

Abstract
NAI-107 is a novel lantibiotic compound with potent in vitro activity against Gram-positive bacteria, including methicillin-resistant Staphylococcus aureus (MRSA). The purpose of this study was to examine the activity of NAI-107 against S. aureus strains, including MRSA, in the neutropenic murine thigh infection model. Serum pharmacokinetics were determined and time-kill studies were performed following administration of single subcutaneous doses of 5, 20, and 80 mg/kg body weight. The dose fractionation included total doses ranging from 1.56 to 400 mg/kg/72 h, divided into 1, 2, 3, or 6 doses. Studies of treatment effects against 9 S. aureus strains (4 methicillin-susceptible Staphylococcus aureus [MSSA] and 5 MRSA) using a 12-h dosing interval and total dose range of 1.56 to 400 mg/kg/72 h were also performed. A maximum effect (Emax) model was used to determine the pharmacokinetic/pharmacodynamic (PK/PD) index that best described the dose-response data and to estimate the doses required to achieve a net bacteriostatic dose (SD) and a 1-log reduction in CFU/thigh. The pharmacokinetic studies demonstrated an area under the concentration-time curve (AUC) range of 26.8 to 276 mg · h/liter and half-lives of 4.2 to 8.2 h. MICs ranged from 0.125 to 0.5 μg/ml. The 2 highest single doses produced more than a 2-log kill and prolonged postantibiotic effects (PAEs) ranging from 36 to >72 h. The dose fractionation-response curves were similar, and the AUC/MIC ratio was the most predictive PD index (AUC/MIC, coefficient of determination [R2] = 0.89; maximum concentration of drug in serum [Cmax]/MIC, R2 = 0.79; time [T] > MIC, R2 = 0.63). A ≥2-log kill was observed against all 9 S. aureus strains. The total drug 24-h AUC/MIC values associated with stasis and a 1-log kill for the 9 S. aureus strains were 371 ± 130 and 510 ± 227, respectively. NAI-107 demonstrated concentration-dependent killing and prolonged PAEs. The AUC/MIC ratio was the predictive PD index. Extensive killing was observed for S. aureus organisms, independent of the MRSA status. The AUC/MIC target should be useful for the design of clinical dosing regimens.
INTRODUCTION
Bacterial resistance to antimicrobial agents is a pervasive problem worldwide. Of chief concern for hospitalized patients is the emergence of a group of organisms termed the ESKAPE pathogens (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species), which escape the activity of most available antibacterials (1). It is hoped that the discovery and development of novel antibiotic classes with unique mechanisms of action will stem this epidemic. However, very few new drug classes have been developed in the past decade (1–5).
Lantibiotics are natural product peptide antibiotics with a broad Gram-positive spectrum of activity that includes multidrug-resistant (MDR) S. aureus (6, 7). The mechanism of action, as deduced in the studies of the prototype lantibiotic, nisin, is inhibition of the growth of Gram-positive bacteria by interfering with peptidoglycan synthesis by binding to lipid II (8–12). This novel mechanistic site is distinct from that of other antibiotics, and cross-resistance has not been described (13, 14).
The following studies were designed to characterize the in vivo pharmacokinetic-pharmacodynamic (PK/PD) characteristics of NAI-107. Specifically, the impact of the dose and dosing regimen on the in vivo efficacy of this drug in experimental thigh infection in neutropenic mice was assessed. The studies included those designed to (i) investigate the pharmacodynamic characteristics of NAI-107 via time-kill and postantibiotic effect studies, (ii) determine which pharmacokinetic index (peak serum level, area under the concentration-time curve [AUC], or duration of time serum levels exceed the MIC) is most closely linked to the efficacy of NAI-107 via dose fractionation and pharmacodynamic modeling, and (iii) identify the magnitude of the PK/PD index required for efficacy among multiple S. aureus isolates, including beta-lactam-resistant strains.
MATERIALS AND METHODS
Organisms, media, and antibiotics.
Nine isolates of Staphylococcus aureus were used for these studies, including 4 that are methicillin susceptible and 5 that are methicillin resistant (Table 1). The methicillin-resistant strains included hospital- and community-acquired isolates and three U.S. genotypes. Organisms were grown, subcultured, and quantified using Mueller-Hinton broth (MHB) and agar (Difco Laboratories, Detroit, MI). NAI-107 was supplied by the sponsor, Sentinella Pharmaceuticals, Inc.
TABLE 1.
In vitro activity of NAI-107 against select S. aureus isolates using CLSI methods
| S. aureus strain | NAI-107 MIC (μg/ml) | Oxacillin MIC (μg/ml) | Commenta |
|---|---|---|---|
| ATCC 29213 | 0.25 | 0.125 | |
| ATCC 33591 | 0.5 | >16 | U.S. 200 |
| 307109 | 0.5 | >16 | |
| MW2 | 0.5 | >16 | U.S. 400 |
| R-2527 | 0.5 | >16 | U.S. 300 |
| ATCC 25923 | 0.5 | 0.25 | |
| 6538P | 0.125 | 0.50 | |
| Smith | 0.25 | 0.25 | |
| WIS-1 | 0.5 | >16 |
aPulsed-field gel electrophoresis genetic lineage.
In vitro susceptibility testing.
MICs were determined in MHB using a modification of the CLSI microdilution technique (15). Specifically, the polystyrene microtiter wells were coated with 0.02% bovine serum albumin to reduce drug binding to the plate. All MICs were measured in triplicate on at least three occasions. The geometric mean MICs are presented in Table 1.
Murine thigh infection model.
Animals were maintained in accordance with the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) criteria (16). All animal studies were approved by the animal research committees of the William S. Middleton Memorial VA Hospital and the University of Wisconsin. Six-week-old, specific pathogen-free, female ICR/Swiss mice weighing 24 to 27 g were used for all studies (Harlan Sprague-Dawley, Indianapolis, IN). Mice were rendered neutropenic (neutrophils, <100/mm3) by injection of cyclophosphamide (Mead Johnson Pharmaceuticals, Evansville, IN) intraperitoneally 4 days (150 mg/kg body weight) and 1 day (100 mg/kg) before thigh infection. Previous studies have shown that this regimen produces neutropenia in this model for 5 days (17). Broth cultures of freshly plated bacteria were grown to the logarithmic phase overnight to an absorbance of 0.3 at 580 nm (Spectronic 88; Bausch and Lomb, Rochester, NY). After a 1:10 dilution into fresh MHB, bacterial counts of the inoculum ranged from 106.3 to 106.9 CFU/ml. Thigh infections with each of the isolates were produced by injection of 0.1 ml of inoculum into the thighs of isoflurane-anesthetized mice 2 h before therapy with NAI-107.
Drug pharmacokinetics.
Single-dose serum pharmacokinetics of NAI-107 were determined in thigh-infected mice. Animals were administered a single subcutaneous dose (0.2 ml/dose) of NAI-107 at dose levels of 5, 20, and 80 mg/kg. Groups of three mice were sampled at each time point (6 or 7 time points) and dose level. Sampling times ranged from 3 to 72 h over a 72-h period. Serum concentrations were determined by the sponsor using liquid chromatography-tandem mass spectrometry (LC-MS/MS) techniques. The lower and upper limits of quantification were 8.93 and 6,660 ng/ml, respectively. The pharmacokinetic constants (± standard deviation), including the elimination half-life (t1/2), AUC, and maximum concentration of drug in serum (Cmax), were calculated using a noncompartmental model. The half-life of NAI-107 was determined by linear least-squares regression. The AUC was calculated from the mean concentrations using the trapezoidal rule. The pharmacokinetic estimates for dose levels that were not measured were calculated using linear interpolation for dose levels between those with measured kinetics (e.g., between 5 and 20 mg/kg) and linear extrapolation for dose levels greater than or less than the highest and lowest dose levels with kinetic measurements (i.e., 5 and 80 mg/kg).
In vivo time kill and PAE.
Two hours after thigh infection with S. aureus ATCC 25923, mice were treated with single subcutaneous doses of NAI-107 (5, 20, and 80 mg/kg). Groups of two treated and untreated mice were sacrificed at eight time points, every 3 to 24 h over a 72-h study period. The thighs (four per treatment group) were immediately removed upon euthanasia and processed for CFU determination. The burden of organisms in the thigh was measured by viable plate counts of tissue homogenates. The impact of each dose on the burden of organisms over time was measured. The time that drug concentrations would be expected to exceed the MIC was determined from the pharmacokinetic data. The postantibiotic effect (PAE) was calculated by subtracting the time that it took for organisms to increase 1 log in level in the thighs of saline-treated animals from the time that it took organisms to grow the same amount in treated animals after serum levels fell below the MIC for the infecting organism (PAE = T − C, where C is the time for 1-log10 control growth and T is the time for 1-log10 treatment growth after drug levels have fallen below the MIC).
Pharmacokinetic/pharmacodynamic index determination.
Neutropenic mice were infected with S. aureus ATCC 25923 as described above. Treatment with NAI-107 was initiated 2 h after infection and included 20 dosing regimens administered over a 72-h study period using 12-, 24-, 36-, and 72-h dosing intervals. Four thigh infections were included in each dosing group. The five total doses of NAI-107 ranged from 1.56 to 400 mg/kg/72 h. The drug doses were administered subcutaneously. Most of the mice were euthanized after 72 h of therapy, and the thighs were removed and processed for CFU determination. A few mice that received the lowest drug doses were euthanized earlier than 72 h because of the development of signs of distress that required early euthanasia.
To determine which PK/PD index was most closely linked with efficacy, the number of bacteria in the thigh at the end of 72 h of therapy (or earlier for some of the lowest doses) was correlated with (i) the Cmax/MIC ratio, (ii) the 24-hour AUC/MIC ratio, and (iii) the percentage of the dosing interval during which serum levels exceeded the MIC for each of the dosage regimens studied. The correlation between efficacy and each of the three PK/PD indices was determined by nonlinear least-squares multivariate regression (SigmaPlot version 12.3; Systat Software, San Jose, CA). The model is derived from the Hill equation: E = (Emax × DN)/(ED50N − DN), where E is the effector, in this case, the log change in CFU per thigh between treated mice and untreated controls after the 72-h period of study, Emax is the maximum effect, D is the 24-h total dose, ED50 is the dose required to achieve 50% of the Emax, and N is the slope of the dose-effect curve. The indices Emax, ED50, and N were calculated using nonlinear least-squares regression. The coefficient of determination (R2) was used to estimate the variance that might be due to regression with each of the PD parameters.
Pharmacokinetic/pharmacodynamic index target for efficacy.
Five (4-fold) increasing doses of NAI-107 were used to treat neutropenic mice with thigh infections produced by 9 strains of S. aureus (4 methicillin-susceptible strains and 5 methicillin-resistant strains). The subcutaneous doses of NAI-107 varied from 1.56 to 400 mg/kg/72 h fractionated into an every-12-h regimen. Four thigh infections were included in each dosing regimen group. Therapy was initiated 2 h after infection. The animals were euthanized at 72 h after infection, and the thighs were removed and immediately processed for CFU determination. A sigmoid dose-response model derived from the four-parameter Hill equation was used to calculate the dose of NAI-107 that produced a net bacteriostatic effect and a 1-log10 kill over 72 h (static and 1-log kill doses). The 24-h AUC/MIC and 72-h AUC/MIC values for the static and 1-log10 kill doses were calculated using the sigmoid Emax model.
RESULTS
In vitro susceptibility testing.
The MICs of NAI-107 for the nine S. aureus strains used in the studies are shown in Table 1 and ranged from 0.125 to 0.5 μg/ml. Resistance to methicillin did not impact the NAI-107 potency.
Serum pharmacokinetics.
Single-dose pharmacokinetics of NAI-107 are shown in Fig. 1. At the doses studied, exposure to NAI-107 increased in a dose-dependent manner across the dose range studied. The Cmax concentrations ranged from 3.6 to 22.3 μg/ml. The AUC values ranged from 26.8 to 276 mg · h/liter. The elimination half-lives ranged from 4.2 to 8.2 h.
FIG 1.
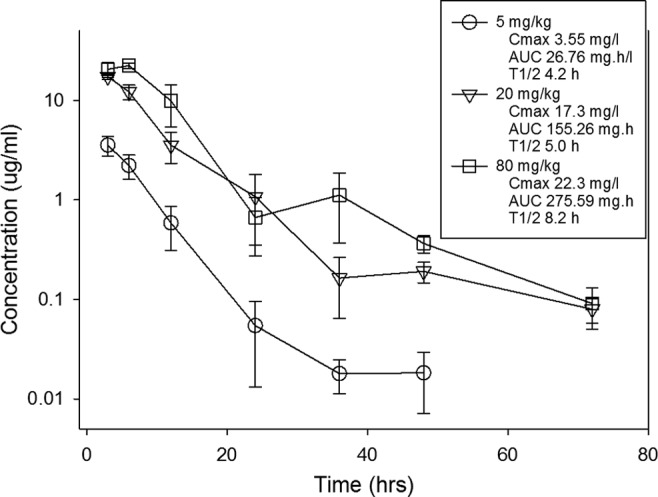
Single-dose serum pharmacokinetics of NAI-107 in neutropenic mice. Three different doses of NAI-107 that varied by 4-fold concentrations on a mg/kg basis were administered by the subcutaneous route. Serum drug concentrations were measured by the sponsor. Groups of three mice were sampled for each time point. Samples were collected every 3 to 24 h over 72 h. Each symbol represents the mean value from three animals. The error bars represent standard deviations. The 72-h time point for the 5 mg/kg dose was below the limit of detection. The PK parameters listed in the box include the maximum drug concentrations (Cmax), the AUC from zero to infinity (AUC), and the elimination half-life (t1/2) for each dose.
In vivo time kill and PAE.
The effects of single doses of NAI-107 at 5, 20, and 80 mg/kg on the in vivo killing and regrowth of a strain of S. aureus (ATCC 25923) are shown in Fig. 2. Rapid, dose-dependent killing of organisms occurred with the two highest dose levels. Prolonged growth inhibition was observed following administration of the lowest dose level. More than a 2-log10 CFU/thigh reduction was observed with two of the three dose levels, and the maximal killing was >3 log10 CFU/thigh for the highest dose relative to the organism burden at the initiation of drug therapy. Organism regrowth was delayed for many hours for all of the dose levels with calculated PAEs ranging from 36 to >72 h.
FIG 2.
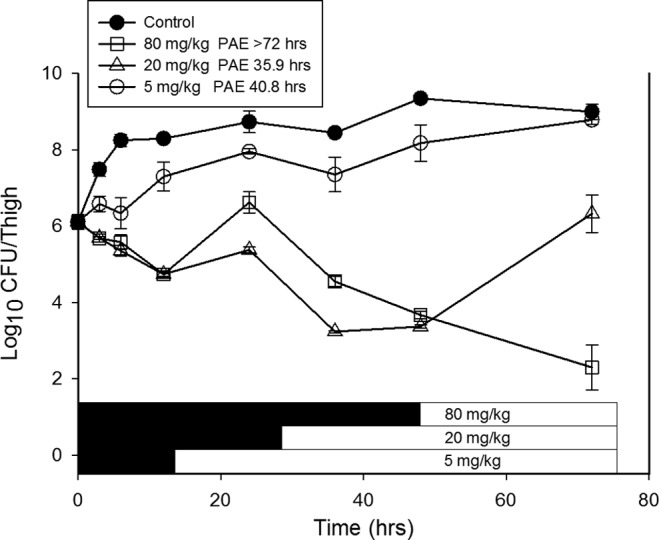
In vivo time-kill experiment with NAI-107 using a neutropenic mouse thigh model. Each symbol represents the mean and standard deviation from four thighs infected with S. aureus ATCC 25923. The error bars represent the standard deviations. Three single subcutaneous doses of NAI-107 were administered to mice. The solid symbols represent the burden of organisms from untreated control animals. The burden of organisms was measured every 3 to 24 h over the 72-h study. The first symbol in time (0 h) represents the burden at the time of dosing. The black horizontal bars represent the time that serum drug concentrations would be estimated to remain above the MIC of the infecting organism. The duration of the postantibiotic effect (PAE) was calculated and is reported at the top of the figure.
Pharmacokinetic/pharmacodynamic index determination.
The relationship among the dose of NAI-107, dosing interval, and effect against S. aureus ATCC 25923 is shown in Fig. 3. The dose-response curves for the every 24-, 36-, and 72-h dosing regimens were very similar. For the two highest dose levels, the every 12-h regimen was shifted somewhat to the left indicating an enhanced effect. The similarity of the dose-response curves among the dosing intervals suggests that the AUC/MIC ratio would be the predictive pharmacodynamic index.
FIG 3.

In vivo dose fractionation with NAI-107 using a neutropenic mouse thigh model. Each symbol represents the mean and standard deviation from four thighs infected with S. aureus ATCC 25923. The error bars represent the standard deviations. Five total drug (mg/kg/72 h) dose levels were fractionated into one of four dosing regimens. The burden of organisms was measured at the start and end of therapy. The study period was 72 h. The horizontal dashed line at 0 represents the burden of organisms in the thighs of mice at the start of therapy. Data points below the line represent killing, and points above the line represent growth.
The relationships between the log10 CFU/thigh and the Cmax/MIC ratio, the AUC/MIC ratio, and the percentage of time serum levels exceeded the MIC are illustrated in Fig. 4A to C for S. aureus ATCC 25923, respectively. Analysis of these complementary analyses suggest the importance of the AUC/MIC ratio based upon visual data fit and the R2 values.
FIG 4.

Impact of pharmacodynamic regression of the in vivo dose fractionation study with NAI-107 against S. aureus ATCC 25923. Each symbol represents the mean and standard deviation from four thighs. The dose data are expressed as either the Cmax/MIC (A), the AUC/MIC (B), or the percentage of time drug concentrations exceeded the MIC over the dosing period (C) (% time above MIC). The R2 represents the coefficient of determination. The ED50 represents the PD index associated with 50% of the maximal effect (Emax), and N is the slope of the relationship or the Hill coefficient. The line drawn through the data points is the best fit line based upon the sigmoid Emax formula. The horizontal dashed line at 0 represents the burden of organisms in the thighs of mice at the start of therapy. Data points below the line represent killing and points above the line represent growth.
Pharmacokinetic/pharmacodynamic index target for efficacy.
To determine whether the AUC/MIC ratios required for effect were similar for multiple pathogens, we studied the activities of the NAI-107 every 12-h dosing regimens against 8 additional strains of S. aureus. The dose-response data for each of the nine S. aureus strains are shown in Fig. 5. The dose-response relationships were quite similar among the strains, which is not surprising, given the relatively narrow MIC range. A 2- to>4-log reduction was observed with this strain collection over the dose-range studies. The doses necessary to produce a bacteriostatic effect and a 1-log reduction in the organism burden as well as in the corresponding AUC/MIC values are shown in Table 2. The static doses varied from 9.6 mg/kg/24 h to 46.1 mg/kg/24 h. The doses associated with a 1-log kill were roughly 2-fold higher than those associated with stasis. The presence of beta-lactam resistance did not alter the pharmacodynamic target required to produce efficacy. The relationships between NAI-107 exposure (expressed as the AUC/MIC ratio) and efficacy against all S. aureus strains are shown in Fig. 6. The relationship among the data for each of the nine strains studied is extremely strong with an R2 value of 0.89. The mean 24-h AUC/MIC associated with stasis was 371 and that needed for a 1-log reduction was nearly 500.
FIG 5.
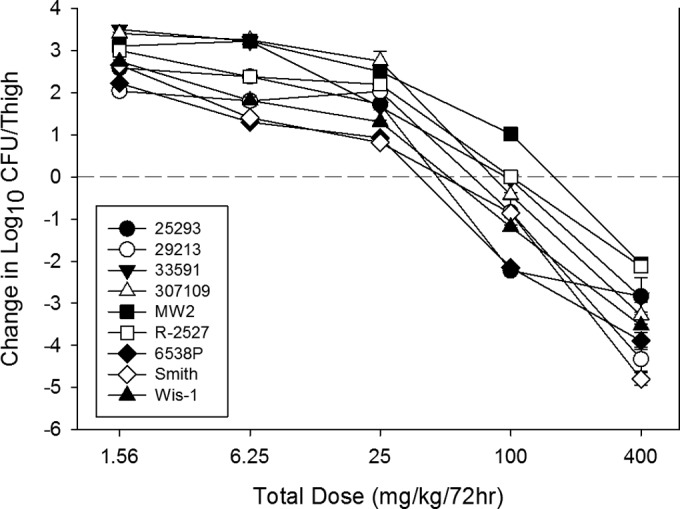
In vivo dose effect of NAI-107 against nine select S. aureus isolates using a neutropenic mouse thigh model. Each symbol represents the mean and standard deviation from four thighs. Five total drug dose levels were fractionated into an every 12-hour regimen. The burden of organisms was measured at the start and end of therapy. The study period was 72 h. The horizontal dashed line at 0 represents the burden of organisms in the thighs of mice at the start of therapy. Data points below the line represent killing and points above the line represent growth.
TABLE 2.
In vitro and in vivo efficacy of NAI-107 against select S. aureus isolates using AUC/MIC as the predictive pharmacodynamic index
| S. aureus strain | MIC (μg/ml) | Static dose (mg/kg/24 h) | 1-Log kill dose (mg/kg/24 h) | AUC/MIC |
|||
|---|---|---|---|---|---|---|---|
| Static dose 24 h | Static dose 72 h | 1-Log kill 24 h | 1-Log kill 72 h | ||||
| 29213 | 0.25 | 25.7 | 35.2 | 623 | 1,080 | 692 | 1,454 |
| 33591 | 0.5 | 25.4 | 44.2 | 310 | 536 | 379 | 913 |
| 307109 | 0.5 | 29.0 | 40.8 | 323 | 600 | 366 | 843 |
| MW2 | 0.5 | 46.1 | 74.5 | 386 | 952 | 490 | 1,539 |
| R-2527 | 0.5 | 35.6 | 64.1 | 348 | 737 | 452 | 1,326 |
| 25923 | 0.5 | 14.6 | 19.7 | 205 | 406 | 286 | 469 |
| 6538P | 0.125 | 9.6 | 17.4 | 500 | 1,382 | 998 | 1,761 |
| Smith | 0.25 | 14.7 | 24.3 | 412 | 814 | 612 | 1,045 |
| WIS-1 | 0.5 | 16.2 | 27.1 | 230 | 425 | 316 | 559 |
| Mean | 24.1 | 38.6 | 371 | 770 | 510 | 1,101 | |
| SD | 11.7 | 19.8 | 130 | 325 | 227 | 447 | |
| Median | 25.4 | 35.2 | 348 | 737 | 452 | 1,045 | |
FIG 6.

In vivo dose effect of NAI-107 against nine S. aureus isolates using a neutropenic mouse thigh model. Each symbol represents the mean and standard deviation from four thighs. Five total drug dose levels were fractionated into an every 12-hour regimen. The study period was 72 h. The NAI-107 exposure is expressed as the total drug 72-h AUC/MIC. The burden of organisms was measured at the start and end of therapy. The horizontal line at 0 represents the burden of organisms in the thighs of mice at the start of therapy. Data points below the line represent killing and points above the line represent growth. The R2 represents the coefficient of determination. The ED50 represents the AUC/MIC associated with 50% of the maximal effect (Emax), and N is the slope of the relationship or the Hill coefficient. The line drawn through the data points is the best fit line based upon the sigmoid Emax formula.
DISCUSSION
The emergence of treatment-resistant pathogens is an escalating threat to public health (18–21). The increasing incidence of drug-resistant bacteria, fungi, parasites, and viruses is evident from contemporary epidemiologic surveys in the United States and worldwide (4, 22–24). This change in microbial ecology is occurring at the same time as our at-risk patient populations continue to grow and effective antimicrobial drug discovery has been on the decline (1, 4, 5, 25–30).
The group of nosocomial bacterial pathogens of greatest concern has been termed the ESKAPE pathogens as they effectively “escape” the effects of common antibacterial drugs (1). This organism collection includes Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species. These bacteria are responsible for the majority of U.S. hospital infections. At least 70% of these hospital-acquired bacterial infections are resistant to at least one commonly used antibiotic (U.S. FDA). Among the 2 million annual infections in hospitalized patients, death results for nearly 100,000 patients (24, 31). For example, more people now die of methicillin-resistant S. aureus (MRSA) in U.S. health care settings than of HIV and tuberculosis combined (32, 33). Additionally, resistance to the last line of antibiotics currently available for most Gram-positive infections (glycopeptides and lipopeptides) are increasing (34–39). Thus, novel agents against Gram-positive pathogens are urgently needed.
The lantibiotic class represents a novel group of naturally derived, ribosomal-synthesized peptides with potent Gram-positive activity. The prototype molecule nisin was discovered in the 1920s and has been used as a food preservative for >40 years (29). The lantibiotics exert their antimicrobial activities by binding to lipid II at a site different from that affected by glycopeptides and are therefore active against MDR Gram-positive pathogens. The result of binding to and sequestering lipid II is inhibition of bacterial cell wall biosynthesis at the transglycosylation step. There are additional proposed mechanisms of action after lipid II binding, which include membrane interaction with or without pore formation (12, 40, 41). Recently, a candidate lantibiotic, NAI-107 (microbisporicin), emerged from a screening program designed to find new bacterial cell wall inhibitors (7, 13).
We report the in vivo pharmacodynamic activity of NAI-107 against S. aureus strains. NAI-107 demonstrated potent in vitro and in vivo efficacy against S. aureus isolates, including those with resistance to beta-lactams. The in vitro MICs varied only 4-fold against NAI-107 in this diverse group of S. aureus isolates despite the oxacillin activity varying by >128-fold. Numerically, the NAI-107 MIC range in the study was lower than those of the comparator drugs vancomycin and linezolid and very similar to those of daptomycin, ceftaroline, and tedizolid. Isolates with demonstrated resistance to these comparator drugs were not tested in the current study, but previous studies have demonstrated a lack of cross-resistance between lantibiotics and glycopeptides (7, 13, 14, 42). The treatment studies demonstrated marked bactericidal activity that was dose dependent. In addition, we observed prolonged growth suppression (PAEs) of >35 h. Dose fractionation studies found that the AUC/MIC was most closely linked to the drug effect. Together, these complementary findings indicate that optimal dosing would include large, infrequent dosing.
An important consideration in PK/PD studies is the inclusion of multiple isolates with various phenotypes and genotypes in order to form a robust PK/PD target estimate. The studies presented here included 9 total isolates, including 5 methicillin-susceptible S. aureus (MSSA) isolates and 4 MRSA isolates, both hospital and community acquired, as well as three different U.S. genotypes. NAI-107 demonstrated potent efficacy in vivo against all of these isolates with a 2- to 5-log10 CFU/thigh drop over a 72-h treatment period with an overall Emax of 8.6 log10 CFU/thigh. This microbiological response in the animal model was similar to or in excess of those for the currently approved MRSA drugs that have been studied in this same model: ceftaroline (43), daptomycin (44–46), oxazolidinones (46, 47), and vancomycin (46). The NAI-107 24-h target AUC/MIC values associated with a net static and a 1-log10 killing effect against S. aureus were nearly 370 and 500, respectively. Once more, the AUC/MIC index was a very strong predictor of efficacy based on regression analysis of the treatment data against a relatively large strain set. One limitation of the current study is that we were unable to perform protein binding studies given the limited drug availability, and therefore only total drug values were considered in these analyses. It will be important in future studies to determine protein binding and perform PK/PD analyses with both total and free-drug exposures. Only one previous study of antimicrobial efficacy of NAI-107 is reported in the literature (42). Jabés and colleagues demonstrated efficacy against MRSA, glycopeptide-intermediate Staphylococcus aureus (GISA), and vancomycin-resistant Enterococcus faecalis (VRE) isolates in a lethal murine model, rat granuloma pouch model, and rat endocarditis model. The S. aureus lethal murine model demonstrated an ED50 of 25 mg/kg/24 h, which is similar to the static dose and ED50 (25 and 30 mg/kg/24 h, respectively) identified in the current study. Besides this single study, which was not optimized to examine pharmacodynamic relationships, there are no other preclinical PK/PD studies for comparison purposes.
In conclusion, these studies demonstrate that NAI-107 has dose-dependent in vivo activity against various strains of S. aureus. The AUC/MIC was the PK/PD index that best predicted efficacy. Both static and killing endpoints were achieved at relatively modest AUC/MIC targets. The targets identified in these studies and preliminary human PK data should be useful for guiding the appropriate dosing regimen design for clinical trials. These findings suggest that NAI-107 is a promising and novel antibiotic candidate for further study and development for the treatment of Gram-positive infections.
ACKNOWLEDGMENT
This study was funded by Sentinella Pharmaceuticals, Inc.
REFERENCES
- 1.Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, Scheld M, Spellberg B, Bartlett J. 2009. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis 48:1–12. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 2.Bassetti M, Merelli M, Temperoni C, Astilean A. 2013. New antibiotics for bad bugs: where are we? Ann Clin Microbiol Antimicrob 12:22. doi: 10.1186/1476-0711-12-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Infectious Diseases Society of America. 2010. The 10 × '20 initiative: pursuing a global commitment to develop 10 new antibacterial drugs by 2020. Clin Infect Dis 50:1081–1083. doi: 10.1086/652237. [DOI] [PubMed] [Google Scholar]
- 4.Spellberg B, Guidos R, Gilbert D, Bradley J, Boucher HW, Scheld WM, Bartlett JG, Edwards J Jr, Infectious Diseases Society of America . 2008. The epidemic of antibiotic-resistant infections: a call to action for the medical community from the Infectious Diseases Society of America. Clin Infect Dis 46:155–164. doi: 10.1086/524891. [DOI] [PubMed] [Google Scholar]
- 5.Infectious Diseases Society of America (IDSA), Spellberg B, Blaser M, Guidos RJ, Boucher HW, Bradley JS, Eisenstein BI, Gerding D, Lynfield R, Reller LB, Rex J, Schwartz D, Septimus E, Tenover FC, Gilbert DN. 2011. Combating antimicrobial resistance: policy recommendations to save lives. Clin Infect Dis 52(Suppl 5):S397–S428. doi: 10.1093/cid/cir153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brumfitt W, Salton MR, Hamilton-Miller JM. 2002. Nisin, alone and combined with peptidoglycan-modulating antibiotics: activity against methicillin-resistant Staphylococcus aureus and vancomycin-resistant enterococci. J Antimicrob Chemother 50:731–734. doi: 10.1093/jac/dkf190. [DOI] [PubMed] [Google Scholar]
- 7.Castiglione F, Lazzarini A, Carrano L, Corti E, Ciciliato I, Gastaldo L, Candiani P, Losi D, Marinelli F, Selva E, Parenti F. 2008. Determining the structure and mode of action of microbisporicin, a potent lantibiotic active against multiresistant pathogens. Chem Biol 15:22–31. doi: 10.1016/j.chembiol.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 8.Breukink E, de Kruijff B. 2006. Lipid II as a target for antibiotics. Nat Rev Drug Discov 5:321–332. doi: 10.1038/nrd2004. [DOI] [PubMed] [Google Scholar]
- 9.Breukink E, Wiedemann I, van Kraaij C, Kuipers OP, Sahl HG, de Kruijff B. 1999. Use of the cell wall precursor lipid II by a pore-forming peptide antibiotic. Science 286:2361–2364. doi: 10.1126/science.286.5448.2361. [DOI] [PubMed] [Google Scholar]
- 10.Brötz H, Josten M, Wiedemann I, Schneider U, Gotz F, Bierbaum G, Sahl HG. 1998. Role of lipid-bound peptidoglycan precursors in the formation of pores by nisin, epidermin and other lantibiotics. Mol Microbiol 30:317–327. doi: 10.1046/j.1365-2958.1998.01065.x. [DOI] [PubMed] [Google Scholar]
- 11.Wiedemann I, Breukink E, van Kraaij C, Kuipers OP, Bierbaum G, de Kruijff B, Sahl HG. 2001. Specific binding of nisin to the peptidoglycan precursor lipid II combines pore formation and inhibition of cell wall biosynthesis for potent antibiotic activity. J Biol Chem 276:1772–1779. doi: 10.1074/jbc.M006770200. [DOI] [PubMed] [Google Scholar]
- 12.Münch D, Muller A, Schneider T, Kohl B, Wenzel M, Bandow JE, Maffioli S, Sosio M, Donadio S, Wimmer R, Sahl HG. 2014. The lantibiotic NAI-107 binds to bactoprenol-bound cell wall precursors and impairs membrane functions. J Biol Chem 289:12063–12076. doi: 10.1074/jbc.M113.537449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Castiglione F, Cavaletti L, Losi D, Lazzarini A, Carrano L, Feroggio M, Ciciliato I, Corti E, Candiani G, Marinelli F, Selva E. 2007. A novel lantibiotic acting on bacterial cell wall synthesis produced by the uncommon actinomycete Planomonospora sp. Biochemistry 46:5884–5895. doi: 10.1021/bi700131x. [DOI] [PubMed] [Google Scholar]
- 14.Cotter PD, Hill C, Ross RP. 2005. Bacterial lantibiotics: strategies to improve therapeutic potential. Curr Protein Pept Sci 6:61–75. doi: 10.2174/1389203053027584. [DOI] [PubMed] [Google Scholar]
- 15.Clinical and Laboratory Standards Institute. 2012. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; approved standard, 9th ed. CLSI document M07-A9. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 16.National Research Council Committee on the Care and Use of Laboratory Animals, Institute of Laboratory Animal Resources and Commission on Life Sciences. 1996. Guide for the care and use of laboratory animals. National Academy Press, Washington, DC. [Google Scholar]
- 17.Andes D, Craig WA. 1998. In vivo activities of amoxicillin and amoxicillin-clavulanate against Streptococcus pneumoniae: application to breakpoint determinations. Antimicrob Agents Chemother 42:2375–2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fauci AS, Morens DM. 2012. The perpetual challenge of infectious diseases. N Engl J Med 366:454–461. doi: 10.1056/NEJMra1108296. [DOI] [PubMed] [Google Scholar]
- 19.Morens DM, Fauci AS. 2012. Emerging infectious diseases in 2012: 20 years after the Institute of Medicine report. mBio 3(6):e00494-12. doi: 10.1128/mBio.00494-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morens DM, Folkers GK, Fauci AS. 2004. The challenge of emerging and reemerging infectious diseases. Nature 430:242–249. doi: 10.1038/nature02759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Taubes G. 2008. The bacteria fight back. Science 321:356–361. doi: 10.1126/science.321.5887.356. [DOI] [PubMed] [Google Scholar]
- 22.Buckingham SC, McDougal LK, Cathey LD, Comeaux K, Craig AS, Fridkin SK, Tenover FC. 2004. Emergence of community-associated methicillin-resistant Staphylococcus aureus at a Memphis, Tennessee children's hospital. Pediatr Infect Dis J 23:619–624. doi: 10.1097/01.inf.0000131981.67342.c4. [DOI] [PubMed] [Google Scholar]
- 23.Burton DC, Edwards JR, Horan TC, Jernigan JA, Fridkin SK. 2009. Methicillin-resistant Staphylococcus aureus central line-associated bloodstream infections in US intensive care units, 1997-2007. JAMA 301:727–736. doi: 10.1001/jama.2009.153. [DOI] [PubMed] [Google Scholar]
- 24.Sievert DM, Ricks P, Edwards JR, Schneider A, Patel J, Srinivasan A, Kallen A, Limbago B, Fridkin S, National Healthcare Safety Network (NHSN) Team and Participating NHSN Facilities . 2013. Antimicrobial-resistant pathogens associated with healthcare-associated infections: summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2009-2010. Infect Control Hosp Epidemiol 34:1–14. doi: 10.1086/668770. [DOI] [PubMed] [Google Scholar]
- 25.Talbot GH. 2008. What is in the pipeline for Gram-negative pathogens? Expert Rev Anti Infect Ther 6:39–49. doi: 10.1586/14787210.6.1.39. [DOI] [PubMed] [Google Scholar]
- 26.Talbot GH, Bradley J, Edwards JE Jr, Gilbert D, Scheld M, Bartlett JG, Antimicrobial Availability Task Force of the Infectious Diseases Society of America . 2006. Bad bugs need drugs: an update on the development pipeline from the Antimicrobial Availability Task Force of the Infectious Diseases Society of America. Clin Infect Dis 42:657–668. doi: 10.1086/499819. [DOI] [PubMed] [Google Scholar]
- 27.Spellberg B. 2008. Antibiotic resistance and antibiotic development. Lancet Infect Dis 8:211−212. doi: 10.1016/S1473-3099(08)70048-3. [DOI] [PubMed] [Google Scholar]
- 28.Spellberg B, Bartlett JG, Gilbert DN. 2013. The future of antibiotics and resistance. N Engl J Med 368:299–302. doi: 10.1056/NEJMp1215093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Spellberg B, Powers JH, Brass EP, Miller LG, Edwards JE Jr. 2004. Trends in antimicrobial drug development: implications for the future. Clin Infect Dis 38:1279–1286. doi: 10.1086/420937. [DOI] [PubMed] [Google Scholar]
- 30.Nathan C. 2004. Antibiotics at the crossroads. Nature 431:899–902. doi: 10.1038/431899a. [DOI] [PubMed] [Google Scholar]
- 31.Klevens RM, Edwards JR, Richards CL Jr, Horan TC, Gaynes RP, Pollock DA, Cardo DM. 2007. Estimating health care-associated infections and deaths in U.S. hospitals, 2002. Public Health Rep 122:160–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klevens RM, Morrison MA, Nadle J, Petit S, Gershman K, Ray S, Harrison LH, Lynfield R, Dumyati G, Townes JM, Craig AS, Zell ER, Fosheim GE, McDougal LK, Carey RB, Fridkin SK, Active Bacterial Core Surveillance (ABCs) MRSA Investigators . 2007. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA 298:1763–1771. doi: 10.1001/jama.298.15.1763. [DOI] [PubMed] [Google Scholar]
- 33.Liu C, Bayer A, Cosgrove SE, Daum RS, Fridkin SK, Gorwitz RJ, Kaplan SL, Karchmer AW, Levine DP, Murray BE, Rybak MJ, Talan DA, Chambers HF. 2011. Clinical practice guidelines by the Infectious Diseases Society of America for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children: executive summary. Clin Infect Dis 52:285–292. doi: 10.1093/cid/cir034. [DOI] [PubMed] [Google Scholar]
- 34.Rice LB. 2012. Mechanisms of resistance and clinical relevance of resistance to beta-lactams, glycopeptides, and fluoroquinolones. Mayo Clin Proc 87:198–208. doi: 10.1016/j.mayocp.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rodvold KA, McConeghy KW. 2014. Methicillin-resistant Staphylococcus aureus therapy: past, present, and future. Clin Infect Dis 58(Suppl 1):S20–S27. doi: 10.1093/cid/cit614. [DOI] [PubMed] [Google Scholar]
- 36.Rubinstein E, Keynan Y. 2013. Vancomycin-resistant enterococci. Crit Care Clin 29:841–852. doi: 10.1016/j.ccc.2013.06.006. [DOI] [PubMed] [Google Scholar]
- 37.Calfee DP. 2012. Methicillin-resistant Staphylococcus aureus and vancomycin-resistant enterococci, and other Gram-positives in healthcare. Curr Opin Infect Dis 25:385–394. doi: 10.1097/QCO.0b013e3283553441. [DOI] [PubMed] [Google Scholar]
- 38.Stryjewski ME, Corey GR. 2014. Methicillin-resistant Staphylococcus aureus: an evolving pathogen. Clin Infect Dis 58(Suppl 1):S10–S19. doi: 10.1093/cid/cit613. [DOI] [PubMed] [Google Scholar]
- 39.Chambers HF, Deleo FR. 2009. Waves of resistance: Staphylococcus aureus in the antibiotic era. Nat Rev Microbiol 7:629–641. doi: 10.1038/nrmicro2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dawson MJ, Scott RW. 2012. New horizons for host defense peptides and lantibiotics. Curr Opin Pharmacol 12:545–550. doi: 10.1016/j.coph.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van Heusden HE, de Kruijff B, Breukink E. 2002. Lipid II induces a transmembrane orientation of the pore-forming peptide lantibiotic nisin. Biochemistry 41:12171–12178. doi: 10.1021/bi026090x. [DOI] [PubMed] [Google Scholar]
- 42.Jabés D, Brunati C, Candiani G, Riva S, Romano G, Donadio S. 2011. Efficacy of the new lantibiotic NAI-107 in experimental infections induced by multidrug-resistant Gram-positive pathogens. Antimicrob Agents Chemother 55:1671–1676. doi: 10.1128/AAC.01288-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Andes D, Craig WA. 2006. Pharmacodynamics of a new cephalosporin, PPI-0903 (TAK-599), active against methicillin-resistant Staphylococcus aureus in murine thigh and lung infection models: identification of an in vivo pharmacokinetic-pharmacodynamic target. Antimicrob Agents Chemother 50:1376–1383. doi: 10.1128/AAC.50.4.1376-1383.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Safdar N, Andes D, Craig WA. 2004. In vivo pharmacodynamic activity of daptomycin. Antimicrob Agents Chemother 48:63–68. doi: 10.1128/AAC.48.1.63-68.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.LaPlante KL, Leonard SN, Andes DR, Craig WA, Rybak MJ. 2008. Activities of clindamycin, daptomycin, doxycycline, linezolid, trimethoprim-sulfamethoxazole, and vancomycin against community-associated methicillin-resistant Staphylococcus aureus with inducible clindamycin resistance in murine thigh infection and in vitro pharmacodynamic models. Antimicrob Agents Chemother 52:2156–2162. doi: 10.1128/AAC.01046-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee DG, Murakami Y, Andes DR, Craig WA. 2013. Inoculum effects of ceftobiprole, daptomycin, linezolid, and vancomycin with Staphylococcus aureus and Streptococcus pneumoniae at inocula of 105 and 107 CFU injected into opposite thighs of neutropenic mice. Antimicrob Agents Chemother 57:1434–1441. doi: 10.1128/AAC.00362-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lepak AJ, Marchillo K, Pichereau S, Craig WA, Andes DR. 2012. Comparative pharmacodynamics of the new oxazolidinone tedizolid phosphate and linezolid in a neutropenic murine Staphylococcus aureus pneumonia model. Antimicrob Agents Chemother 56:5916–5922. doi: 10.1128/AAC.01303-12. [DOI] [PMC free article] [PubMed] [Google Scholar]