

Abstract
Chronicity of hepatitis B virus (HBV) infection is due to the failure of a host to mount a sufficient immune response to clear the virus. The aim of this study was to identify small-molecular agonists of the pattern recognition receptor (PRR)-mediated innate immune response to control HBV infection. To achieve this goal, a coupled mouse macrophage and hepatocyte culture system mimicking the intrahepatic environment was established and used to screen small-molecular compounds that activate macrophages to produce cytokines, which in turn suppress HBV replication in a hepatocyte-derived stable cell line supporting HBV replication in a tetracycline-inducible manner. An agonist of the mouse stimulator of interferon (IFN) genes (STING), 5,6-dimethylxanthenone-4-acetic acid (DMXAA), was found to induce a robust cytokine response in macrophages that efficiently suppressed HBV replication in mouse hepatocytes by reducing the amount of cytoplasmic viral nucleocapsids. Profiling of cytokines induced by DMXAA and agonists of representative Toll-like receptors (TLRs) in mouse macrophages revealed that, unlike TLR agonists that induced a predominant inflammatory cytokine/chemokine response, the STING agonist induced a cytokine response dominated by type I IFNs. Moreover, as demonstrated in an HBV hydrodynamic mouse model, intraperitoneal administration of DMXAA significantly induced the expression of IFN-stimulated genes and reduced HBV DNA replication intermediates in the livers of mice. This study thus proves the concept that activation of the STING pathway induces an antiviral cytokine response against HBV and that the development of small-molecular human STING agonists as immunotherapeutic agents for treatment of chronic hepatitis B is warranted.
INTRODUCTION
Hepatitis B virus (HBV) is a noncytopathic hepadnavirus that chronically infects more than 350 million people worldwide. The outcomes and pathogenesis of HBV infections are largely determined by the nature and magnitude of the host antiviral immune response (1), which is generally related to the person's age at the time of infection. While over 95% of adult-acquired infections are spontaneously cleared within 6 months by a vigorous and polyclonal HBV-specific T cell response, more than 90% of exposed neonates and approximately 30% of children 1 to 5 years old develop a chronic infection (2, 3), which is associated with a weaker and often barely detectable virus-specific T cell response.
Sustained suppression of viral replication by long-term nucleos(t)ide analogue therapy or through a finite duration of pegylated alpha interferon (IFN-α) therapy has been associated with improvement of liver diseases, prevention of liver decompensation, and reduction of hepatocellular carcinoma morbidity and mortality (4, 5). However, a “functional cure,” characterized by normalization of liver function, HBV surface antigen (HBsAg) seroconversion, and durable immune control of HBV replication, is rarely achieved with the current therapies (6, 7). Hence, the restoration of host innate and HBV-specific adaptive immune responses is essential for a functional cure of chronic HBV infection (8).
Although hepatocytes are the primary host cells of HBV, hepatic nonparenchymal cells (NPCs) have been shown to play a critical role in the priming of effective HBV-specific antiviral immunity (9, 10). For instance, it was demonstrated recently that activation of hepatic macrophages induces the expression of a distinct profile of cytokines/chemokines that regulate the priming of a successful immune response against HBV in the livers of mice (10). It is therefore conceivable that pharmacological activation of Kupffer cells and/or intrahepatic dendritic cells with agonists of Toll-like receptors (TLRs) or other pattern recognition receptors (PRRs) may facilitate the intrahepatic priming of an anti-HBV immune response. In support of this hypothesis, it has been demonstrated recently in woodchucks and chimpanzees that administration of TLR7 agonists not only induced an innate cytokine response that suppresses HBV replication but also shaped the adaptive immune response to achieve durable control of HBV infection (11). In addition, studies with HBV transgenic mice also demonstrated that activation of myeloid dendritic cells with an agonistic anti-CD40 antibody not only induced an intrahepatic cytokine response to control HBV replication (12, 13) but also restored HBV-specific, PD-1-mediated CD8+ T cell exhaustion (14).
Stimulator of IFN genes (STING) is the adaptor protein of multiple cytoplasmic DNA receptors and a PRR that recognizes the bacterial second messengers cyclic di-AMP (c-di-AMP) and c-di-GMP (15). It was discovered recently that cytoplasmic DNA activates cyclic GMP-AMP synthase (cGAS) to produce cGAMP, which subsequently binds to STING and induces IFNs and other cytokines (16, 17). The fact that STING can be activated by cyclic dinucleotides implies that, like TLR7/8, STING might be activated by other small molecules and thus be a potential target for pharmacological activation of the innate immune response, as well as priming of an adaptive immune response. Indeed, some previously identified flavonoid IFN inducers, including 10-(carboxymethyl)-9(10H)acridone (CMA) and 5,6-dimethylxanthenone-4-acetic acid (DMXAA), were recently identified as murine STING agonists (18, 19). DMXAA (Vadimezan or ASA404) was initially developed as a vascular disrupting agent with antitumor activity partially through activation of natural killer (NK) cells and tumor-associated macrophages in various mouse models and has been evaluated in phase II clinical trials for treating cancers (18). In studies reported here, we demonstrated that DMXAA activated a STING-dependent signaling pathway to induce a type I IFN-dominant cytokine response in mouse macrophages, which efficiently suppressed HBV replication in cultured murine hepatocytes and in the livers of mice by reducing the amount of cytoplasmic viral nucleocapsids. Our work has thus validated STING as a valuable target for the immunotherapy of chronic hepatitis B, and the development of small-molecular human STING agonists as immunotherapeutics against chronic HBV infections is warranted.
MATERIALS AND METHODS
Cell culture.
Murine macrophage cell line RAW264.7 (ATCC TIB-71) and GP2-293 cells (Clontech) were maintained in Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum. AML12HBV10, an immortalized murine hepatocyte (AML12)-derived cell line supporting a high level of HBV replication in a tetracycline-inducible manner, was maintained as previously described (20, 21).
Reagents.
DMXAA, CMA, and 2,7-bis(2-[diethylamino]ethoxy)-9-fluorenone (tilorone) were purchased from Sigma-Aldrich. The TLR1/2 agonist Pam3CSK4, the TLR3 agonist poly(I:C), the TLR4 agonist lipopolysaccharide (LPS), and the TLR7 agonist Gardiquimod were from InvivoGen. Recombinant murine IFN-α, interleukin-1 (IL-1), IL-6, and tumor necrosis factor alpha (TNF-α) were from PBL InterferonSource. An antibody against the carboxyl-terminal 14 amino acids of the HBV core protein was described previously (21). Antibodies against β-actin and mouse IFNAR-1 were obtained from Sigma-Aldrich and Santa Cruz Biotechnology, respectively. Antibodies against mouse STING, TBK1, S172-phosphorylated TBK1, IkBα, p38, phosphorylated p38, JNK, phosphorylated JNK, ERK, and phosphorylated ERK, were purchased from Cell Signaling Technology. Plasmids pTmcs-HBV1.3 and pCMV-SB were kind gifts of Francis V. Chisari (The Scripps Research Institute, La Jolla, CA).
Analyses of HBV DNA, RNA, and nucleocapsids.
HBV core DNA extraction from AML12HBV10 cells, Southern blot hybridization, and real-time PCR assays were done as described previously (21). Total cellular RNA was extracted with TRIzol reagent (Invitrogen). HBV RNA was analyzed by Northern blot hybridization with an [α-32P]UTP-labeled full-length minus-stranded RNA probe. HBV capsid and capsid-associated viral DNA were analyzed by native agarose gel electrophoresis, followed by transfer to a nitrocellulose membrane. HBV capsids were detected by probing the membrane with an antibody against HBV core protein, followed by visualization with the LI-COR Odyssey system. Capsid-associated DNA was detected by hybridization with radioactively labeled HBV riboprobe (21).
Real-time qRT-PCR assays.
For cytokine gene expression analysis, total RNA was extracted with TRIzol reagent. cDNAs were synthesized with SuperScript III (Invitrogen). Real-time quantitative reverse transcription (qRT)-PCR analysis was performed with a LightCycler 480 instrument II (Roche). Primer sequence information is available upon request.
Generation of a STING knockdown cell line.
A plasmid expressing short hairpin RNA (shRNA) specifically targeting murine STING was constructed by inserting the following cDNA sequence into the pRS vector (Origene): TCAATCAGCTACATAACAACTCGAGTTGTTATGTAGCTGATTGA. A control plasmid expressing scrambled shRNA from the pRS vector was purchased from Origene. Packaging of vesicular stomatitis virus G protein-pseudotyped retroviruses into GP2-293 cells with pCMV/VSV-G and a pRS vector-derived plasmid expressing scrambled shRNA or STING-specific shRNA was done essentially as reported previously (22). RAW264.7 cells were transduced with pseudotyped retroviruses expressing each of the shRNAs as previously described (22).
Antiviral efficacy of DMXAA in HBV DNA hydrodynamic mouse model.
Ten-week-old female NOD/SCID mice were purchased from the Vital River Laboratory Animal Technology Co. Ltd. All experiments were conducted with Institutional Animal Care and Use Committee approval. To establish an HBV hydrodynamic model, 13.5 μg of plasmid pTmcsHBV1.3 expressing 1.3mer HBV genome and 4.5 μg of pCMV-SB expressing the Sleeping Beauty transposase were injected through the tail vein as previously described (23, 24). To test the efficacy of DMXAA in vivo, at 7 days postinjection, mice were treated with either DMXAA (in phosphate-buffered saline with 7.5% sodium bicarbonate) at 25 mg/kg or the vehicle via intraperitoneal injection. Mice were sacrificed at 24 h after treatment. Intrahepatic HBV core DNA was extracted and quantified by with a real-time PCR assay. Intrahepatic total RNA was extracted, and the IFN-stimulated gene (ISG) OAS1b and viperin mRNAs were determined by real-time RT-PCR assay. The weights of individual mice were measured before and 24 h after treatment.
RESULTS
Establishment of a cell culture system for evaluation of PRR agonist-induced antiviral response against HBV.
Unlike RIG-I-like receptors that are ubiquitously expressed in many types of somatic cells, the expression of other PRRs, such as TLRs and cGAS, is usually restricted to macrophages, dendritic cells, and a few other cell types. For instance, because of the lack or low-level expression of TLRs in hepatocytes, direct treatment of hepatocytes with TLR agonists induces a negligible cytokine response. However, some hepatic NPCs, particularly Kupffer cells and dendritic cells, express high levels of TLRs (25, 26). Hence, our strategy to achieve a “functional cure” of chronic HBV infection is to activate intrahepatic macrophages and/or dendritic cells with the agonist of TLRs or other PRRs, which should not only induce an antiviral cytokine response to suppress HBV replication in hepatocytes (26–28) but also promote the priming and activation of an HBV-specific adaptive immune response to ultimately control the virus infection (25, 26, 29).
Accordingly, we have developed a cell-based assay mimicking the intrahepatic environment to screen small-molecular PRR agonists for the treatment of chronic hepatitis B. Specifically, as depicted in Fig. 1A, mouse macrophages (RAW264.7) were treated with test compounds and the conditioned media of treated macrophages were then applied to immortalized mouse hepatocytes harboring HBV (AML12HBV10) to test the compound-induced antiviral cytokine response in macrophages. The assay system was first validated with known TLR agonists. As shown in Fig. 1B, while direct treatment of AML12HBV10 cells with the agonist of TLR1/2, TLR3, TLR4, or TLR7 did not apparently inhibit HBV DNA replication, treatment of AML12HBV10 cells with the conditioned media harvested from TLR agonist-treated macrophages (indirect treatment) effectively inhibited HBV DNA replication. The results thus confirmed and extended previous findings that, although they failed to directly activate an antiviral response in hepatocytes, TLR agonists induced macrophages to produce soluble factors that suppress HBV replication in hepatocytes (25, 29).
FIG 1.
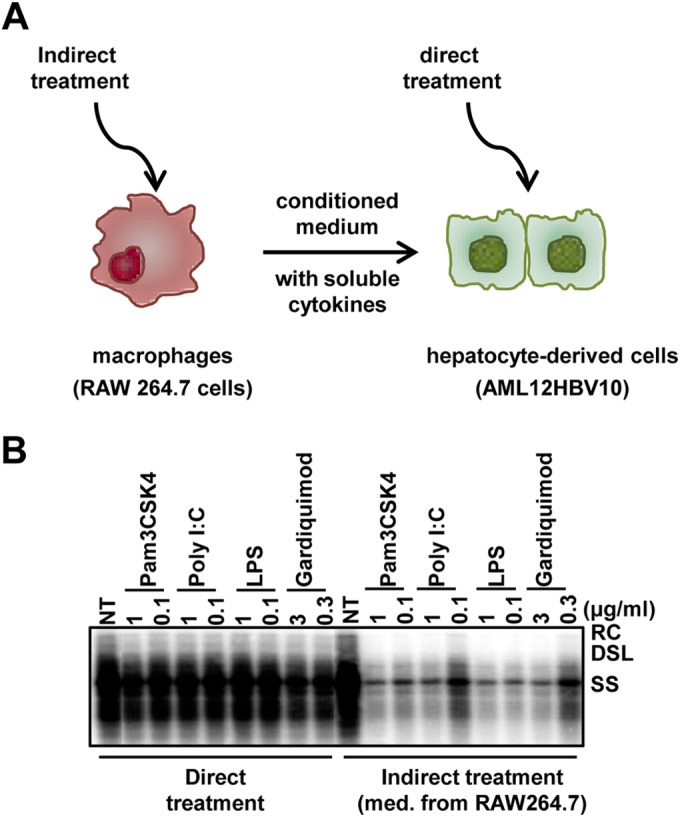
Suppression of HBV replication in hepatocytes by TLR response activation in macrophages. (A) Schematic representation of the assay strategy used. (B) AML12HBV10 cells were seeded into a 12-well plate at a density of 1 × 105/well and cultured in the absence of tetracycline. Twenty-four hours later, the cells were treated with the TLR agonist at the indicated concentrations (direct treatment). Alternatively, AML12HBV10 cells were treated with media containing 50% of the conditioned media harvested from TLR agonist-treated RAW264.7 cells (cultured in a 12-well plate at a density of 5 × 105/well and treated for 12 h with the TLR agonist at the indicated concentrations) (indirect treatment). Cytoplasmic HBV core DNA was analyzed by Southern blot hybridization 2 days posttreatment. RC, relaxed circular DNA; DSL, double-stranded linear DNA; SS, single-stranded DNA; NT, no-treatment control, which consisted of samples from mock-treated AML12HBV10 cells (direct-treatment control) or from AML12HBV10 cells treated with 50% of the media from mock-treated RAW264.7 cells (indirect-treatment control).
DMXAA-induced antiviral response in macrophages suppressed HBV replication in hepatocytes.
Using the assay system described above, we initially screened 75 flavonoids in the MicroSource compound collection (MicroSource Discovery Systems, Inc.) and identified a flavonoid, 7,2′-dihydroxyflavone, that demonstrated indirect, but not direct, antiviral activity against HBV (data not shown). Encouraged by this finding, we performed a literature search for additional flavonoids with IFN-inducing activity that revealed three compounds, tilorone, CMA, and DMXAA (Fig. 2A), that were identified as potent IFN inducers in mice several decades ago (30–32). While tilorone neither directly nor indirectly inhibited HBV replication, both CMA and DMXAA demonstrated strong indirect anti-HBV activity in our assay system (Fig. 2B). Interestingly, it was recently reported by several groups that both CMA and DMXAA are mouse STING agonists (18, 19, 33). In support of this notion, we show in Fig. 2C that DMXAA, but not LPS, efficiently induces phosphorylation of STING in RAW264.7 cells. Consistent with the fact that AML12HBV10 cells express undetectable levels of STING (Fig. 2C), direct treatment of these hepatocytes with DMXAA did not inhibit HBV DNA replication. However, the conditioned media harvested from DMXAA-treated RAW264.7 cells inhibited HBV DNA replication in mouse hepatocytes in a dose-dependent manner, with an estimated 50% effective dose (EC50) of 10 μM (Fig. 2D). The cytotoxicity of DMXAA in both mouse macrophage (RWA246.7) and hepatocyte (AML12HBV10) cell lines has been tested under the conditions described for indirect and direct treatments by microscopic inspection and 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide assay. No cytotoxic effect was observed at concentrations of up to 500 μM DMXAA.
FIG 2.

DMXAA and CMA, but not tilorone, activated antiviral cytokine response in macrophages that suppressed HBV replication in hepatocytes. (A) Structures of three mouse IFN inducers. (B) AML12HBV10 cells, seeded and cultured as described in the legend to Fig. 1B, were treated for 2 days with the indicated concentrations of DMXAA (upper panel), CMA (middle panel), or tilorone (lower panel) (direct treatment) or 50% of the conditioned media harvested from RAW264.7 cells (treated with each compound for 12 h) (indirect treatment). Cytoplasmic HBV core DNA was analyzed by Southern blot hybridization. NT controls were as described in the legend to Fig. 1B. (C) RAW264.7 and AML12HBV10 cells were mock treated or treated with 125 μM DMXAA or 1 μg/ml of LPS for 30 and 60 min. Expression and activation of STING were determined by Western blot assay. Arrowheads indicate the shift in gel mobility that resulted from STING phosphorylation. β-Actin served as a loading control. (D) AML12HBV10 cells cultured in the absence of tetracycline were directly or indirectly treated as described for panel B, except that lower doses of DMXAA were used. AML12HBV10 cells treated for 2 days with the concentrations of mouse IFN-α indicated served as positive controls. Cytoplasmic HBV core DNA was analyzed by Southern blot hybridization.
DMXAA-induced antiviral response reduced the amount of cytoplasmic HBV capsids.
In order to map the HBV replication step(s) being inhibited by the DMXAA-induced antiviral response in macrophages, AML12HBV10 cells were treated for 2 days with conditioned media harvested from TLR agonist- or DMXAA-treated RAW264.7 cells. Under this experimental condition, covalently closed circular DNA (cccDNA) is undetectable and HBV replication is fully supported by pregenomic RNA transcribed from the integrated copy of the HBV genome in the host cell chromosome. The amounts of HBV mRNA, capsid protein, total capsids, and capsid-associated HBV DNA in the treated AML12HBV10 cells were detected by Northern blot hybridization, Western blot assay, native agarose gel-based viral particle assay, and Southern blot hybridization, respectively. As shown in Fig. 3, the antiviral response induced by DMXAA, and to lesser extents by TLR1/2 and TLR3 agonists, in macrophages posttranscriptionally reduced the amounts of HBV capsid protein and, to a greater extent, the assembled capsids (Fig. 3A to C). Consequently, the amounts of HBV DNA replication intermediates were also decreased (Fig. 3D and E). This antiviral mechanism is similar to that observed in AML12HBV10 cells treated with IFN-α or IFN-γ, which reduced HBV nucleocapsids by accelerating their decay (21). These results thus support the hypothesis that both STING and TLR agonist-induced antiviral responses against HBV in macrophages may be primarily mediated by IFNs (25, 26).
FIG 3.
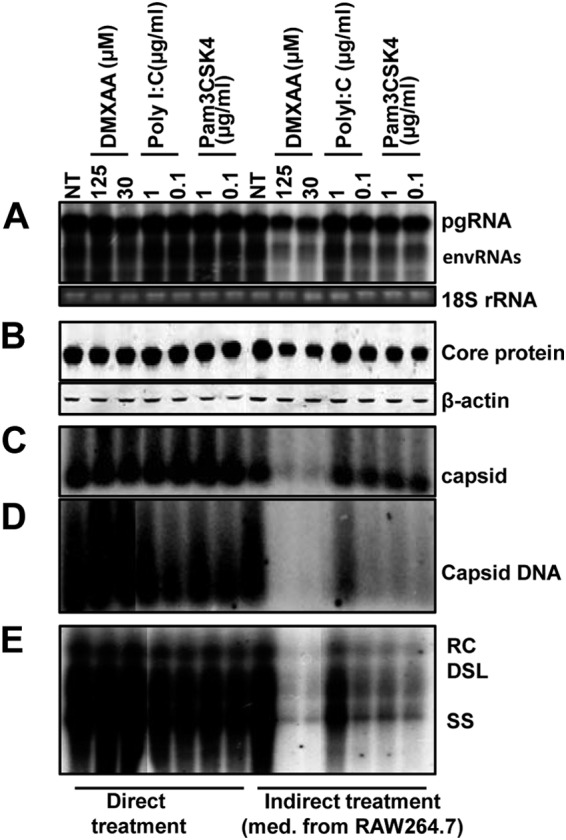
DMXAA-induced antiviral response in macrophages reduced the HBV nucelocapsids in a hepatocyte cell line. AML12HBV10 cells cultured in the absence of tetracycline for 1 day were either directly treated with the indicated concentrations of DMXAA, poly(I:C), or Pam3CSK4 or indirectly treated with 50% of the conditioned media harvested from RAW264.7 cells (treated with the indicated concentrations of DMXAA or TLR agonists for 12 h). The AML12HBV10 cells were harvested 2 days after treatment for the following analyses. (A) Intracellular HBV RNA was determined by Northern blotting. pgRNA, pregenomic RNA; envRNAs, viral mRNAs specifying envelope proteins. 18S rRNA served as a loading control. (B) HBV core protein was determined by Western blot assay with total cell lysate. β-Actin served as a loading control. The amounts of HBV capsids (C) and capsid-associated HBV DNA (D) were determined by a native agarose gel assay. (E) Cytoplasmic HBV core DNA was determined by Southern blot hybridization.
DMXAA induced a distinct profile of cytokine responses in macrophages.
To characterize the cytokine response induced by DMXAA, we first compared the signaling pathway activated and cytokine profiles induced by DMXAA, as well as three representative TLR agonists, in RAW264.7 cells. As shown in Fig. 4, only DMXAA, and not the TLR1/2, TLR7, TLR3, and TLR4 agonists, induced STING phosphorylation, which was detectable in cells treated with DMXAA for more than 30 min. However, DMXAA and all of the TLR agonists tested efficiently induced the phosphorylation of TBK1, a kinase essential for IRF3 phosphorylation and induction of IFN-β in both the STING and TLR pathways. Also, as expected, DMXAA and all of the TLR agonists tested induced degradation of IκBα. Interestingly, while the activation of all three mitogen-activated protein kinase (MAPK) pathways, as demonstrated by the increase in phosphorylated p38α, JNK, and ERK, was evident as early as 15 min in the cells treated with any one of the four TLR agonists, activation of the MAPK pathways was delayed and only became detectable at 90 min after DMXAA treatment.
FIG 4.
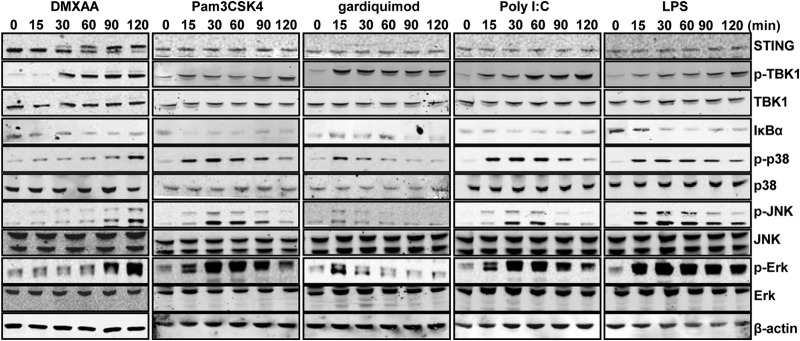
Signaling pathways activated by DMXAA and TLR agonists in mouse macrophage cells. RAW264.7 cells were treated with 125 μM DMXAA, 1 μg/ml of Pam3CSK4 (TLR1/2 agonist), 10 μg/ml of poly(I:C) (TLR3 agonist), 1 μg/ml of LPS (TLR4 agonist), or 3 μM Gardiquimod (TLR7 agonist) for the times indicated. Total cellular proteins were fractioned by SDS-PAGE and transferred onto polyvinylidene difluoride membranes. Total and phosphorylated STING, TBK1, p38, JNK, and Erk, as well as IkB-α, were detected by Western blot assays with the specific antibodies. β-Actin served as a loading control.
The cytokine profiles induced by DMXAA and TLR agonists in RAW264.7 cells were determined by qRT-PCR assays. DMXAA induced approximately 100-fold more IFN-β mRNA expression than the TLR1/2, TLR3, or TLR7 agonist did at 2 h posttreatment (Fig. 5A). On the contrary, compared to DMXAA, the three TLR agonists tested generally induced stronger proinflammatory cytokine (Fig. 5B to F) and chemokine (Fig. 5G) expression. The weaker cytokine response induced by DMXAA might be due, at least in part, to its slower and weaker activation of MAPK pathways.
FIG 5.
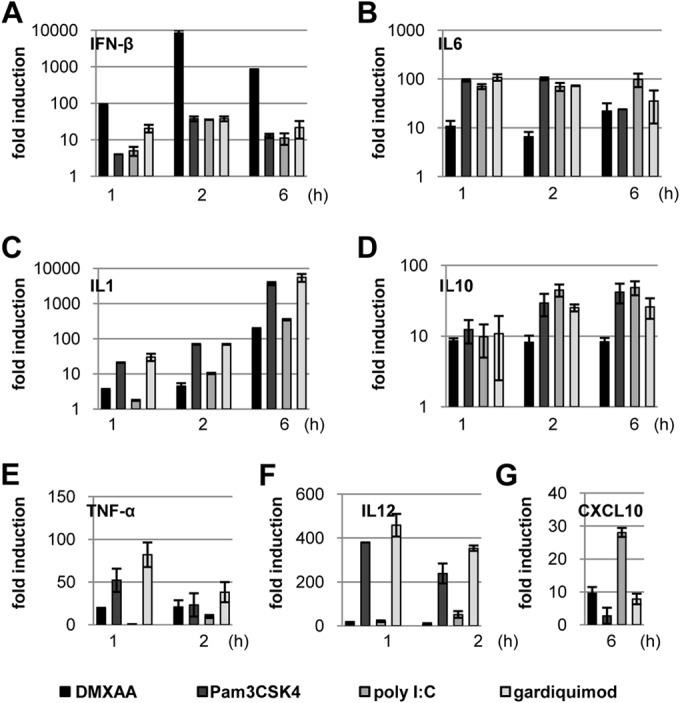
DMXAA induced a distinct cytokine response profile. RAW264.7 cells were treated with 125 μM DMXAA, 1 μg/ml of Pam3CSK4 (TLR1/2 agonist), 10 μg/ml of poly(I:C) (TLR3 agonist), or 3 μM Gardiquimod (TLR7 agonist) for the times indicated. (A to G) The amounts of mRNAs specifying the specific cytokines and chemokines were quantified by real-time RT-PCR assays. Data (mean values ± standard deviations; n = 3) are expressed as fold induction of gene expression relative to that in untreated controls.
DMXAA-induced cytokine and antiviral responses were STING dependent.
We have thus far demonstrated that DMXAA treatment of RAW264.7 cells activated STING and induced an antiviral cytokine response to suppress HBV replication in mouse hepatocytes. In order to further determine the role of STING in the DMXAA-induced antiviral cytokine response, we established RAW264.7-derived stable cell lines expressing either scrambled shRNA or shRNA specifically targeting mouse STING mRNA. Reduction of STING mRNA and protein expression in the cells expressing STING-specific shRNA was validated by qRT-PCR (Fig. 6A) and Western blot assay (Fig. 6B), respectively. DMXAA-induced, but not LPS-induced, TBK1 activation was significantly compromised in RAW264.7 cells expressing STING mRNA-specific shRNA (Fig. 6B). Moreover, DMXAA-induced IFN-β expression in RAW264.7 cells where STING expression was partially knocked down by shRNA (Fig. 6C) and its indirect antiviral activity against HBV in AML12HBV10 cells (Fig. 6D) were significantly attenuated. Taken together, these results and those presented above collectively support the notion that DMXAA does not directly inhibit HBV replication in hepatocytes but induces a STING-dependent cytokine response in mouse macrophages and acts indirectly on hepatocytes to suppress HBV replication.
FIG 6.

DMXAA-induced cytokine and antiviral responses were STING dependent. (A) Total RNAs were extracted from parental wild-type (WT) RAW264.7 cells and RAW264.7-derived stable cell lines that express scrambled shRNA (shcontrol) or shRNA targeting mouse STING mRNA (shSTING). STING mRNA levels were determined by real-time RT-PCR, and data (mean values ± standard deviations; n = 3) are expressed as percentages of the STING mRNA in wild-type RAW264.7 cells. (B) RAW264.7 cells expressing shcontrol or shSTING were mock treated or treated with 125 μM DMXAA for 30 min. Expression and activation of STING and TBK1 were determined by Western blot assays. Cells treated with 1 μg/ml of LPS served as controls. β-Actin served as a loading control. (C) Knockdown of STING abrogated DMXAA-induced IFN-β gene expression. RAW264.7-derived shcontrol and shSTING cells were treated with 125 μM DMXAA for 3 h. IFN-β mRNA was quantified by a real-time RT-PCR assay. Data (mean values ± standard deviations; n = 3) are expressed as fold induction of gene expression relative to that in untreated controls. (D) Knockdown of STING compromised DMXAA-induced antiviral activity. AML12HBV10 cells cultured in the absence of tetracycline for 1 day were mock treated or treated for 2 days with 50% of the conditioned media harvested from RAW264.7-derived shcontrol or shSTING cells, which were treated for 12 h with the concentrations of DMXAA, LPS, or Gardiquimod indicated. Cytoplasmic HBV core DNA was extracted and quantified by a real-time PCR assay. AML12HBV10 cells treated with conditioned media from mock-treated RAW267.4 cells served as a control (NT). HBV DNA levels (mean values ± standard deviations; n = 3) are expressed as percentages of the NT control value. Data were statistically analyzed by Student's t test. *, P < 0.05.
Type I IFNs were the primary mediators of the DMXAA-induced antiviral response against HBV.
Among the DMXAA-induced cytokines in macrophages, we have demonstrated previously that treatment of AML12HBV10 cells with type I IFN potently inhibits HBV replication (21). To determine the role of type I IFNs and other cytokines in the DMXAA-induced antiviral response against HBV, we first examined if blockade of the type I IFN response with a monoclonal antibody specifically recognizing the type I IFN receptor would attenuate the antiviral activity induced by DMXAA in macrophages. As expected, blockade of the type I IFN receptor in AML12HBV10 cells significantly reduced the antiviral response induced by IFN-α (Fig. 7A). Treatment of AML12HBV10 cells with the type I IFN receptor antibody also significantly attenuated the antiviral response induced by conditioned media from DMXAA-treated RAW264.7 cells (Fig. 7B), suggesting that type I IFNs are likely to be the primary mediators of the DMXAA-induced antiviral response against HBV. To determine the role of other cytokines in the DMXAA-induced antiviral response, we tested the antiviral effects of IL-1, IL-6, and TNF-α and demonstrated that only TNF-α inhibited HBV DNA replication and thus may also play a role in the DMXAA-induced antiviral response against HBV (Fig. 7C). This result is consistent with the fact that anti-TNF therapy of rheumatoid arthritis, inflammatory bowel diseases, and psoriasis reactivates HBV infection in inactive carriers and suggests that TNF-α plays an important role in the immune control of HBV infection in humans (34).
FIG 7.
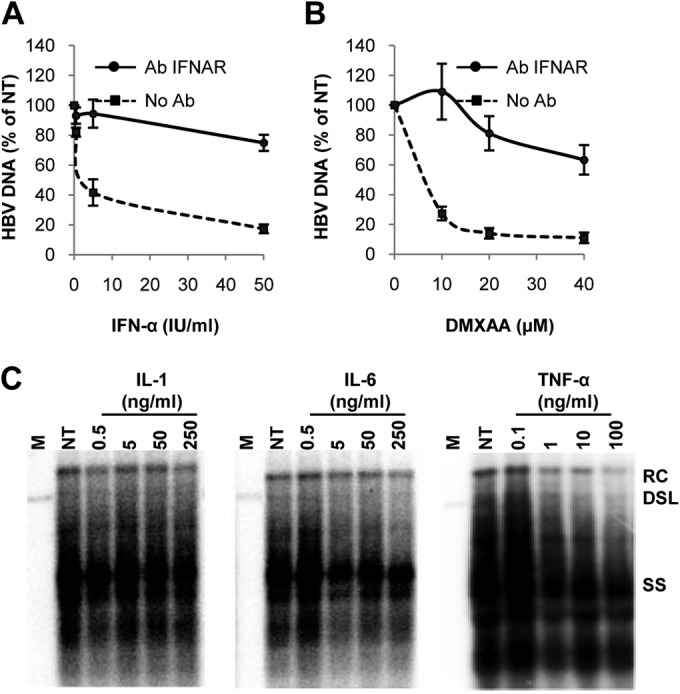
Identification of cytokines mediating the DMXAA-induced antiviral response. AML12HBV10 cells were cultured in the absence of tetracycline for 1 day either with or without preincubation with 10 μg/ml of a monoclonal antibody against type I IFN receptor IFNAR1 (Ab INFAR) at 37°C for 1 h, followed by treatment for 2 days with the concentrations of mouse IFN-α indicated (A) or 50% of the conditioned media harvested from RAW264.7 cells (treated with the indicated concentrations of DMXAA for 12 h) (B). Cytoplasmic HBV DNA were quantified by a real-time PCR assay, and data (mean values ± standard deviations; n = 4) are presented as percentages of the mock-treated control (NT) value. (C) AML12HBV10 cells cultured in the absence of tetracycline were treated for 4 days with the concentrations of IL-1, IL-6, or TNF-α indicated. Cytoplasmic HBV core DNA was analyzed by Southern blot hybridization. Lanes M contained molecular size markers.
DMXAA inhibits HBV replication in mice.
To further validate the antiviral effects of DMXAA in vivo, NOD/SCID mice were hydrodynamically injected with an HBV 1.3mer plasmid to establish HBV replication in hepatocytes. As shown in Fig. 8A, single-dose DMXAA treatment produced a 1.3-log reduction in intrahepatic HBV core DNA, compared to that of the vehicle-treated control group, at 24 h after treatment. In agreement with the anticipated antiviral mechanism, the expression of representative ISGs, such as those for OAS1b and viperin, was significantly induced in the livers of DMXAA-treated animals (Fig. 8B and C). Noticeably, the average body weight of DMXAA-treated mice was reduced by approximately 8% (data not shown), which may indicate the side effect of the DMXAA-induced cytokine response and could be used as a surrogate marker for STING agonist-induced pharmacological and toxicological responses in vivo.
FIG 8.
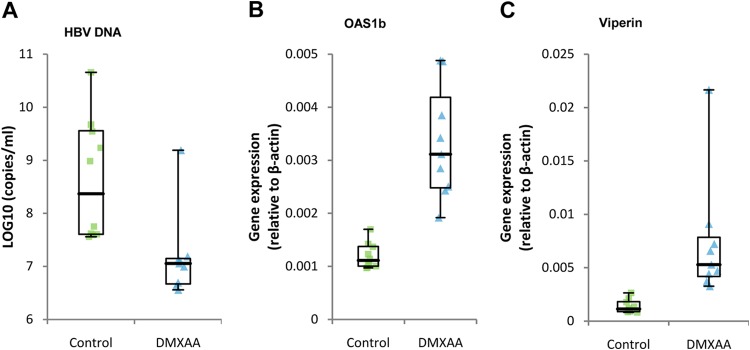
In vivo efficacy of DMXAA in an HBV hydrodynamic mouse model. Seven days after hydrodynamic injection of HBV 1.3mer plasmid, mice were treated with a single dose of either DMXAA at 25 mg/kg or the vehicle by intraperitoneal injection. (A) At 24 h posttreatment, HBV core DNA was extracted from their livers and analyzed by a real-time PCR assay. Ten mice were included in the control group, and nine mice were included in the treatment group. Plots represent numbers of HBV DNA copies/ml from each animal after subtraction of the input plasmid copy number. (B and C) Total RNA was extracted from livers, and levels of OAS1b and viperin mRNAs were analyzed by real-time RT-PCR assay. Plots represent the mRNA levels of each animal. All of the data are presented in box plots showing medians, interquartiles, and ranges (minimum and maximum values) and were statistically analyzed by Student's t test (P < 0.05).
DISCUSSION
Hepatocytes are the primary, if not the only, host cells of HBV (35). Although the liver is generally considered an organ with unique immune regulatory functions favoring the induction of immune tolerance rather than immunity, the vast majority of adulthood HBV infection is able to induce a sufficient immune response to resolve the infection (36). Evidence accumulated in recent studies suggests that both the unique hepatic microenvironment and liver-resident cells, including both hepatocytes and NPCs, actively modulate immune responses locally in the liver and thereby determine the outcome of hepatic immune responses (4, 9, 10). As the host cells of HBV, hepatocytes not only produce antigens that induce adaptive immune responses and molecular patterns that activate innate immunity but are also the target cells of host innate and adaptive immune responses against HBV. Particularly, it has been convincingly demonstrated in woodchuck hepatitis virus-infected woodchucks and HBV-infected chimpanzees that, in addition to killing virus-infected hepatocytes by virus-specific cytotoxic T lymphocytes (37–39), cytokine-mediated antiviral immunity is able to noncytolytically cure HBV-infected hepatocytes (27, 28, 40). Pharmacological induction of such an intrahepatic cytokine response is thus one of the ideal curative approaches to chronic hepatitis B (8). Unlike IFN therapy, therapeutic induction of the intrahepatic antiviral cytokine response not only avoids the side effects of systematic application of IFN-α but also suppresses HBV infection via coordinated actions of multiple cytokines (Fig. 7), which may be essential for more efficient immune control of HBV infection and achievement of a functional cure (8).
Moreover, while liver-resident NK cells and NKT cells have been shown to restrict HBV infection in vivo (41–43), accumulating evidence suggests that liver-resident macrophages (Kupffer cells) and dendritic cells play essential roles in the priming and activation of the host adaptive immune response to HBV by shaping intrahepatic lymphoid organization, antigen presentation, expression of costimulating cell surface proteins, and secretion of inflammatory cytokines (4, 9, 10). Although the mechanism by which HBV fails to induce a sufficient immune response in chronic carriers remains to be elucidated, it is conceivable that intrahepatic activation of the TLR response or the STING pathway in liver sinusoidal endothelial cells, Kupffer cells, and/or dendritic cells may promote the priming and activation of an anti-HBV adaptive immune response, as well as induce an antiviral cytokine response (44, 45). These effects not only suppress HBV replication but also reduce the viral antigen load through the suppression of cccDNA transcription (46) or even promotion of cccDNA decay (47, 48), which may attenuate the T cell exhaustion and favor the restoration of the HBV-specific T cell response.
Indeed, we demonstrated in this study that activation of TLRs, as well as STING, in macrophages by their cognate agonists induced strong antiviral cytokine responses to potently suppress HBV replication in hepatocytes. However, detailed characterization of the antiviral cytokine responses induced by TLR and STING agonists revealed distinct characteristics. Consistent with its strong activation of TBK1 and delayed activation of MAPK pathways (Fig. 4), DMXAA induced a cytokine response in macrophages that was dominated by IFN-β (Fig. 5) and demonstrated a more potent antiviral activity against HBV in hepatocytes (Fig. 3). Accordingly, if only considering the property of inducing a noncytolytic antiviral cytokine response, intrahepatic activation of the STING pathway seems to be superior to the activation of TLR pathways. This is because STING agonists induce not only a more potent antiviral response but also a less proinflammatory cytokine response, which may result in less inflammation and tissue damage. However, whether STING or TLR agonists more efficiently prime and activate an HBV-specific adaptive immune response in the liver can only be evaluated in vivo in chronically HBV-infected animals, such as HBV transgenic mice, adeno-associated virus vector-mediated HBV-transduced mice, or recently developed humanized mice with human immune system and liver cells that are susceptible to HBV infection (49–51). Encouragingly, it was shown recently that activation of the cGAS-STING pathway did boost antigen-specific T cell activation and antibody production in mice (52).
Nevertheless, our studies have convincingly demonstrated that STING is a potential target for immunotherapy of chronic hepatitis B. Similar to immunotherapies with TLR agonists, which are limited by the side effects of systematic activation of cytokine responses (11, 53, 54), systematic administration of STING agonists, despite the weaker induction of proinflammatory cytokines, may also induce detrimental effects. In principle, this obstacle could be circumvented by targeted activation of the STING pathway in liver macrophages or other NPCs through liposomal delivery of STING agonists, which are cleared from the circulation primarily by Kupffer cells (55).
ACKNOWLEDGMENTS
We thank Julia Ma (Drexel University), John M. Taylor (Fox Chase Cancer Center), and David Horn (Mid-Atlantic BioTherapeutics, Inc.) for critical reading of the manuscript and Yanming Du (Baruch S. Blumberg Institute, Hepatitis B Foundation) for comments and suggestions on this project.
This work was supported in part by NIH grant R01AI113267 and the Hepatitis B Foundation through an appropriation from the Commonwealth of Pennsylvania. The animal study was supported by the National Natural Science Foundation of China (81321004), State Mega Programs (2012ZX09301002-003/006), the Beijing Key Laboratory of New Drug Mechanisms and Pharmacological Evaluation Study (BZ0150), and the 973 project (2012CB911103).
REFERENCES
- 1.Chisari FV, Ferrari C. 1995. Hepatitis B virus immunopathogenesis. Annu Rev Immunol 13:29–60. doi: 10.1146/annurev.iy.13.040195.000333. [DOI] [PubMed] [Google Scholar]
- 2.Lee WM. 1997. Hepatitis B virus infection. N Engl J Med 337:1733–1745. doi: 10.1056/NEJM199712113372406. [DOI] [PubMed] [Google Scholar]
- 3.Liang TJ. 2009. Hepatitis B: the virus and disease. Hepatology 49:S13–S21. doi: 10.1002/hep.22881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dienstag JL. 2009. Benefits and risks of nucleoside analog therapy for hepatitis B. Hepatology 49:S112–S121. doi: 10.1002/hep.22920. [DOI] [PubMed] [Google Scholar]
- 5.Perrillo R. 2009. Benefits and risks of interferon therapy for hepatitis B. Hepatology 49:S103–S111. doi: 10.1002/hep.22956. [DOI] [PubMed] [Google Scholar]
- 6.Rehermann B, Ferrari C, Pasquinelli C, Chisari FV. 1996. The hepatitis B virus persists for decades after patients' recovery from acute viral hepatitis despite active maintenance of a cytotoxic T-lymphocyte response. Nat Med 2:1104–1108. doi: 10.1038/nm1096-1104. [DOI] [PubMed] [Google Scholar]
- 7.Block TM, Gish R, Guo H, Mehta A, Cuconati A, Thomas London W, Guo JT. 2013. Chronic hepatitis B: what should be the goal for new therapies? Antiviral Res 98:27–34. doi: 10.1016/j.antiviral.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chang J, Block TM, Guo JT. 2012. The innate immune response to hepatitis B virus infection: implications for pathogenesis and therapy. Antiviral Res 96:405–413. doi: 10.1016/j.antiviral.2012.10.001. [DOI] [PubMed] [Google Scholar]
- 9.Publicover J, Goodsell A, Nishimura S, Vilarinho S, Wang ZE, Avanesyan L, Spolski R, Leonard WJ, Cooper S, Baron JL. 2011. IL-21 is pivotal in determining age-dependent effectiveness of immune responses in a mouse model of human hepatitis B. J Clin Invest 121:1154–1162. doi: 10.1172/JCI44198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Publicover J, Gaggar A, Nishimura S, Van Horn CM, Goodsell A, Muench MO, Reinhardt RL, van Rooijen N, Wakil AE, Peters M, Cyster JG, Erle DJ, Rosenthal P, Cooper S, Baron JL. 2013. Age-dependent hepatic lymphoid organization directs successful immunity to hepatitis B. J Clin Invest 123:3728–3739. doi: 10.1172/JCI68182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lanford RE, Guerra B, Chavez D, Giavedoni L, Hodara VL, Brasky KM, Fosdick A, Frey CR, Zheng J, Wolfgang G, Halcomb RL, Tumas DB. 2013. GS-9620, an oral agonist of Toll-like receptor-7, induces prolonged suppression of hepatitis B virus in chronically infected chimpanzees. Gastroenterology 144:1508–1517, 1517 e1501–1510. doi: 10.1053/j.gastro.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kimura K, Moriwaki H, Nagaki M, Saio M, Nakamoto Y, Naito M, Kuwata K, Chisari FV. 2006. Pathogenic role of B cells in anti-CD40-induced necroinflammatory liver disease. Am J Pathol 168:786–795. doi: 10.2353/ajpath.2006.050314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kimura K, Kakimi K, Wieland S, Guidotti LG, Chisari FV. 2002. Activated intrahepatic antigen-presenting cells inhibit hepatitis B virus replication in the liver of transgenic mice. J Immunol 169:5188–5195. doi: 10.4049/jimmunol.169.9.5188. [DOI] [PubMed] [Google Scholar]
- 14.Isogawa M, Chung J, Murata Y, Kakimi K, Chisari FV. 2013. CD40 activation rescues antiviral CD8(+) T cells from PD-1-mediated exhaustion. PLoS Pathog 9:e1003490. doi: 10.1371/journal.ppat.1003490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burdette DL, Vance RE. 2013. STING and the innate immune response to nucleic acids in the cytosol. Nat Immunol 14:19–26. doi: 10.1038/ni.2491. [DOI] [PubMed] [Google Scholar]
- 16.Gao D, Wu J, Wu YT, Du F, Aroh C, Yan N, Sun L, Chen ZJ. 2013. Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science 341:903–906. doi: 10.1126/science.1240933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ishikawa H, Barber GN. 2008. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455:674–678. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Conlon J, Burdette DL, Sharma S, Bhat N, Thompson M, Jiang Z, Rathinam VA, Monks B, Jin T, Xiao TS, Vogel SN, Vance RE, Fitzgerald KA. 2013. Mouse, but not human STING, binds and signals in response to the vascular disrupting agent 5,6-dimethylxanthenone-4-acetic acid. J Immunol 190:5216–5225. doi: 10.4049/jimmunol.1300097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cavlar T, Deimling T, Ablasser A, Hopfner KP, Hornung V. 2013. Species-specific detection of the antiviral small-molecule compound CMA by STING. EMBO J 32:1440–1450. doi: 10.1038/emboj.2013.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Campagna MR, Liu F, Mao R, Mills C, Cai D, Guo F, Zhao X, Ye H, Cuconati A, Guo H, Chang J, Xu X, Block TM, Guo JT. 2013. Sulfamoylbenzamide derivatives inhibit the assembly of hepatitis B virus nucleocapsids. J Virol 87:6931–6942. doi: 10.1128/JVI.00582-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu C, Guo H, Pan XB, Mao R, Yu W, Xu X, Wei L, Chang J, Block TM, Guo JT. 2010. Interferons accelerate decay of replication-competent nucleocapsids of hepatitis B virus. J Virol 84:9332–9340. doi: 10.1128/JVI.00918-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao X, Guo F, Liu F, Cuconati A, Chang J, Block TM, Guo JT. 2014. Interferon induction of IFITM proteins promotes infection by human coronavirus OC43. Proc Natl Acad Sci U S A 111:6756–6761. doi: 10.1073/pnas.1320856111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chang J, Sigal LJ, Lerro A, Taylor J. 2001. Replication of the human hepatitis delta virus genome is initiated in mouse hepatocytes following intravenous injection of naked DNA or RNA sequences. J Virol 75:3469–3473. doi: 10.1128/JVI.75.7.3469-3473.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang PL, Althage A, Chung J, Chisari FV. 2002. Hydrodynamic injection of viral DNA: a mouse model of acute hepatitis B virus infection. Proc Natl Acad Sci U S A 99:13825–13830. doi: 10.1073/pnas.202398599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu J, Lu M, Meng Z, Trippler M, Broering R, Szczeponek A, Krux F, Dittmer U, Roggendorf M, Gerken G, Schlaak JF. 2007. Toll-like receptor-mediated control of HBV replication by nonparenchymal liver cells in mice. Hepatology 46:1769–1778. doi: 10.1002/hep.21897. [DOI] [PubMed] [Google Scholar]
- 26.Isogawa M, Robek MD, Furuichi Y, Chisari FV. 2005. Toll-like receptor signaling inhibits hepatitis B virus replication in vivo. J Virol 79:7269–7272. doi: 10.1128/JVI.79.11.7269-7272.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guidotti LG, Ishikawa T, Hobbs MV, Matzke B, Schreiber R, Chisari FV. 1996. Intracellular inactivation of the hepatitis B virus by cytotoxic T lymphocytes. Immunity 4:25–36. doi: 10.1016/S1074-7613(00)80295-2. [DOI] [PubMed] [Google Scholar]
- 28.Guidotti LG, Rochford R, Chung J, Shapiro M, Purcell R, Chisari FV. 1999. Viral clearance without destruction of infected cells during acute HBV infection. Science 284:825–829. doi: 10.1126/science.284.5415.825. [DOI] [PubMed] [Google Scholar]
- 29.Wu J, Meng Z, Jiang M, Pei R, Trippler M, Broering R, Bucchi A, Sowa JP, Dittmer U, Yang D, Roggendorf M, Gerken G, Lu M, Schlaak JF. 2009. Hepatitis B virus suppresses Toll-like receptor-mediated innate immune responses in murine parenchymal and nonparenchymal liver cells. Hepatology 49:1132–1140. doi: 10.1002/hep.22751. [DOI] [PubMed] [Google Scholar]
- 30.Stringfellow DA. 1977. Comparation interferon-inducing and antiviral properties of 2-amino-5-bromo-6-methyl-4-pyrimidinol (U-25,166), tilorone hydrochloride, and polyinosinic-polycytidylic acid. Antimicrob Agents Chemother 11:984–992. doi: 10.1128/AAC.11.6.984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ching LM, Young HA, Eberly K, Yu CR. 1999. Induction of STAT and NFkappaB activation by the antitumor agents 5,6-dimethylxanthenone-4-acetic acid and flavone acetic acid in a murine macrophage cell line. Biochem Pharmacol 58:1173–1181. doi: 10.1016/S0006-2952(99)00194-X. [DOI] [PubMed] [Google Scholar]
- 32.Baguley BC. 2001. Small-molecule cytokine inducers causing tumor necrosis. Curr Opin Investig Drugs 2:967–975. [PubMed] [Google Scholar]
- 33.Kim S, Li L, Maliga Z, Yin Q, Wu H, Mitchison TJ. 2013. Anticancer flavonoids are mouse-selective STING agonists. ACS Chem Biol 8:1396–1401. doi: 10.1021/cb400264n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Viganò M, Degasperi E, Aghemo A, Lampertico P, Colombo M. 2012. Anti-TNF drugs in patients with hepatitis B or C virus infection: safety and clinical management. Expert Opin Biol Ther 12:193–207. doi: 10.1517/14712598.2012.646986. [DOI] [PubMed] [Google Scholar]
- 35.Seeger C, Mason WS. 2000. Hepatitis B virus biology. Microbiol Mol Biol Rev 64:51–68. doi: 10.1128/MMBR.64.1.51-68.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Knolle PA, Thimme R. 2014. Hepatic immune regulation and its involvement in viral hepatitis infection. Gastroenterology 146:1193–1207. doi: 10.1053/j.gastro.2013.12.036. [DOI] [PubMed] [Google Scholar]
- 37.Summers J, Jilbert AR, Yang W, Aldrich CE, Saputelli J, Litwin S, Toll E, Mason WS. 2003. Hepatocyte turnover during resolution of a transient hepadnaviral infection. Proc Natl Acad Sci U S A 100:11652–11659. doi: 10.1073/pnas.1635109100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kajino K, Jilbert AR, Saputelli J, Aldrich CE, Cullen J, Mason WS. 1994. Woodchuck hepatitis virus infections: very rapid recovery after a prolonged viremia and infection of virtually every hepatocyte. J Virol 68:5792–5803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rehermann B, Lau D, Hoofnagle JH, Chisari FV. 1996. Cytotoxic T lymphocyte responsiveness after resolution of chronic hepatitis B virus infection. J Clin Invest 97:1655–1665. doi: 10.1172/JCI118592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guo JT, Zhou H, Liu C, Aldrich C, Saputelli J, Whitaker T, Barrasa MI, Mason WS, Seeger C. 2000. Apoptosis and regeneration of hepatocytes during recovery from transient hepadnavirus infections. J Virol 74:1495–1505. doi: 10.1128/JVI.74.3.1495-1505.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zeissig S, Murata K, Sweet L, Publicover J, Hu Z, Kaser A, Bosse E, Iqbal J, Hussain MM, Balschun K, Rocken C, Arlt A, Gunther R, Hampe J, Schreiber S, Baron JL, Moody DB, Liang TJ, Blumberg RS. 2012. Hepatitis B virus-induced lipid alterations contribute to natural killer T cell-dependent protective immunity. Nat Med 18:1060–1068. doi: 10.1038/nm.2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baron JL, Gardiner L, Nishimura S, Shinkai K, Locksley R, Ganem D. 2002. Activation of a nonclassical NKT cell subset in a transgenic mouse model of hepatitis B virus infection. Immunity 16:583–594. doi: 10.1016/S1074-7613(02)00305-9. [DOI] [PubMed] [Google Scholar]
- 43.Edlich B, Ahlenstiel G, Zabaleta Azpiroz A, Stoltzfus J, Noureddin M, Serti E, Feld JJ, Liang TJ, Rotman Y, Rehermann B. 2012. Early changes in interferon signaling define natural killer cell response and refractoriness to interferon-based therapy of hepatitis C patients. Hepatology 55:39–48. doi: 10.1002/hep.24628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Verrier ER, Gack MU, Baumert TF. 2014. Cyclic guanosine monophosphate/adenosine monophosphate synthase (cGAS), innate immune responses, and viral hepatitis. Hepatology 60:1098–1100. doi: 10.1002/hep.27187. [DOI] [PubMed] [Google Scholar]
- 45.Schoggins JW, MacDuff DA, Imanaka N, Gainey MD, Shrestha B, Eitson JL, Mar KB, Richardson RB, Ratushny AV, Litvak V, Dabelic R, Manicassamy B, Aitchison JD, Aderem A, Elliott RM, Garcia-Sastre A, Racaniello V, Snijder EJ, Yokoyama WM, Diamond MS, Virgin HW, Rice CM. 2014. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature 505:691–695. doi: 10.1038/nature12862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu F, Campagna M, Qi Y, Zhao X, Guo F, Xu C, Li S, Li W, Block TM, Chang J, Guo JT. 2013. Alpha-interferon suppresses hepadnavirus transcription by altering epigenetic modification of cccDNA minichromosomes. PLoS Pathog 9:e1003613. doi: 10.1371/journal.ppat.1003613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lucifora J, Xia Y, Reisinger F, Zhang K, Stadler D, Cheng X, Sprinzl MF, Koppensteiner H, Makowska Z, Volz T, Remouchamps C, Chou WM, Thasler WE, Huser N, Durantel D, Liang TJ, Munk C, Heim MH, Browning JL, Dejardin E, Dandri M, Schindler M, Heikenwalder M, Protzer U. 2014. Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA. Science 343:1221–1228. doi: 10.1126/science.1243462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ding S, Robek MD. 2014. Cytidine deamination and cccDNA degradation: a new approach for curing HBV? Hepatology 60:2118–2121. doi: 10.1002/hep.27386. [DOI] [PubMed] [Google Scholar]
- 49.Yang D, Liu L, Zhu D, Peng H, Su L, Fu YX, Zhang L. 2014. A mouse model for HBV immunotolerance and immunotherapy. Cell Mol Immunol 11:71–78. doi: 10.1038/cmi.2013.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bility MT, Cheng L, Zhang Z, Luan Y, Li F, Chi L, Zhang L, Tu Z, Gao Y, Fu Y, Niu J, Wang F, Su L. 2014. Hepatitis B virus infection and immunopathogenesis in a humanized mouse model: induction of human-specific liver fibrosis and M2-like macrophages. PLoS Pathog 10:e1004032. doi: 10.1371/journal.ppat.1004032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guidotti LG, Matzke B, Schaller H, Chisari FV. 1995. High-level hepatitis B virus replication in transgenic mice. J Virol 69:6158–6169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li XD, Wu J, Gao D, Wang H, Sun L, Chen ZJ. 2013. Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science 341:1390–1394. doi: 10.1126/science.1244040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kanzler H, Barrat FJ, Hessel EM, Coffman RL. 2007. Therapeutic targeting of innate immunity with Toll-like receptor agonists and antagonists. Nat Med 13:552–559. doi: 10.1038/nm1589. [DOI] [PubMed] [Google Scholar]
- 54.Horsmans Y, Berg T, Desager JP, Mueller T, Schott E, Fletcher SP, Steffy KR, Bauman LA, Kerr BM, Averett DR. 2005. Isatoribine, an agonist of TLR7, reduces plasma virus concentration in chronic hepatitis C infection. Hepatology 42:724–731. doi: 10.1002/hep.20839. [DOI] [PubMed] [Google Scholar]
- 55.Ponnappa BC, Israel Y. 2002. Targeting Kupffer cells with antisense oligonucleotides. Front Biosci 7:e223–233. doi: 10.2741/ponnappa. [DOI] [PubMed] [Google Scholar]