 0.8 | 100 | ||||
| M28T | 1/23 | 4/21 | 4/24 | 24,475 ± 5,710 | 8,965 | 100 | |
| M28V | 10/23 | 159 ± 10 | 58 | 87 | |||
| Q30R | 4/23 | 4/21 | 8/24 | 2,184 ± 687 | 800 | 60 | |
| H58D | 1/21 | 664 ± 222 | 243 | 66 | |||
| Y93C | 4/23 | 5/21 | 3/24 | 4,573 ± 2,362 | 1,675 | 24 | |
| Y93H | 4/23 | 5/21 | 9/24 | 112,975 ± 47,977 | 41,383 | 18 | |
| Y93N | 1/21 | 182,200 ± 7,212 | 66,740 | 25 | |||
| Y93S | 1/21 | 1,013c | |||||
| Q30H | 7.7 ± 3.9 | 3 | 64 | ||||
| L31M | 4.8 ± 2.1 | 2 | 141 | ||||
| Q30L + Y93H | 1,137 ± 100 | 416 | 30 | ||||
| Q30L + Y93S | 596 ± 81 | 218 | 1 | ||||
| 1b | Wild type | 0.79 ± 0.25 | 100 | ||||
| L28T | 2/8d | 522 ± 279 | 661 | 17 | |||
| L31F | 4/23 | 7.6 ± 3.8 | 10 | 127 | |||
| L31V | 2/23 | 2/23 | 6.6 ± 1.2 | 8 | 86 | ||
| Y93H | 12/23 | 9/23 | 1/8 | 60 ± 22 | 77 | 73 | |
| L28M + L31F | 1/23 | 1/8 | 569c | ||||
| L28V + L31F | 1/8 | 2,170c | |||||
| L31F + P58L | 1/23 | 78c | |||||
| L31F + P58S | 1/23 | NDe | ND | ND | |||
| L28M + Y93N | 1/23 | ND | ND | ND | |||
| L28M + Y93H | 2/23 | 328 ± 231 | 415 | 104 | |||
| R30Q + Y93H | 1/23 | 7/23 | 224 ± 51 | 284 | 60 | ||
| L31F + Y93H | 2/8 | 8,115 ± 2,070 | 10,272 | 35 | |||
| L31V + Y93H | 1/8 | 9,739 ± 1,122 | 12,328 | 24 | |||
| P58A + Y93H | 2/23 | 967 ± 230 | 1,226 | 11 | |||
| P58L + Y93H | 1/23 | 107 ± 69 | 135 | 8 | |||
| L28M | 1.3 ± 0.1 | 2 | 114 | ||||
| R30Q | 0.31 ± 0.23 | 0.4 | 60 | ||||
| L31M | 0.7 ± 0.3 | 0.9 | 119 | ||||
Number of times that this variant was found/ total number of colonies analyzed.
EC50 of ombitasvir in replicon containing the variant in NS5A determined using transient luciferase assay.
Fold resistance was evaluated in a stable chimeric replicon cell line harboring the amino acid substitution; therefore, the EC50 in transient assay is not shown.
Only 8 colonies (out of 1 × 106 cells) survived exposure to 1,000-fold the EC50 of ombitasvir.
ND, not determined.
TABLE 3.
Selection of resistant variants in NS5A by ombitasvir in genotype 2 to 6 subgenomic chimeric replicon cell lines
| Genotype | Variant | Prevalence in replicon assaysa | Mean EC50 (pM) ± SDb | Fold resistance |
|---|---|---|---|---|
| 2ac | Wild type | 1.3 ± 0.1 | ||
| T24A | 8/15 | 50 ± 6.9 | 38 | |
| T24A (L31) | 20 ± 0.37 | 15 | ||
| F28S | 4/15 | 4,710d | ||
| 2bc | Wild type | 0.71 ± 0.37 | ||
| L28F | 1/24 | 33 ± 6.1 | 47 | |
| L28F (M31) | 176 ± 30 | 247 | ||
| L31V | 8/24 | 361 ± 70 | 511 | |
| Y93H | 13/24 | NA | ||
| 3a | wt | 4.4 ± 2.4 | ||
| M28T | 3/24 | 2,934 ± 1,618 | 659 | |
| L31F | 3/24 | 28d | ||
| Y93H | 14/24 | 29,940 ± 368 | 6,728 | |
| 4a | Wild type | 0.35 ± 0.07 | ||
| L28V | 21/21 | 8.0 ± 2.8 | 23 | |
| 5a | Wild type | 0.91 ± 0.34 | ||
| L28I | 6/23 | 72 ± 25 | 79 | |
| L31V | 12/23 | 220 ± 105 | 243 | |
| L31F | 6/23 | 263 ± 117 | 289 | |
| 6a | Wild type | 82 ± 58 | ||
| L31V | 4/24 | 5,572 ± 734 | 68 | |
| T58N | 4/24 | 8,354 ± 1,248 | 101 | |
| T58A | 8/24 | 1,452 ± 599 | 18 | |
| T58S | 6/24 | 1,504 ± 319 | 18 |
Number of times this variant was found/total number of colonies analyzed.
EC50 of ombitasvir in replicon containing the variant in NS5A determined using transient luciferase assay. NA, not available due to low replication efficiency of the variant.
The genotype 2a replicon cell line contains Met at position 31; the genotype 2b replicon cell line contains Leu at position 31.
F28S in GT 2a and L31F in GT 3a replicated poorly in a transient assay; therefore, the EC50 and fold resistance were evaluated in a stable chimeric replicon cell line.
Resistance selection was conducted at 50-fold above the EC50 in genotypes 2 to 5 and at 10-fold above the ombitasvir EC50 in the genotype 6a cell line, as at concentrations above these values, replicon cells did not survive in the presence of G418, indicating that the cells had been cleared of replicons.
In genotype 2a, the predominant variants selected were T24A and F28S, and in genotype 2b, the predominant variants selected were L31V and Y93H, while the L28F variant was observed in only one out of the 24 clones. In the European HCV database (41), amino acid position 31 in genotypes 2a and 2b is polymorphic with a prevalence of both methionine and leucine. Therefore, some of the resistant variants were constructed in the background of M31 as well as L31. In genotype 2a, both T24A (plus M31) and T24A (plus L31) were found to confer similar levels of resistance to ombitasvir. The genotype 2b L28F (plus L31) and L31V variants conferred 47-fold and 511-fold resistance, respectively; however, variant L28F in an M31 background was found to confer 248-fold resistance to ombitasvir. The predominant resistance-associated variant detected in genotype 3a was Y93H, which conferred 6728-fold resistance to ombitasvir. In genotype 4a, the only variant selected was L28V, and it conferred 23-fold resistance to ombitasvir. In genotype 5a, variants L28I, L31F, and L31V were observed, of which L28I conferred 79-fold resistance to ombitasvir, while both the L31F and L31V variants conferred over 240-fold resistance. In genotype 6a, L31V and several variants at T58 were selected, and these conferred 18- to 101-fold resistance to ombitasvir.
In summary, the key resistance-associated amino acid positions observed across genotypes 1 to 6 were 28, 30, 31, 58, and 93 in NS5A; however, the resistance conferred by variants at these amino acid positions to ombitasvir varied by genotype. Figure 2 shows an alignment of amino acids 1 to 100 of NS5A in the wild-type genotype 1 to 6 replicons, highlighting the signature resistance-associated amino acid positions in each genotype.
FIG 2.

Alignment of amino acids 1 to 100 of NS5A in the replicon cell lines. Amino acid changes relative to the 1b-Con1 sequence are indicated. Amino acids within each genotype where variants resistant to ombitasvir were selected are highlighted in gray.
Activity of ombitasvir against a panel of HCV genotype 1 to 6 isolates.
Given the genetic diversity of HCV and the degree of amino acid polymorphisms within the N-terminal region of NS5A, the activity of ombitasvir was evaluated against a panel of treatment-naive genotype 1 to 6 isolates in order to characterize its breadth of coverage (Table 4). The variability at signature resistance-associated amino acid positions relative to the consensus in the European HCV database was also analyzed by population sequencing (41), and the polymorphisms observed in the isolates are shown in Table 4. A total of 69 genotype 1 to 6 isolates were included in the panel. The EC50 of ombitasvir ranged from 0.1 to 15.1 pM against NS5A from 66 genotype 1 to 5 isolates. Polymorphisms at amino acid position 31 in NS5A in genotypes 2a and 2b or at amino acid position 28 in genotype 4a had no impact on activity of ombitasvir. In addition, ombitasvir was active against the genotype 2a JFH-1 replicon, with an EC50 of 0.82 pM. The EC50 of ombitasvir was higher against one genotype 2b sample with L28F plus M31 and one genotype 3a sample with the A30K variant. Only one genotype 6a sample, containing L28, was available for analysis. In order to better represent polymorphisms in genotype 6a isolates, L28F was introduced into the available genotype 6a replicon. The EC50s of ombitasvir were 42 pM and 68 pM against the L28 and F28 variants of this genotype 6a replicon, respectively.
TABLE 4.
Antiviral activity of ombitasvir in transient assays with HCV replicons containing NS5A genes from treatment-naive HCV-infected patients
| Genotype | No. isolates | EC50 (pM) |
|
|---|---|---|---|
| Range | Mean ± SDa | ||
| 1a | 11 | 0.35–0.88 | 0.66 ± 0.14 |
| 1b | 11 | 0.74–1.5 | 1.0 ± 0.23 |
| 2ab | 9 | 0.87–11 | 3.9 ± 3.0 |
| 2bc | 14 | 0.54–45 | 1.1 ± 1.2 |
| 3a | 13 | 0.90–55 | 4.5 ± 4.3 |
| 4ad | 9 | 0.10–0.36 | 0.24 ± 0.08 |
| 5a | 1 | 0.67 | |
| 6ae | 2 | 42–68 | 55 ± 18 |
Outliers due to presence of known resistance-associated variants were excluded when calculating the mean EC50s.
All the genotype 2a isolates had M31.
Of the genotype 2b isolates, 8 had L31 and 6 had M31.
Two of the genotype 4a isolates had M28 and 7 had L28.
Only 1 genotype 6a isolate was available. L28F, a common polymorphism, was introduced into this isolate.
Three-day monotherapy pharmacokinetics and antiviral efficacy.
In study M12-116, six treatment-naive patients infected with HCV genotype 1 were in each dose group (5, 50 or 200 mg once daily [QD]), with 4 patients administered active drug and 2 patients receiving a matched placebo for 3 days. Sixteen of the 18 patients enrolled in this study were infected with HCV genotype 1a, while 2 patients were infected with HCV genotype 1b, both of whom were randomized to the placebo group. Two of the patients randomized to the 50-mg group actually received 25 mg during monotherapy due to a dosing error. At baseline, the mean HCV RNA in the 18 patients was 6.32 log10 IU/ml. On day 3, ombitasvir dose-normalized maximum concentration (Cmax) and area under the concentration-time curve (AUC) values were similar across all doses. The Cmax ranged from 5.7 to 442 ng/ml, and the half-life ranged from 25.5 to 32.0 h (40). Figure 3 shows the individual HCV RNA viral load declines for the 12 HCV-infected patients who were administered ombitasvir. Similar robust antiviral responses were observed across all doses studied, with mean maximum decreases in HCV RNA of up to 3.10 log10 IU/ml observed during the 3-day monotherapy, while the mean decrease in HCV RNA was 0.15 log10 IU/ml in the placebo arm of the study. In general, ombitasvir was well tolerated across all doses administered. Most adverse events were mild, self-limiting, and of short duration and were considered not related or probably not related to ombitasvir. There was no dose-responsive pattern to the adverse events. No deaths or serious adverse events were reported (40).
FIG 3.
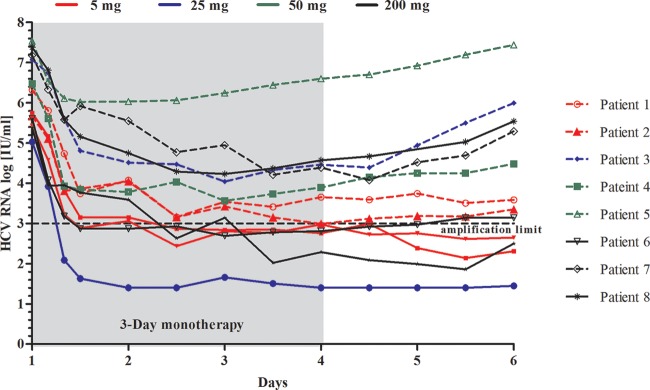
HCV RNA viral load during 3-day monotherapy with ombitasvir in HCV genotype 1-infected treatment-naive patients. Samples with HCV RNA levels of ≥1,000 IU/ml (amplification limit) at day 3 and day 6 were sequenced. Patients with available postbaseline sequencing data are indicated.
Evaluation of in vivo resistance development.
Clonal sequencing and phenotypic analysis of the NS5A-coding region were performed on all patients who received ombitasvir in this study for whom the viral titer at baseline, day 3, and/or day 6 was ≥1,000 IU/ml (Fig. 3 and Table 5). All 12 patients given ombitasvir in the 3 dose groups were infected with genotype 1a virus, and variants at resistance-associated amino acid position 28, 30, 31, 58, or 93 were not detected at baseline in any patient by clonal sequence analysis.
TABLE 5.
Phenotypic analysis and prevalence of signature NS5A resistance-conferring amino acid variants relative to baseline by clonal sequencing
| Dose QD (mg) and patient | Visit day | EC50 (pM) | Fold resistancea | Treatment-emergent variants at signature resistance-conferring amino acid positions in NS5A (% frequency)b |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Multiple variants | M28 |
Q30 |
Y93 |
|||||||||
| T | V | E | R | C | H | N | S | |||||
| 5 | ||||||||||||
| 1 | 3 | 19.4 | 41 | 100 | ||||||||
| 6 | 46.2 | 98 | 26 | 31 | 30 | |||||||
| 2 | 3 | 1.7 | 2.7 | 21 | 8 | |||||||
| 6 | 2.3 | 3.7 | 15 | 6 | ||||||||
| 25 | ||||||||||||
| 3 | 3 | 12.2 | 18 | 16 | 24 | 37 | 15 | |||||
| 6 | 24.5 | 36 | 16 | 52 | 14 | |||||||
| 50 | ||||||||||||
| 4 | 3 | 75.8 | 105 | 46 | 22 | 5 | 16 | |||||
| 6 | 254 | 353 | 49 | 13 | 25 | 5 | ||||||
| 5 | 3 | 180 | 219 | 62c | 10 | 22 | ||||||
| 6 | 333 | 406 | 44c | 11 | 39 | |||||||
| 200 | ||||||||||||
| 6 | 6 | 1,477 | 2,238 | 65 | 22 | 8 | ||||||
| 7 | 3 | 103 | 198 | 18 | 21 | 17 | 19 | |||||
| 6 | 482 | 927 | 40 | 35 | 6 | 6 | ||||||
| 8 | 3 | 963 | 2,006 | 34 | 55 | 5 | ||||||
| 6 | 892 | 1,858 | 40 | 50 | 5 | |||||||
Fold change in EC50 of ombitasvir in samples from day 3 or day 6 relative to the EC50 in the respective baseline sample.
Variants observed in ≥5% of the clones are shown.
Q30L + Y93H was the major double variant, and Q30L + Y93S was the minor variant.
In the 5-mg QD dose group, 2 of the 4 patients had HCV RNA viral loads of ≥1,000 IU/ml at both day 3 and day 6. Patient 1 had predominantly M28V at day 3 but had a mixture of M28T, M28V, and Q30R at day 6. Patient 2 had predominantly wild-type virus with M28V and Q30R as minor variants at both day 3 and day 6. Of the 2 patients who received the 25-mg dose, one (patient 3) had HCV RNA viral loads of ≥1,000 IU/ml at day 3 and day 6. In the sample from this patient, M28T, M28V, Q30R, and Y93C were observed on day 3; however, the M28V variant was no longer detected on day 6. Consistent with these sequencing observations, day 6 samples from patients 1, 2, and 3 conferred 98-, 4-, and 36-fold resistance to ombitasvir, respectively, in the phenotypic assay.
Of the 2 patients receiving the 50-mg dose of ombitasvir, M28T was the predominant variant in patient 4 at both days 3 and 6, although M28V, Q30E, Q30R, and Y93C were also detected. Patient 5 in the 50-mg dose group had an HCV RNA viral load nadir at 36 h after initiation of ombitasvir dosing, which was only a 1.52 log10 IU/ml decrease from the baseline value. Although preexisting resistance-associated variants were not detected at baseline, patient 5 had a complex mixture of variants at day 3 and day 6. Y93S and Q30L plus Y93H were the predominant variants, while Y93H and Q30L plus Y93S were also detected as minor variants. In vitro analysis of these variants in the HCV genotype 1a-H77 replicon (Table 2) indicated that resistance conferred by Q30L plus Y93H (416-fold) or Q30L plus Y93S (218-fold) was lower that that conferred by Y93H or Y93S alone. Consistent with the presence of resistant variants, day 6 samples from patients 4 and 5 conferred 353- and 406-fold resistance to ombitasvir, respectively, in the phenotypic assay.
In the 200-mg dose group, 3 of the 4 patients dosed with active drug had viral loads of ≥1,000 IU/ml HCV RNA at day 3 and/or day 6. M28T and Q30R were the predominant variants in all 3 patients at both day 3 and day 6, while Y93C and Y93H were observed as minor variants. Consistent with the presence of resistant variants, day 6 samples from patients 6, 7, and 8 conferred 927- to 2,238-fold resistance to ombitasvir in the phenotypic assay.
The clinical samples at days 3 or 6 usually contained a mixture of NS5A variants (including the wild type). Each of these variants may have differing replication fitness and variable susceptibility to ombitasvir, and the EC50 against clinical isolates is therefore a reflection of the multiple variants present in the quasispecies. The EC50s from reference replicons harboring single or multiple defined resistant variants are, in many cases, different from those for the clinical isolates.
DISCUSSION
Ombitasvir is an NS5A inhibitor with picomolar potency against genotype 1 to 6 replicons. In addition, ombitasvir retained picomolar levels of potency against a panel of 69 HCV genotype 1 to 6 clinical isolates, indicating that genetic diversity in the wild-type virus did not affect susceptibility to ombitasvir. Amino acid position 31 is known to be polymorphic in genotype 2, with both methionine and leucine prevalent in the HCV sequence database (41). While the presence of M31 in NS5A in genotype 2 is known to confer reduced susceptibility to NS5A inhibitors such as daclatasvir (>100-fold loss) and samatasvir (75-fold loss) (42, 43), ombitasvir retained activity against genotype 2 clinical isolates harboring either L31 or M31 in NS5A. The activity of ombitasvir against genotype 6a was attenuated 114-fold relative to the activity against genotype 5a. Both genotypes 5a and 6a have threonine at position 93 in NS5A rather than tyrosine; however, as genotype 6a also has threonine at position 58 versus proline in genotype 5a, it is possible that the T58 alone, or T58 in combination with T93, results in the reduced activity of ombitasvir against genotype 6a.
In the in vitro replicon system, the resistance-conferring variants selected by ombitasvir in genotype 1a were M28T, M28V, Q30R, Y93C, and Y93H. Of these, M28V conferred 58-fold resistance, whereas the other variants conferred greater than 800-fold resistance to ombitasvir. Consistent with this observation, M28V was not selected by exposure to 100-fold the EC50 of ombitasvir. In genotype 1b, Y93H, which conferred 77-fold resistance to ombitasvir, was the predominant variant. At exposures greater than 100-fold the EC50 of ombitasvir, resistant colonies with double variants, most commonly Y93H in combination with variants at amino acid position 28, 30, or 31, were selected. Overall, the most prevalent resistance-associated variants resulting from replicon assays with ombitasvir across all genotypes included variants at positions 28, 30, 31, 58, and 93. Variants Q30H (genotype 1a) and L31M (genotype 1a and 1b), which are known to confer resistance to daclatasvir and ledipasvir (43–45), remained susceptible to ombitasvir, as shown in Table 2. In the European HCV database, the preexisting prevalence of Q30H and L31M in genotype 1 ranges between 2.5 and 6%; the preexisting prevalence of the M28V variant in genotype 1a is around 6%, whereas the prevalence of M28T, Q30R, Y93C, and Y93H is each less than 1% (41). The preexisting prevalence of the Y93H variant in genotype 1b is approximately 5% (41). Thus, it would be expected that the most prevalent preexisting variants in genotype 1a (M28V) and genotype 1b (Y93H) would be suppressed in vivo with an ombitasvir drug level of 60- to 80-fold over the protein-adjusted wild-type EC50, which would be equivalent to 10 to 13 ng/ml.
Ombitasvir was evaluated in the dose-ranging clinical study M12-116, in which the maximum change from baseline in HCV RNA was evaluated along with a thorough resistance analysis of samples obtained within the first few days after initiation of ombitasvir dosing. In M12-116, 12 treatment-naive patients infected with HCV genotype 1 were administered 5, 25, 50, or 200 mg once daily of ombitasvir for 3 days, followed by an optional regimen of pegIFN and RBV for 48 weeks. All ombitasvir-treated patients were genotype 1a infected, and the mean maximum declines in HCV RNA observed across the doses studied were up to 3.10 log10 IU/ml during the 3-day monotherapy. None of the patients had preexisting resistance-conferring variants at signature amino acid position 28, 30, 31, 58, or 93 in NS5A detectable by clonal sequencing. Postbaseline samples from 8 patients were available for resistance analyses. Variants M28T, M28V, and Q30R in NS5A were the predominant treatment-emergent variants, while Y93C and Y93H were detected as minor variants. On day 3 of treatment or day 6 (48 h posttreatment), greater than 90% of the clones from each of the patient samples contained variants at signature resistance-conferring amino acid positions, indicating that in most patients, the wild-type virus had been suppressed. At the 5-mg dose, M28V was detected in two patients at day 3 and day 6; however, at higher doses where the Cmax values also increased, M28T or Q30R was the predominant variant. This is consistent with the observation in vitro that the M28V variant, which confers 58-fold resistance to ombitasvir, can be suppressed at higher doses of ombitasvir. In line with the genotypic observations, phenotypic analysis indicated that samples from patients in the 5-mg and 25-mg dose groups conferred 1- to <100-fold resistance to ombitasvir, while most samples from patients receiving 50 or 200 mg of ombitasvir conferred >500-fold resistance to ombitasvir. Although ombitasvir demonstrated similarly robust antiviral responses across all doses studied, the in vivo resistance profile suggest a benefit of using a dose higher than 5 mg of ombitasvir in order to suppress prevalent preexisting variants such as M28V in genotype 1a or Y93H in genotype 1b.
The in vitro profile of ombitasvir and the results in the 3-day monotherapy study M12-116 provided the basis for investigating the combination of ombitasvir with the NS3/4A protease inhibitor ABT-450 and nonnucleoside NS5B polymerase inhibitor dasabuvir (ABT-333) in treatment of chronic genotype 1 HCV infection. The combination of these 3 DAAs provides a barrier to resistance in patients, as evidenced by the high SVR rate in the six phase 3 clinical trials using an interferon-free combination of ombitasvir/ABT-450/ritonavir and dasabuvir with or without ribavirin (30–34).
ACKNOWLEDGMENTS
We thank the trial participants, investigators, and coordinators who made this study possible. We thank Barbara McGovern for critical review of the manuscript.
The design, study conduct, and financial support for this study were provided by AbbVie.
AbbVie participated in the interpretation of data, review, and approval of the publication. All authors are employees of AbbVie and may own AbbVie stock.
REFERENCES
- 1.Lavanchy D. 2011. Evolving epidemiology of hepatitis C virus. Clin Microbiol Infect 17:107–115. doi: 10.1111/j.1469-0691.2010.03432.x. [DOI] [PubMed] [Google Scholar]
- 2.Mohd Hanafiah K, Groeger J, Flaxman AD, Wiersma ST. 2013. Global epidemiology of hepatitis C virus infection: new estimates of age-specific antibody to HCV seroprevalence. Hepatology 57:1333–1342. doi: 10.1002/hep.26141. [DOI] [PubMed] [Google Scholar]
- 3.Smith DB, Bukh J, Kuiken C, Muerhoff AS, Rice CM, Stapleton JT, Simmonds P. 2014. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: updated criteria and genotype assignment Web resource. Hepatology 59:318–327. doi: 10.1002/hep.26744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blatt LM, Mutchnick MG, Tong MJ, Klion FM, Lebovics E, Freilich B, Bach N, Smith C, Herrera J, Tobias H, Conrad A, Schmid P, McHutchison JG. 2000. Assessment of hepatitis C virus RNA and genotype from 6807 patients with chronic hepatitis C in the United States. J Viral Hepat 7:196–202. doi: 10.1046/j.1365-2893.2000.00221.x. [DOI] [PubMed] [Google Scholar]
- 5.Zein NN. 2000. Clinical significance of hepatitis C virus genotypes. Clin Microbiol Rev 13:223–235. doi: 10.1128/CMR.13.2.223-235.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Toyoda H, Kumada T, Takaguchi K, Shimada N, Tanaka J. 2014. Changes in hepatitis C virus genotype distribution in Japan. Epidemiol Infect 142:2624–2628. doi: 10.1017/S0950268814000478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zein NN, Persing DH. 1996. Hepatitis C genotypes: current trends and future implications. Mayo Clin Proc 71:458–462. doi: 10.4065/71.5.458. [DOI] [PubMed] [Google Scholar]
- 8.Nousbaum JB. 1998. Genomic subtypes of hepatitis C virus: epidemiology, diagnosis and clinical consequences. Bull Soc Pathol Exot 91:29–33. [PubMed] [Google Scholar]
- 9.Adams NJ, Chamberlain RW, Taylor LA, Davidson F, Lin CK, Elliott RM, Simmonds P. 1997. Complete coding sequence of hepatitis C virus genotype 6a. Biochem Biophys Res Commun 234:393–396. doi: 10.1006/bbrc.1997.6627. [DOI] [PubMed] [Google Scholar]
- 10.Chamberlain RW, Adams N, Saeed AA, Simmonds P, Elliott RM. 1997. Complete nucleotide sequence of a type 4 hepatitis C virus variant, the predominant genotype in the Middle East. J Gen Virol 78:1341–1347. [DOI] [PubMed] [Google Scholar]
- 11.Chamberlain RW, Adams NJ, Taylor LA, Simmonds P, Elliott RM. 1997. The complete coding sequence of hepatitis C virus genotype 5a, the predominant genotype in South Africa. Biochem Biophys Res Commun 236:44–49. doi: 10.1006/bbrc.1997.6902. [DOI] [PubMed] [Google Scholar]
- 12.Simmonds P. 2004. Genetic diversity and evolution of hepatitis C virus—15 years on. J Gen Virol 85:3173–3188. doi: 10.1099/vir.0.80401-0. [DOI] [PubMed] [Google Scholar]
- 13.Guedj J, Dahari H, Rong L, Sansone ND, Nettles RE, Cotler SJ, Layden TJ, Uprichard SL, Perelson AS. 2013. Modeling shows that the NS5A inhibitor daclatasvir has two modes of action and yields a shorter estimate of the hepatitis C virus half-life. Proc Natl Acad Sci U S A 110:3991–3996. doi: 10.1073/pnas.1203110110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pawlotsky JM. 2012. The science of direct-acting antiviral and host-targeted agent therapy. Antivir Ther 17:1109–1117. doi: 10.3851/IMP2423. [DOI] [PubMed] [Google Scholar]
- 15.Pawlotsky JM. 2014. New hepatitis C therapies: the toolbox, strategies, and challenges. Gastroenterology 146:1176–1192. doi: 10.1053/j.gastro.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 16.Jacobson IM, Pawlotsky JM, Afdhal NH, Dusheiko GM, Forns X, Jensen DM, Poordad F, Schulz J. 2012. A practical guide for the use of boceprevir and telaprevir for the treatment of hepatitis C. J Viral Hepat 19(Suppl 2):S1–S26. doi: 10.1111/j.1365-2893.2012.01590.x. [DOI] [PubMed] [Google Scholar]
- 17.Jacobson IM, McHutchison JG, Dusheiko G, Di Bisceglie AM, Reddy KR, Bzowej NH, Marcellin P, Muir AJ, Ferenci P, Flisiak R, George J, Rizzetto M, Shouval D, Sola R, Terg RA, Yoshida EM, Adda N, Bengtsson L, Sankoh AJ, Kieffer TL, George S, Kauffman RS, Zeuzem S. 2011. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med 364:2405–2416. doi: 10.1056/NEJMoa1012912. [DOI] [PubMed] [Google Scholar]
- 18.Jacobson I, Dore G, Foster G, Fried M, Radu M, Rafalskiy V, Moroz I, Craxi A, Peeters M, Lenz O, Ouwerkerk-Mahadevan S, Kalmeijer R, Beumont-Mauviel M. 2013. Simeprevir (TMC435) with peginterferon/ribavirin for chronic HCV genotype-1 infection in treatment-naive parients: results from QUEST-1, a phase III trial. J Hepatol 58:S574. doi: 10.1016/S0168-8278(13)61424-5. [DOI] [Google Scholar]
- 19.Manns M, Marcellin P, Poordad F, Stanislau Affonso de Araujo E, Buti M, Horsmans Y, Janczewska E, Villamil F, Peeters M, Lenz O, Ouwerkerk-Mahadevan S, Kalmeijer R, Beumont-Mauviel M. 2013. Simeprevir (TMC435) with perinterferon/ribavirin for treatment of chronic HCV genotype-1 infection in treatment-naive patients: results from QUEST-2, a phase III trial. J Hepatol 58:S568. doi: 10.1016/S0168-8278(13)61413-0. [DOI] [Google Scholar]
- 20.Lawitz E, Mangia A, Wyles D, Rodriguez-Torres M, Hassanein T, Gordon SC, Schultz M, Davis MN, Kayali Z, Reddy KR, Jacobson IM, Kowdley KV, Nyberg L, Subramanian GM, Hyland RH, Arterburn S, Jiang D, McNally J, Brainard D, Symonds WT, McHutchison JG, Sheikh AM, Younossi Z, Gane EJ. 2013. Sofosbuvir for previously untreated chronic hepatitis C infection. N Engl J Med 368:1878–1887. doi: 10.1056/NEJMoa1214853. [DOI] [PubMed] [Google Scholar]
- 21.Tellinghuisen TL, Foss KL, Treadaway J. 2008. Regulation of hepatitis C virion production via phosphorylation of the NS5A protein. PLoS Pathog 4:e1000032. doi: 10.1371/journal.ppat.1000032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Macdonald A, Harris M. 2004. Hepatitis C virus NS5A: tales of a promiscuous protein. J Gen Virol 85:2485–2502. doi: 10.1099/vir.0.80204-0. [DOI] [PubMed] [Google Scholar]
- 23.Huang L, Hwang J, Sharma SD, Hargittai MR, Chen Y, Arnold JJ, Raney KD, Cameron CE. 2005. Hepatitis C virus nonstructural protein 5A (NS5A) is an RNA-binding protein. J Biol Chem 280:36417–36428. doi: 10.1074/jbc.M508175200. [DOI] [PubMed] [Google Scholar]
- 24.Hughes M, Griffin S, Harris M. 2009. Domain III of NS5A contributes to both RNA replication and assembly of hepatitis C virus particles. J Gen Virol 90:1329–1334. doi: 10.1099/vir.0.009332-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pawlotsky JM. 2013. NS5A inhibitors in the treatment of hepatitis C. J Hepatol 59:375–382. doi: 10.1016/j.jhep.2013.03.030. [DOI] [PubMed] [Google Scholar]
- 26.Krueger AC, Madigan DL, Beno DW, Betebenner DA, Carrick R, Green BE, He W, Liu D, Maring CJ, McDaniel KF, Mo H, Molla A, Motter CE, Pilot-Matias TJ, Tufano MD, Kempf DJ. 2012. Novel hepatitis C virus replicon inhibitors: synthesis and structure-activity relationships of fused pyrimidine derivatives. Bioorg Med Chem Lett 22:2212–2215. doi: 10.1016/j.bmcl.2012.01.096. [DOI] [PubMed] [Google Scholar]
- 27.DeGoey DA, Grampovnik DJ, Liu D, Pratt JK, Krishnan P, Pilot-Matias TJ, Marsh KC, Molla A, Kempf DJ, Maring CJ. 2013. Discovery of pyrido[2,3-d]pyrimidine-based inhibitors of HCV NS5A. Bioorg Med Chem Lett 23:3627–3630. doi: 10.1016/j.bmcl.2013.04.009. [DOI] [PubMed] [Google Scholar]
- 28.DeGoey DA, Randolph JT, Liu D, Pratt J, Hutchins C, Donner P, Krueger AC, Matulenko M, Patel S, Motter CE, Nelson L, Keddy R, Tufano M, Caspi DD, Krishnan P, Mistry N, Koev G, Reisch TJ, Mondal R, Pilot-Matias T, Gao Y, Beno DW, Maring CJ, Molla A, Dumas E, Campbell A, Williams L, Collins C, Wagner R, Kati WM. 2014. Discovery of ABT-267, a pan-genotypic inhibitor of HCV NS5A. J Med Chem 57:2047–2057. doi: 10.1021/jm401398x. [DOI] [PubMed] [Google Scholar]
- 29.Pilot-Matias T, Tripathi R, Cohen D, Gaultier I, Dekhtyar T, Lu L, Reisch T, Irvin M, Hopkins T, Pithawalla R, Middleton T, Ng T, McDaniel K, Or YS, Menon R, Kempf D, Molla A, Collins C. 2015. In vitro and in vivo antiviral activity and resistance profile of the hepatitis C virus NS3/4A protease inhibitor ABT-450. Antimicrob Agents Chemother 59:988–997. doi: 10.1128/AAC.04227-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Feld JJ, Kowdley KV, Coakley E, Sigal S, Nelson DR, Crawford D, Weiland O, Aguilar H, Xiong J, Pilot-Matias T, DaSilva-Tillmann B, Larsen L, Podsadecki T, Bernstein B. 2014. Treatment of HCV with ABT-450/r-ombitasvir and dasabuvir with ribavirin. N Engl J Med 370:1594–1603. doi: 10.1056/NEJMoa1315722. [DOI] [PubMed] [Google Scholar]
- 31.Ferenci P, Bernstein D, Lalezari J, Cohen D, Luo Y, Cooper C, Tam E, Marinho RT, Tsai N, Nyberg A, Box TD, Younes Z, Enayati P, Green S, Baruch Y, Bhandari BR, Caruntu FA, Sepe T, Chulanov V, Janczewska E, Rizzardini G, Gervain J, Planas R, Moreno C, Hassanein T, Xie W, King M, Podsadecki T, Reddy KR. 2014. ABT-450/r-ombitasvir and dasabuvir with or without ribavirin for HCV. N Engl J Med 370:1983–1992. doi: 10.1056/NEJMoa1402338. [DOI] [PubMed] [Google Scholar]
- 32.Poordad F, Hezode C, Trinh R, Kowdley KV, Zeuzem S, Agarwal K, Shiffman ML, Wedemeyer H, Berg T, Yoshida EM, Forns X, Lovell SS, Da Silva-Tillmann B, Collins CA, Campbell AL, Podsadecki T, Bernstein B. 2014. ABT-450/r-ombitasvir and dasabuvir with ribavirin for hepatitis C with cirrhosis. N Engl J Med 370:1973–1982. doi: 10.1056/NEJMoa1402869. [DOI] [PubMed] [Google Scholar]
- 33.Zeuzem S, Jacobson IM, Baykal T, Marinho RT, Poordad F, Bourliere M, Sulkowski MS, Wedemeyer H, Tam E, Desmond P, Jensen DM, Di Bisceglie AM, Varunok P, Hassanein T, Xiong J, Pilot-Matias T, DaSilva-Tillmann B, Larsen L, Podsadecki T, Bernstein B. 2014. Retreatment of HCV with ABT-450/r-ombitasvir and dasabuvir with ribavirin. N Engl J Med 370:1604–1614. doi: 10.1056/NEJMoa1401561. [DOI] [PubMed] [Google Scholar]
- 34.Andreone P, Colombo MG, Enejosa JV, Koksal I, Ferenci P, Maieron A, Müllhaupt B, Horsmans Y, Weiland O, Reesink HW, Rodrigues L, Hu YB, Podsadecki T, Bernstein B. 9 May 2014. ABT-450, ritonavir, ombitasvir, and dasabuvir achieves 97% and 100% sustained virologic response with or without ribavirin in treatment-experienced patients with HCV genotype 1b infection. Gastroenterology doi: 10.1053/j.gastro.2014.04.045. [DOI] [PubMed] [Google Scholar]
- 35.Halfman CJ. 1981. Concentrations of binding protein and labeled analyte that are appropriate for measuring at any analyte concentration range in radioimmunoassays. Methods Enzymol 74:481–497. doi: 10.1016/0076-6879(81)74034-5. [DOI] [PubMed] [Google Scholar]
- 36.Kato T, Date T, Miyamoto M, Furusaka A, Tokushige K, Mizokami M, Wakita T. 2003. Efficient replication of the genotype 2a hepatitis C virus subgenomic replicon. Gastroenterology 125:1808–1817. doi: 10.1053/j.gastro.2003.09.023. [DOI] [PubMed] [Google Scholar]
- 37.Hopkins S, Scorneaux B, Huang Z, Murray MG, Wring S, Smitley C, Harris R, Erdmann F, Fisher G, Ribeill Y. 2010. A novel nonimmunosuppressive analog of cyclosporin a that exhibits potent inhibition of hepatitis C virus RNA replication in vitro. Antimicrob Agents Chemother 54:660–672. doi: 10.1128/AAC.00660-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tripathi RL, Krishnan P, He Y, Middleton T, Pilot-Matias T, Chen CM, Lau DT, Lemon SM, Mo H, Kati W, Molla A. 2007. Replication efficiency of chimeric replicon containing NS5A-5B genes derived from HCV-infected patient sera. Antiviral Res 73:40–49. doi: 10.1016/j.antiviral.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 39.Mo H, Lu L, Pilot-Matias T, Pithawalla R, Mondal R, Masse S, Dekhtyar T, Ng T, Koev G, Stoll V, Stewart KD, Pratt J, Donner P, Rockway T, Maring C, Molla A. 2005. Mutations conferring resistance to a hepatitis C virus (HCV) RNA-dependent RNA polymerase inhibitor alone or in combination with an HCV serine protease inhibitor in vitro. Antimicrob Agents Chemother 49:4305–4314. doi: 10.1128/AAC.49.10.4305-4314.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lawitz E, Marbury T, Campbell A, Dumas E, Pilot-Matias T, Krishnan P, Setze S, Wangang X, Podsadecki T, Bernstein B, Williams L. 2012. Safety and antiviral activity of ABT-267, a novel NS5A inhibitor, during 3-day monotherapy: first study in HCV genotype-1 (GT1)-infected treatment-naive subjects. J Hepatol 56:S469–S470. doi: 10.1016/S0168-8278(12)61198-2. [DOI] [Google Scholar]
- 41.Combet C, Garnier N, Charavay C, Grando D, Crisan D, Lopez J, Dehne-Garcia A, Geourjon C, Bettler E, Hulo C, Le Mercier P, Bartenschlager R, Diepolder H, Moradpour D, Pawlotsky JM, Rice CM, Trepo C, Penin F, Deleage G. 2007. euHCVdb: the European hepatitis C virus database. Nucleic Acids Res 35:D363–D366. doi: 10.1093/nar/gkl970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Scheel TK, Gottwein JM, Mikkelsen LS, Jensen TB, Bukh J. 2011. Recombinant HCV variants with NS5A from genotypes 1-7 have different sensitivities to an NS5A inhibitor but not interferon-alpha. Gastroenterology 140:1032–1042. doi: 10.1053/j.gastro.2010.11.036. [DOI] [PubMed] [Google Scholar]
- 43.McCarville J, Seifer M, Standring D, Mayers D. 2013. Treatment-emergent variants following 3 days of monotherapy with IDX719, a potent, pan-genotypic NS5A inhibitor, in subjects infected with HCV genotypes 1-4. J Hepatol 58:S491–S492. doi: 10.1016/S0168-8278(13)61210-6. [DOI] [Google Scholar]
- 44.Fridell RA, Qiu D, Wang C, Valera L, Gao M. 2010. Resistance analysis of the hepatitis C virus NS5A inhibitor BMS-790052 in an in vitro replicon system. Antimicrob Agents Chemother 54:3641–3650. doi: 10.1128/AAC.00556-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wong KA, Worth A, Martin R, Svarovskaia E, Brainard DM, Lawitz E, Miller MD, Mo H. 2013. Characterization of hepatitis C virus resistance from a multiple-dose clinical trial of the novel NS5A inhibitor GS-5885. Antimicrob Agents Chemother 57:6333–6340. doi: 10.1128/AAC.02193-12. [DOI] [PMC free article] [PubMed] [Google Scholar]