

Abstract
Multidrug resistance constitutes a threat to the medical achievements of the last 50 years. In this study, we demonstrated the abilities of two de novo engineered cationic antibiotic peptides (eCAPs), WLBU2 and WR12, to overcome resistance from 142 clinical isolates representing the most common multidrug-resistant (MDR) pathogens and to display a lower propensity to select for resistant bacteria in vitro compared to that with colistin and LL37. The results warrant an exploration of eCAPs for use in clinical settings.
TEXT
Very few if any medical discoveries have had a larger impact on modern medicine than the discovery and development of antibiotics (1–3). However, the success of this medical achievement is being threatened due to the increasing frequency of antibiotic resistance (4, 5). Alarmingly, the development of novel classes of antibiotics has been limited in the last 4 decades (6, 7). As a result, the diversity of bacterial resistance mechanisms has largely outperformed our current classes of antibiotics. Currently, the Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species (ESKAPE) pathogens, the most common multidrug-resistant (MDR) and extensively drug-resistant (XDR) bacteria (8), represent a serious threat to patients who are frequently in an immunocompromised state (e.g., those undergoing transplantation, cancer, and critically ill patients) (7, 9–11). Hence, more antimicrobial research is required to overcome this critical deficit.
Cationic antimicrobial peptides (AMPs) (12) are ubiquitous and structurally diverse effector molecules representing the first line of defense against microbial pathogens (13–15). AMPs generally recognize microbial organisms by electrostatic interactions with highly electronegative bacterial surface lipids (e.g., lipid A in Gram-negative bacteria and lipoteichoic acid [LTA] in Gram-positive bacteria) (16–18). These nonreceptor-mediated interactions commonly resulting in pore-forming (among other) mechanisms, coupled with a rapid killing kinetics, are largely responsible for the broad success of AMPs against bacterial pathogens (19).
We previously demonstrated that two lead engineered cationic antibiotic peptides (eCAPs), WLBU2 (a 24-mer containing Arg, Val, and Trp) and WR12 (a 12-mer containing Arg and Trp only) (Fig. 1), form amphipathic α-helices in a hydrophobic environment and display broad activities in vitro against diverse Gram-negative and Gram-positive bacteria, including several highly drug-resistant strains (20–22). Importantly, WLBU2 is the first synthetic AMP to demonstrate an ability to eradicate otherwise lethal P. aeruginosa septicemia in vivo in a systemic treatment model in mice (23, 24).
FIG 1.
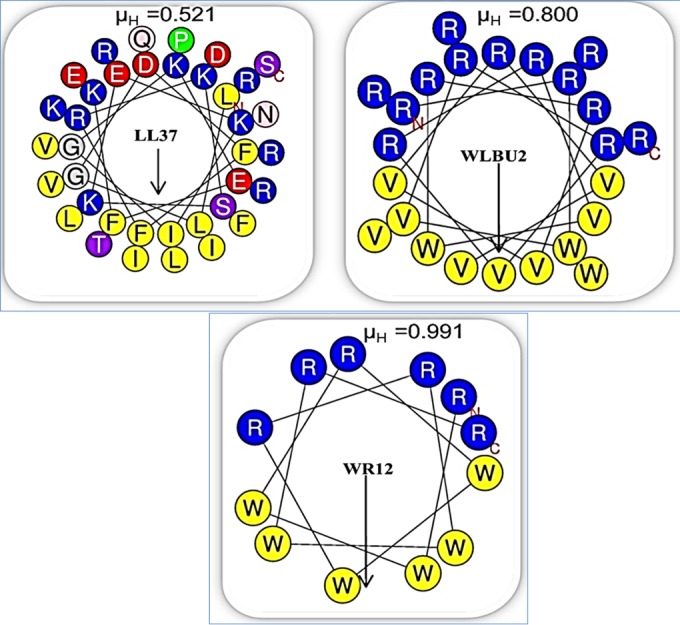
Helical wheel analysis of the amphipathic structures of LL37, WLBU2, and WR12 using HeliQuest (http://heliquest.ipmc.cnrs.fr/). The primary sequence of LL37 displays 14 different amino acids distributed as an imperfect cationic amphipathic structure. In contrast, nature-inspired but sequence-optimized eCAPs are composed of only three (WLBU2) or two (WR12) different amino acids modeled to form idealized amphipathic helices. The arrows indicate the directions and magnitudes of the hydrophobic moments (μH) determined for each peptide; N, amino terminus; C, carboxy terminus.
Although previous studies indicated the unique clinical potential of the lead eCAPs, their efficacy against the most common MDR/XDR pathogens and the propensity of eCAPs to invoke selection of bacterial resistance remain uncharacterized. Thus, we hypothesized that a de novo rational sequence optimization of AMP amphipathic motifs, as demonstrated in WLBU2 and WR12, would substantially potentiate their activities against MDR/XDR pathogens and result in a lower propensity to invoke resistance than that of conventional antimicrobial agents. To address this prediction, we compared the activities of WLBU2 and WR12 with those of the natural AMP LL37 and colistin, a bacterium-derived cationic agent that is often used against hard-to-treat Gram-negative infections when other less toxic agents are inactive (25–29). As shown in Fig. 1, the primary sequence of LL37 displays 14 different amino acids distributed as an imperfect cationic amphipathic structure; in contrast, nature-inspired but sequence-optimized eCAPs are composed of only three (WLBU2) or two (WR12) different amino acids modeled to form idealized amphipathic helices.
In a reference panel of 142 clinical isolates (see Table S1A and B in the supplemental material) that we tested, 58% were the most common MDR/XDR (ESKAPE) pathogens: 100 isolates were from pediatric cystic fibrosis (CF) patients at Seattle Children's Hospital (SCH) with chronic pulmonary infections, and 42 isolates were from hospitalized adult patients at the University of Pittsburgh Medical Center (UPMC). These included 32 Gram-positive and 110 Gram-negative clinical isolates. We first determined the MIC for each individual isolate, determined from three independent experimental trials (for reproducibility) in Mueller-Hinton broth (MHB), and then compared the mean MIC distributions of all four cationic peptides, using 32 μM as the maximum concentration (Fig. 2). As expected, colistin and LL37 were more effective against Gram-negative (Fig. 2B) than against Gram-positive (Fig. 2A) isolates, with mean MICs between 20 and 30 μM for the Gram-positive clinical isolates and 10 to 20 μM for the Gram-negative clinical isolates. In contrast, the mean MICs for the eCAPs were ≤10 μM for both Gram-positive and Gram-negative strains, with statistically significant differences indicated by a P value of <0.0001 compared to those for colistin and LL37 (Fig. 2A and B). Further examination of the breadth of activity, based on susceptibility defined as an MIC of <32 μM, revealed no significant differences between colistin and LL37 (P > 0.05), as they each inhibited 51% (73/142) of the clinical isolates (Table 1). Colistin inhibited the growth of 63% of the Gram-negative bacteria and 13% of the Gram-positive bacteria, while LL37 inhibited 56% of the Gram-negative and 31% of the Gram-positive bacteria, with no statistically significant difference between the activities of colistin and LL37 (Table 1). In marked contrast, the eCAPs displayed activities of 87% for WR12 and 91% for WLBU2. Of note, the difference in bacterial resistance between WR12 (13%) and WLBU2 (8%) is mainly due to the resistance demonstrated by the 20 Burkholderia cepacia complex isolates, of which 10 were resistant to WR12, and 4 were resistant to WLBU2. Interestingly, most colistin-resistant strains were also resistant to LL37 (75%), in sharp contrast to the inhibition of 80 to 86% of the colistin- and LL37-resistant isolates by eCAPs (Table 2; see also Table SA2 in the supplemental material). In addition, almost all eCAP-resistant strains also displayed resistance to colistin and LL37: 10 of the 11 WLBU2- and 14 of the 18 WR12-resistant isolates were also resistant to colistin and LL37 (Table 2). As the 142 isolates were selected because of their MDR/XDR properties (see Table SA1 in the supplemental material), it is quite interesting that colistin and LL37 were active against 51% of these isolates, particularly considering that these isolates have probably been exposed to LL37 in the context of an infected patient with an underlying pathophysiological condition, which reflects environment-dependent efficacy (20, 30). With regard to colistin, the data are consistent with its frequent use as a last-resort antibiotic against MDR bacterial pathogens, given the fact that the panel of clinical isolates used in this study is skewed toward MDR/XDR phenotypes (31, 32).
FIG 2.
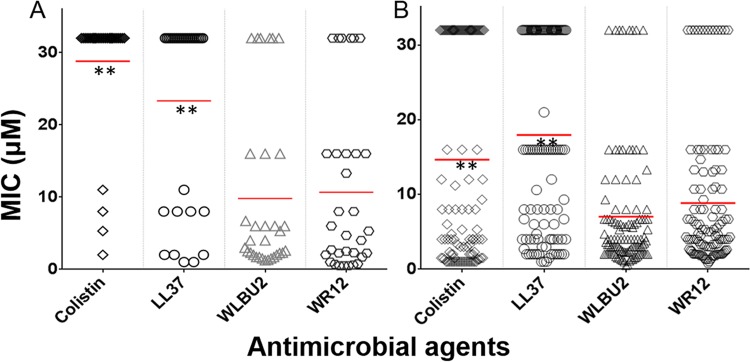
In vitro activities of colistin, LL37, WLBU2, and WR12 against 142 MDR/XDR clinical strains from CF (n = 100) and hospitalized (n = 42) patients. Shown are the mean MIC distributions against Gram-positive (A) and Gram-negative (B) bacterial isolates. **, statistically significant differences between the mean MICs of eCAPs (WLBU2 and WR12) and those of colistin and LL37 (P < 0.0001) using one-way analysis of variance (ANOVA) with multiple comparison tests; there are no significant differences between colistin and LL37 (P > 0.05).
TABLE 1.
Spectrum of activities of colistin, LL37, WLBU2, and WR12 against the MDR/XDR clinical strainsa
| Bacterial strainsb | No. of strains that are susceptible to the indicated antimicrobial agent/total no. of strains |
|||
|---|---|---|---|---|
| Colistin | LL37 | WLBU2 | WR12 | |
| Gram positive | 4/32 | 10/32 | 30/32 | 29/32 |
| MRSA | 4/26 | 4/26 | 25/26 | 25/26 |
| VRE | 0/6 | 6/6 | 5/6 | 4/6 |
| Gram negative | 69/110 | 62/110 | 101/110 | 95/110 |
| Enterobacteriaceae | 14/18 | 13/18 | 17/18 | 18/18 |
| Acinetobacter baumannii | 2/6 | 6/6 | 6/6 | 6/6 |
| Achromobacter spp. | 17/20 | 9/20 | 20/20 | 19/20 |
| S. maltophilia | 13/20 | 9/20 | 17/20 | 17/20 |
| B. cepacia complex | 3/20 | 4/20 | 16/20 | 10/20 |
| P. aeruginosa | 20/26 | 21/26 | 25/26 | 25/26 |
| Total | 73/142 | 72/142 | 131/142 | 124/142 |
Susceptibility defined as mean MIC of <32 μM.
MRSA, methicillin-resistant S. aureus; VRE, vancomycin-resistant enterococci. Enterobacteriaceae includes K. pneumoniae, Enterobacter aerogenes/E. cloacae, and E. coli; Achromobacter spp. includes A. xylosoxidans.
TABLE 2.
Spectrum of activities of colistin, LL37, WLBU2, and WR12 against the MDR/XDR clinical strains
| Cross-resistance | No. of clinical strains that display cross-resistance to cationic antimicrobial agents |
|||
|---|---|---|---|---|
| Colistin | LL37 | WLBU2 | WR12 | |
| Colistin-Ra | 52/69 | 10/69 | 14/69 | |
| LL37-R | 52/70 | 10/70 | 14/70 | |
| WLBU2-R | 10/11 | 9/11 | 9/11 | |
| WR12-R | 14/18 | 14/18 | 9/18 | |
-R, cross-resistance.
To further examine the difference in antimicrobial effectiveness against MDR/XDR organisms between eCAPs and other antibiotics, we compared rifampin, colistin, LL37, and eCAPs for their propensity to select for bacterial resistance phenotypes in vitro. Thus, we serially passaged three different P. aeruginosa strains (PAO1 and 2 clinical isolates) in the presence of subinhibitory concentrations (0.5× the MIC) of the respective test agents and monitored the MIC daily, using standard growth inhibition assays. As shown in Fig. 3, all three P. aeruginosa strains developed resistance to rifampin (fold MIC, >10) within the first 3 days of antibiotic exposure, while the development of LL37 and colistin resistance phenotypes emerged by 9 and 13 days, respectively. In contrast, resistance to eCAPs required up to 25 to 30 days to appear (Fig. 3A). We next examined whether the development of resistance against one antimicrobial agent induced cross-resistance to the other antibiotics. As expected from the different modes of action, rifampin shared no cross-resistance with the cationic peptides (Fig. 3B). Importantly, the induction of resistance to colistin resulted in cross-resistance to LL37 in two of the three P. aeruginosa strains, consistent with a recent report (33), but had no effect on the susceptibilities of any of the three strains to eCAPs. In contrast to the cross-resistance patterns observed in the clinical isolates (Table 1), the selection of resistance against LL37 in vitro unexpectedly resulted in eCAP- and colistin-resistant phenotypes as well. Similarly to the resistance patterns observed in the clinical isolates (Table 2; see also Table S2 in the supplemental material), however, experimentally derived resistance to eCAPs led to resistances to the other cationic antibiotics LL37 and colistin (Fig. 3B).
FIG 3.

(A) Selection of resistance invoked by rifampin, LL37, colistin, WR12, and WLBU2 by serial passages in MHB at 50% of their corresponding MIC, using three P. aeruginosa strains (PAO1 and two clinical isolates); the shaded areas indicate the development of resistance to the corresponding antimicrobial agents (fold MIC, ≥10). (B) Cross-resistance displayed by experimentally derived resistant strains. S, susceptible; R, resistant.
Taken together, the current data demonstrate that de novo structural optimization leading to two idealized amphipathic peptides significantly enhances the in vitro antimicrobial spectrum against the most common clinical isolates of ESKAPE pathogens compared to that with colistin and LL37. In addition, eCAPs are less likely to invoke resistance when bacteria are exposed to subinhibitory antibiotic concentrations. Importantly, the experimentally derived resistance against the cationic antibiotic colistin had no obvious effects on susceptibilities to the eCAPs, indicating distinct eCAP interactions with target bacteria compared to those with colistin.
The prevalence of MDR/XDR bacteria requires the development of both preventive and anti-infective countermeasures. It is increasingly evident that the membrane-targeting antimicrobial mechanisms of AMPs make them likely candidates for overcoming bacterial drug resistance properties and warrant consideration for development as effective antibiotics. Here, we report that the optimization of AMP amphipathic structure, as shown in eCAPs, results in the ability to overcome most AMP resistance by common MDR/XDR bacterial (ESKAPE) pathogens. The data also suggest that further optimization is needed to address less common resistance mechanisms, which can be achieved once those mechanisms are elucidated, a current goal of our laboratory. While a single sequence-optimized eCAP is not predicted to be effective against all MDR/XDR bacteria, continuing AMP or eCAP development will result in more effective and potentially less toxic therapeutic options than what is seen with the currently available agents.
Supplementary Material
ACKNOWLEDGMENTS
We thank Kazi Islam and the staff of the University of Pittsburgh Peptide Synthesis facility for their technical expertise in peptide synthesis.
This project was supported in part by NIH grant 5P30 DK072506 awarded to the Cystic Fibrosis Research Center of the University of Pittsburgh and by funds from the Center for Vaccine Research of the University of Pittsburgh. Additional support was also provided by NIH grants R01AI104895, R21AI107302, and P30 DK089507 (awarded to Seattle Children's Hospital).
R.C.M., J.D.S., and J.K.C. hold stock in Peptilogics. R.C.M. serves on an advisory board for Peptilogics, and J.D.S. has a management role at Peptilogics. Although a financial conflict of interest was identified based on the authors' relationship with Peptilogics, the research findings included in this publication may not necessarily be related to the interests of Peptilogics.
R.C.M. is the principal investigator and provided study objectives, guidance, and primary edits, in addition to final review of the manuscript. J.D.S. and J.K.C. contributed to the final manuscript with data presentation (figures), discussions of data interpretation, review, and edits. Y.D. and J.L.B. are collaborators who provided all the clinical strains and associated antibiotic susceptibility profiles, useful guidance, and reviewed and edited the manuscript. B.D. designed and performed the experiments, performed the primary analysis of the data, and assumed primary responsibility for the manuscript preparation.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.03937-14.
REFERENCES
- 1.Wright JD, Hassan K, Ananth CV, Herzog TJ, Lewin SN, Burke WM, Lu YS, Neugut AI, Hershman DL. 2013. Use of guideline-based antibiotic prophylaxis in women undergoing gynecologic surgery. Obstet Gynecol 122:1145–1153. doi: 10.1097/AOG.0b013e3182a8a36a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Luft D, Lemmen S, Geipel U, Meerbach D, Scheithauer S, Jäger M, Eckmann C, Dettenkofer M. 2012. Infection control in the operating room: preventive measures and isolation precautions in cases of multidrug resistant pathogens]. Zentralbl Chir 137:284–292. (In German.) doi: 10.1055/s-0031-1271525. [DOI] [PubMed] [Google Scholar]
- 3.Shaffer WO, Baisden JL, Fernand R, Matz PG, North American Spine Society . 2013. An evidence-based clinical guideline for antibiotic prophylaxis in spine surgery. Spine J 13:1387–1392. doi: 10.1016/j.spinee.2013.06.030. [DOI] [PubMed] [Google Scholar]
- 4.Rolain JM, Canton R, Cornaglia G. 2012. Emergence of antibiotic resistance: need for a new paradigm. Clin Microbiol Infect 18:615–616. doi: 10.1111/j.1469-0691.2012.03902.x. [DOI] [PubMed] [Google Scholar]
- 5.Il'ina TS. 2012. Mobile ISCR elements: structure, functions, and role in the emergence, increasing and spreading of blocks of bacterial genes of multiple antibiotic resistance. Mol Gen Mikrobiol Virusol (4):3–13. (In Russian.) doi: 10.3103/S0891416812040040. [DOI] [PubMed] [Google Scholar]
- 6.Alemayehu D, Quinn J, Cook J, Kunkel M, Knirsch CA. 2012. A paradigm shift in drug development for treatment of rare multidrug-resistant Gram-negative pathogens. Clin Infect Dis 55:562–567. doi: 10.1093/cid/cis503. [DOI] [PubMed] [Google Scholar]
- 7.Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, Scheld M, Spellberg B, Bartlett J. 2009. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis 48:1–12. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 8.Magiorakos AP, Srinivasan A, Carey RB, Carmeli Y, Falagas ME, Giske CG, Harbarth S, Hindler JF, Kahlmeter G, Olsson-Liljequist B, Paterson DL, Rice LB, Stelling J, Struelens MJ, Vatopoulos A, Weber JT, Monnet DL. 2012. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: an international expert proposal for interim standard definitions for acquired resistance. Clin Microbiol Infect 18:268–281. doi: 10.1111/j.1469-0691.2011.03570.x. [DOI] [PubMed] [Google Scholar]
- 9.Bow EJ. 2013. There should be no ESKAPE for febrile neutropenic cancer patients: the dearth of effective antibacterial drugs threatens anticancer efficacy. J Antimicrob Chemother 68:492–495. doi: 10.1093/jac/dks512. [DOI] [PubMed] [Google Scholar]
- 10.Ho JY, Cira NJ, Crooks JA, Baeza J, Weibel DB. 2012. Rapid identification of ESKAPE bacterial strains using an autonomous microfluidic device. PLoS One 7:e41245. doi: 10.1371/journal.pone.0041245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pendleton JN, Gorman SP, Gilmore BF. 2013. Clinical relevance of the ESKAPE pathogens. Expert Rev Anti Infect Ther 11:297–308. doi: 10.1586/eri.13.12. [DOI] [PubMed] [Google Scholar]
- 12.Tan CM, Therien AG, Lu J, Lee SH, Caron A, Gill CJ, Lebeau-Jacob C, Benton-Perdomo L, Monteiro JM, Pereira PM, Elsen NL, Wu J, Deschamps K, Petcu M, Wong S, Daigneault E, Kramer S, Liang L, Maxwell E, Claveau D, Vaillancourt J, Skorey K, Tam J, Wang H, Meredith TC, Sillaots S, Wang-Jarantow L, Ramtohul Y, Langlois E, Landry F, Reid JC, Parthasarathy G, Sharma S, Baryshnikova A, Lumb KJ, Pinho MG, Soisson SM, Roemer T. 2012. Restoring methicillin-resistant Staphylococcus aureus susceptibility to β-lactam antibiotics. Sci Transl Med 4:126ra135. doi: 10.1126/scitranslmed.3003592. [DOI] [PubMed] [Google Scholar]
- 13.Hancock RE. 2001. Cationic peptides: effectors in innate immunity and novel antimicrobials. Lancet Infect Dis 1:156–164. doi: 10.1016/S1473-3099(01)00092-5. [DOI] [PubMed] [Google Scholar]
- 14.Pasupuleti M, Schmidtchen A, Malmsten M. 2012. Antimicrobial peptides: key components of the innate immune system. Crit Rev Biotechnol 32:143–171. doi: 10.3109/07388551.2011.594423. [DOI] [PubMed] [Google Scholar]
- 15.Phadke SM, Deslouches B, Hileman SE, Montelaro RC, Wiesenfeld HC, Mietzner TA. 2005. Antimicrobial peptides in mucosal secretions: the importance of local secretions in mitigating infection. J Nutr 135:1289–1293. [DOI] [PubMed] [Google Scholar]
- 16.Brogden KA. 2005. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat Rev Microbiol 3:238–250. doi: 10.1038/nrmicro1098. [DOI] [PubMed] [Google Scholar]
- 17.Mihajlovic M, Lazaridis T. 2010. Antimicrobial peptides in toroidal and cylindrical pores. Biochim Biophys Acta 1798:1485–1493. doi: 10.1016/j.bbamem.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Y, Shi W, Tang S, Li J, Yin S, Gao X, Wang L, Zou L, Zhao J, Huang Y, Shan L, Gounni AS, Wu Y, Yuan F, Zhang J. 2013. The influence of cathelicidin LL37 in human anti-neutrophils cytoplasmic antibody (ANCA)-associated vasculitis. Arthritis Res Ther 15:R161. doi: 10.1186/ar4344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hale JD, Hancock RE. 2007. Alternative mechanisms of action of cationic antimicrobial peptides on bacteria. Expert Rev Anti Infect Ther 5:951–959. doi: 10.1586/14787210.5.6.951. [DOI] [PubMed] [Google Scholar]
- 20.Deslouches B, Steckbeck JD, Craigo JK, Doi Y, Mietzner TA, Montelaro RC. 2013. Rational design of engineered cationic antimicrobial peptides consisting exclusively of arginine and tryptophan, and their activity against multidrug-resistant pathogens. Antimicrob Agents Chemother 57:2511–2521. doi: 10.1128/AAC.02218-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deslouches B, Phadke SM, Lazarevic V, Cascio M, Islam K, Montelaro RC, Mietzner TA. 2005. De novo generation of cationic antimicrobial peptides: influence of length and tryptophan substitution on antimicrobial activity. Antimicrob Agents Chemother 49:316–322. doi: 10.1128/AAC.49.1.316-322.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deslouches B, Islam K, Craigo JK, Paranjape SM, Montelaro RC, Mietzner TA. 2005. Activity of the de novo engineered antimicrobial peptide WLBU2 against Pseudomonas aeruginosa in human serum and whole blood: implications for systemic applications. Antimicrob Agents Chemother 49:3208–3216. doi: 10.1128/AAC.49.8.3208-3216.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deslouches B, Gonzalez IA, DeAlmeida D, Islam K, Steele C, Montelaro RC, Mietzner TA. 2007. De novo-derived cationic antimicrobial peptide activity in a murine model of Pseudomonas aeruginosa bacteraemia. J Antimicrob Chemother 60:669–672. doi: 10.1093/jac/dkm253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paranjape SM, Lauer TW, Montelaro RC, Mietzner TA, Vij N. 2013. Modulation of proinflammatory activity by the engineered cationic antimicrobial peptide WLBU-2. F1000Res 2:36. doi: 10.12688/f1000research.2-36.v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shields RK, Kwak EJ, Potoski BA, Doi Y, Adams-Haduch JM, Silviera FP, Toyoda Y, Pilewski JM, Crespo M, Pasculle AW, Clancy CJ, Nguyen MH. 2011. High mortality rates among solid organ transplant recipients infected with extensively drug-resistant Acinetobacter baumannii: using in vitro antibiotic combination testing to identify the combination of a carbapenem and colistin as an effective treatment regimen. Diagn Microbiol Infect Dis 70:246–252. doi: 10.1016/j.diagmicrobio.2010.12.023. [DOI] [PubMed] [Google Scholar]
- 26.Jean SS, Hsueh PR. 2011. Current review of antimicrobial treatment of nosocomial pneumonia caused by multidrug-resistant pathogens. Expert Opin Pharmacother 12:2145–2148. doi: 10.1517/14656566.2011.599320. [DOI] [PubMed] [Google Scholar]
- 27.Michalopoulos A, Falagas ME. 2008. Colistin and polymyxin B in critical care. Crit Care Clin 24:377–391, x. doi: 10.1016/j.ccc.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 28.Bowdish DM, Davidson DJ, Lau YE, Lee K, Scott MG, Hancock REW. 2005. Impact of LL-37 on anti-infective immunity. J Leukoc Biol 77:451–459. doi: 10.1189/jlb.0704380. [DOI] [PubMed] [Google Scholar]
- 29.Scott MG, Davidson DJ, Gold MR, Bowdish D, Hancock RE. 2002. The human antimicrobial peptide LL-37 is a multifunctional modulator of innate immune responses. J Immunol 169:3883–3891. doi: 10.4049/jimmunol.169.7.3883. [DOI] [PubMed] [Google Scholar]
- 30.Pezzulo AA, Tang XX, Hoegger MJ, Alaiwa MH, Ramachandran S, Moninger TO, Karp PH, Wohlford-Lenane CL, Haagsman HP, van Eijk M, Bánfi B, Horswill AR, Stoltz DA, McCray PB Jr, Welsh MJ, Zabner J. 2012. Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature 487:109–113. doi: 10.1038/nature11130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.O'Hara JA, Ambe LA, Casella LG, Townsend BM, Pelletier MR, Ernst RK, Shanks RM, Doi Y. 2013. Activities of vancomycin-containing regimens against colistin-resistant Acinetobacter baumannii clinical strains. Antimicrob Agents Chemother 57:2103–2108. doi: 10.1128/AAC.02501-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Henry R, Vithanage N, Harrison P, Seemann T, Coutts S, Moffatt JH, Nation RL, Li J, Harper M, Adler B, Boyce JD. 2012. Colistin-resistant, lipopolysaccharide-deficient Acinetobacter baumannii responds to lipopolysaccharide loss through increased expression of genes involved in the synthesis and transport of lipoproteins, phospholipids, and poly-β-1,6-N-acetylglucosamine. Antimicrob Agents Chemother 56:59–69. doi: 10.1128/AAC.05191-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Napier BA, Burd EM, Satola SW, Cagle SM, Ray SM, McGann P, Pohl J, Lesho EP, Weiss DS. 2013. Clinical use of colistin induces cross-resistance to host antimicrobials in Acinetobacter baumannii. mBio 4:e00021–13. doi: 10.1128/mBio.00021-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.