

Abstract
Clostridium difficile infections (CDIs) are the leading cause of hospital-acquired infectious diarrhea and primarily involve two exotoxins, TcdA and TcdB. Actoxumab and bezlotoxumab are human monoclonal antibodies that neutralize the cytotoxic/cytopathic effects of TcdA and TcdB, respectively. In a phase II clinical study, the actoxumab-bezlotoxumab combination reduced the rate of CDI recurrence in patients who were also treated with standard-of-care antibiotics. However, it is not known whether the antibody combination will be effective against a broad range of C. difficile strains. As a first step toward addressing this, we tested the ability of actoxumab and bezlotoxumab to neutralize the activities of toxins from a number of clinically relevant and geographically diverse strains of C. difficile. Neutralization potencies, as measured in a cell growth/survival assay with purified toxins from various C. difficile strains, correlated well with antibody/toxin binding affinities. Actoxumab and bezlotoxumab neutralized toxins from culture supernatants of all clinical isolates tested, including multiple isolates of the BI/NAP1/027 and BK/NAP7/078 strains, at antibody concentrations well below plasma levels observed in humans. We compared the bezlotoxumab epitopes in the TcdB receptor binding domain across known TcdB sequences and found that key substitutions within the bezlotoxumab epitopes correlated with the relative differences in potencies of bezlotoxumab against TcdB of some strains, including ribotypes 027 and 078. Combined with in vitro neutralization data, epitope modeling will enhance our ability to predict the coverage of new and emerging strains by actoxumab-bezlotoxumab in the clinic.
INTRODUCTION
Infection with the Gram-positive, spore-forming, anaerobic bacterium Clostridium difficile is the leading cause of hospital-acquired infectious diarrhea in the developed world and can have potentially life-threatening effects. In the United States, approximately 14,000 deaths per year are attributed to C. difficile infections (CDIs), with an additional 250,000 patients per year requiring hospitalization or an increased length of hospital stay due to infection. As a result, it is estimated that more than $1 billion per year are spent in excess medical costs for treatment of CDIs in the United States (1, 2).
C. difficile is transmitted by spores through the fecal-oral route, often in a hospital or health care facility setting. Treatment with broad-spectrum antibiotics, which suppress the normal gut flora, is the primary risk factor for development of CDIs. In the absence of bacterial competition, C. difficile is able to thrive and to colonize the large intestine, leading to symptoms that can include mild to severe diarrhea, fever, pseudomembranous colitis, and toxic megacolon (2). While primary CDIs are generally successfully treated with the current standard-of-care antibiotics vancomycin, metronidazole, and most recently fidaxomicin, over the past decade there has been an increase in antibiotic-resistant and so-called hypervirulent strains. As a result, the rate of CDI recurrence has increased, with 25 to 30% of patients treated with antibiotics having a recurrence of disease after cessation of the initial symptoms (1). The threat of C. difficile infection and its associated persistent health effects and costs have caused the Centers for Disease Control to classify C. difficile as an urgent public health threat requiring immediate action (http://www.cdc.gov/drugresistance/threat-report-2013).
C. difficile produces and secretes the exotoxins TcdA and TcdB, which are part of the large clostridial glucosylating toxin family and are predominantly responsible for the pathogenic effects of C. difficile infection (3–5). The two toxins are organized in a similar manner, with a glucosyltransferase domain at the amino terminus, followed by a cysteine protease domain, a translocation domain, and a receptor binding domain, also called the combined repetitive oligopeptide (CROP) domain, at the carboxy terminus. TcdA and TcdB enter host cells and glucosylate and inactivate small Rho-type GTPases such as Rac, Rho, and Cdc42, leading to disruption of the host cell cytoskeletal architecture, cell rounding, and cell death. Due to their causative role in the virulence of C. difficile, TcdA and TcdB have been a main focus in the development of non-antibiotic-based therapies for treatment and prevention of CDIs, including the combination of toxin-neutralizing monoclonal antibodies actoxumab and bezlotoxumab (6, 7). Actoxumab (MK-3415, GS-CDA1, and MDX-066) and bezlotoxumab (MK-6072, MBL-CDB1, and MDX-1388) are human monoclonal antibodies that target TcdA and TcdB, respectively (6). We showed previously that bezlotoxumab binds to two separate sites in the CROP domain of TcdB, partially overlapping with putative receptor binding pockets and blocking binding of the toxin to host cells (8); actoxumab, which binds to the CROP domain of TcdA (6), is presumed to work in a similar manner. In a relatively small phase II study that enrolled 200 patients in the United States and Canada, the actoxumab-bezlotoxumab combination, when given with standard-of-care therapy, reduced the rate of CDI recurrence, compared to placebo (7% and 25% recurrence, respectively) (7). Two large multinational phase III trials are under way to confirm these findings.
As mentioned above, hypervirulent/epidemic C. difficile strains have recently emerged, including the BI/NAP1/027 strain, which has been associated with localized outbreaks in the United States, the United Kingdom, and Canada, followed by dissemination throughout these regions (9, 10). This strain is just one of hundreds of genetically distinct strains of C. difficile (11–13), whose toxin sequence identities at the amino acid level can be as low as 66% (83% within the CROP domain) across known TcdB sequences and 98% (96% in the CROP domain) across known TcdA sequences. The existence of C. difficile strains with distinct TcdA and TcdB sequences has raised the question of whether the actoxumab-bezlotoxumab combination will be efficacious against a broad range of C. difficile strains. In this study, we test the ability of actoxumab and bezlotoxumab to bind to and neutralize the activities of TcdA and TcdB from numerous geographically diverse and clinically important strains of C. difficile. We also compare the epitopes of bezlotoxumab in the TcdB CROP domain across known TcdB sequences and show a correlation between epitope conservation and bezlotoxumab neutralization potency. These results, combined with clinical and preclinical studies with actoxumab and bezlotoxumab, allow us to make predictions regarding the clinical efficacy of this novel treatment in preventing recurrence among patients infected with various strains of C. difficile.
MATERIALS AND METHODS
Cell lines, bacterial strains, and purified toxins.
Vero and T84 cells were purchased from the American Type Culture Collection (ATCC) (Rockville, MD) and grown at 37°C in 5% CO2. Vero cells were maintained in Eagle's minimal essential medium (MEM) supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin, and 100 U/ml streptomycin. T84 cells were maintained in F-12K/Dulbecco's modified Eagle's medium (DMEM) (1:1 ratio) supplemented with 5% FBS, 2 mM l-glutamine, 100 U/ml penicillin, and 100 U/ml streptomycin. The C. difficile VPI 10463 strain (ribotype 087) was purchased from the ATCC. Clinical isolates of C. difficile were obtained from M. Wilcox (United Kingdom), M. Miller (Canada), D. Gerding (United States), H. Kato (Japan), or tgcBIOMICS (western Europe) (for a complete list, see Table S1 in the supplemental material). Purified native TcdA and TcdB from ribotypes 087, 001, 002, 014, 017, 027, 036, 078, and 106 were purchased from tgcBIOMICS (Bingen, Germany). Purified native TcdA for ribotypes 087, 027, and 078 was also purchased from the Native Antigen Co. (Upper Heyford, United Kingdom).
Culture supernatants, toxin quantitation, and TcdB immunodepletion.
C. difficile strains were inoculated into chopped meat medium (Anaerobe Systems, Morgan Hill, CA) and grown under anaerobic conditions at 37°C. After 72 to 96 h, culture supernatants were collected, filtered 2 times through 0.22-μm filters, and stored at 4°C. TcdA and TcdB levels were quantitated using a pan-strain enzyme-linked immunosorbent assay (ELISA) (tgcBIOMICS, Bingen, Germany). Statistical significance was determined by converting data to log values and comparing each toxinotype/strain group to the group of toxinotype 0 strains (ribotypes 001, 002, 014, 087, and 106), using one-way analysis of variance (ANOVA) with Dunnett's correction (excluding the single strain of ribotype 081 from the calculation). For TcdB immunodepletion, culture supernatants were incubated at 4°C with bezlotoxumab coupled to protein A-agarose beads (Thermo Scientific). After 4 to 6 h of incubation with mixing, the beads were removed by centrifugation. Supernatants were collected, filtered through 0.22-μm filters, and stored at 4°C. TcdB depletion was confirmed by Western blotting and ELISA (data not shown).
Competition binding assay.
Apparent dissociation constants (K′d values) for the binding of actoxumab and bezlotoxumab to various purified toxins were determined by competition ELISA, as described previously (14). Briefly, high-protein-binding ELISA plates (Fisher Scientific, Waltham, MA) were coated overnight at 4°C with 0.5 μg/ml TcdA or TcdB (List Biological Laboratories, Campbell, CA), washed, and subsequently blocked for 1 h at 37°C. Samples containing 2.5 ng/ml actoxumab or bezlotoxumab previously incubated for 18 h at room temperature alone or in the presence of increasing concentrations of purified toxins from various ribotypes (tgcBIOMICS, Bingen, Germany) were added to each well, and free antibody was allowed to bind to immobilized toxins for 90 min at 37°C. Plates were washed and developed using horseradish peroxidase (HRP)-conjugated secondary anti-human IgG F(ab′)2 fragments and Ultra TMB (3,3′,5,5′-tetramethylbenzidine) substrate (both from Thermo Scientific), according to the manufacturer's instructions. Data were analyzed as described by Friguet et al. (14) to determine the K′d for each toxin ribotype. Statistical significance was determined by converting data to log values and comparing each ribotype to ribotype 087 by using a one-way ANOVA with Dunnett's correction.
Toxin neutralization assay.
The effects of toxins on cell growth and survival and the neutralization thereof by actoxumab and bezlotoxumab were assessed with the sulforhodamine B (SRB) assay, which measures total cellular protein as a surrogate measure of cell number (15). Vero and T84 cells were seeded at 2,000 and 3,000 cells/well, respectively, in 96-well dishes and incubated overnight. Purified toxins or culture supernatants were serially diluted in Vero or T84 cell complete medium, incubated at 37°C for 2 h, and then added to cells. After 24 h, the medium was aspirated and the plates were washed 2 times with phosphate-buffered saline (PBS). Two hundred microliters/well of complete medium was added, and the plates were incubated for an additional 48 h (Vero cells) or 72 h (T84 cells) before the medium was removed and the cells were fixed with 100 μl/well of 10% cold trichloroacetic acid (TCA). After 60 min at 4°C, the TCA was removed and the plates were washed 4 times with distilled water. One hundred microliters/well of 100 μg/ml sulforhodamine B (Sigma, St. Louis, MO) in 10% acetic acid was added. The plates were incubated for 15 min at room temperature, washed 4 times with 10% acetic acid, and air dried; 150 μl/well of 10 mM Tris was added, and the plates were incubated at room temperature for 10 min, with shaking. The plates were read in a SpectraMax plate reader (Molecular Biosystems) at an absorbance wavelength of 570 nm. Quantitation of cells relative to untreated controls was calculated by comparing wells of treated and untreated cells, and 90% lethal concentration (LC90) (i.e., toxin concentration or supernatant dilution required to cause a 90% reduction in cell number) values were calculated. To measure antibody-mediated toxin neutralization, LC90 levels of purified toxins or supernatants were combined with serially diluted actoxumab or bezlotoxumab antibodies for 2 h at 37°C and then added to Vero or T84 cells as described above. After 24 h, cells were washed 2 times with PBS and treated and analyzed as described above. Statistical significance was determined by converting data to log values and comparing each toxinotype/ribotype group to the group of toxinotype 0 strains by using one-way ANOVA with Dunnett's correction (excluding the single strain of ribotype 081 from the calculation).
Molecular modeling and binding free energy calculations.
Modeling of bezlotoxumab-TcdB interactions was based on our previously described structure of Fab fragments of bezlotoxumab bound to two distinct epitopes (E1 and E2) within the CROP domain of TcdB (8). Amino acids within E1 and E2 that differ between TcdB of ribotype 087 (VPI 10463, against which bezlotoxumab was raised) and known TcdB sequences of other ribotypes were mapped onto the 087 B2-Fab X-ray crystal structure using Maestro (Schrodinger LLC). Minimization of proton positions was carried out using MacroModel (Schrodinger LLC). The molecular mechanics-generalized Born surface area (MM-GBSA) method in Prime (Schrodinger LLC) was used to compute the binding free energy of TcdB from each ribotype with the bezlotoxumab Fab fragments. In modeling of bezlotoxumab binding to the various ribotype sequences, the side chain conformations of the original (VPI 10463) residues were copied onto the mutants; repacking or refinement of side chain and backbone atoms of the substituted residues was not allowed.
RESULTS
Potencies and affinities of actoxumab and bezlotoxumab against purified toxins.
Actoxumab and bezlotoxumab were raised against TcdA and TcdB, respectively, from the VPI 10463 strain (ribotype 087) (6), and their abilities to bind to and neutralize toxins from genetically distinct strains of C. difficile are unknown. To address this, we first tested the efficacy of the antibodies against purified toxins from clinically relevant C. difficile ribotypes. Included in this test group were toxins from the reference strain VPI 10463 (ribotype 087) and from strains of ribotypes 001, 002, 014, 017, 036, and 106, as well as from the so-called hypervirulent ribotypes 027 and 078.
As a first step, the concentration at which a 90% reduction in cell number was achieved (90% lethal concentration [LC90]) was determined on Vero cells for each purified toxin by using the SRB assay, as described in Materials and Methods (Fig. 1A and B for ribotypes 027, 078, and 087; data not shown for other ribotypes). As observed previously (16), Vero cells are significantly more sensitive to TcdB than to TcdA. At LC90 levels, actoxumab fully neutralized TcdA from all ribotypes tested (Fig. 1C); however, the neutralization potencies against TcdA purified from ribotypes 027 and 078 were significantly lower than those against TcdA from all other ribotypes (50% effective concentrations [EC50s] of 1,900 and 1,500 ng/ml, respectively, versus a range of 16 to 38 ng/ml for all other strains) (Table 1). A similar trend was observed with TcdB and bezlotoxumab (Fig. 1D). At LC90 toxin levels, bezlotoxumab neutralized TcdB from ribotypes 027, 078, and 036 at EC50s higher than those for other ribotypes (960, 130, and 890 ng/ml, respectively, versus 1.7 to 5.2 ng/ml) (Table 1). Notably, ribotype 036 (strain 8864) is a unique strain and therefore is of little clinical relevance, but it was included here as the first TcdA−/TcdB+ strain ever isolated (17).
FIG 1.

Neutralization of TcdA- and TcdB-mediated effects on Vero cell growth and survival by actoxumab and bezlotoxumab. (A and B) TcdA-dependent (A) and TcdB-dependent (B) reductions in Vero cell growth and survival using purified toxins from ribotypes 087 (red circles), 027 (green squares), and 078 (blue diamonds). (C and D) Actoxumab-mediated (C) and bezlotoxumab-mediated (D) neutralization of TcdA and TcdB, respectively, purified from strains of various ribotypes (red circles, ribotype 087; dark green squares, ribotype 027; dark blue diamonds, ribotype 078; purple triangles, ribotype 001; light green inverted triangles, ribotype 002; light blue diamonds, ribotype 014; orange circles, ribotype 106; brown squares, ribotype 017; yellow triangles, ribotype 036). Representative experiments are shown.
TABLE 1.
Neutralization potencies and apparent binding affinities of actoxumab and bezlotoxumab against purified toxins of different ribotypes
| Strain | Actoxumab/TcdA |
Bezlotoxumab/TcdB |
||
|---|---|---|---|---|
| K′d (ng/ml)a | EC50 (ng/ml)b | K′d (ng/ml) | EC50 (ng/ml) | |
| 087c | 3.1 ± 1.2 | 18 | 8.1 ± 2.8 | 5.2 |
| 001 | 3.1 ± 1.7 | 16 | 5.8 ± 0.9 | 4.5 |
| 002 | 4.1 ± 1.0 | 22 | 6.8 ± 1.3 | 4.5 |
| 014 | 5.3 ± 1.5 | 38 | 62.3 ± 0.5d | 5.1 |
| 017 | NAe | NA | 4.4 ± 0.7 | 1.7 |
| 027 | 699 ± 70f | 1,900 | 291 ± 11f | 960 |
| 036 | NA | NA | 300 ± 197f | 890 |
| 078 | 637 ± 633f | 1,500 | 173 ± 111f | 130 |
| 106 | 15.7 ± 1.9d | 26 | 5.6 ± 1.9 | 2.0 |
K′d values are means ± standard deviations of at least two separate determinations.
EC50 values are from representative experiments.
Strain 087 is VPI 10463.
P < 0.05, compared to ribotype 087.
NA, not applicable; ribotypes 017 and 036 do not express TcdA.
P < 0.0001, compared to ribotype 087.
To gain insight into whether the differences observed across ribotypes are related to differences in the affinities of the antibodies for the toxins, we measured the apparent binding affinities of actoxumab and bezlotoxumab for the purified toxins using a competition ELISA approach, as described previously by Friguet et al. (14) (Table 1) (see Materials and Methods). The binding data show that actoxumab and bezlotoxumab bind with lower apparent affinities to toxins of ribotypes 027, 036, and 078 than to toxins of other ribotypes (Table 1). When plotted against the antibody neutralization potencies (EC50 values), the apparent binding affinities (K′d values) correlated strongly with the neutralization potencies of both antibodies (Fig. 2), confirming that potencies measured in whole-cell neutralization assays largely reflected the affinities of actoxumab and bezlotoxumab for their respective toxins. Since the Friguet approach is an indirect approach for determining K′d, we confirmed the lower affinities of actoxumab and bezlotoxumab for toxins from ribotypes 027 and 078 versus ribotype 087 using surface plasmon resonance (Table 2). The K′d values of actoxumab and bezlotoxumab were 8- to 15-fold higher for ribotype 027 and 078 toxins than for ribotype 087 toxins, consistent with the competition ELISA and neutralization potency data. As observed previously, the data for bezlotoxumab binding to TcdB of ribotype 087 were best fitted to a two-binding-site model, likely reflecting the cooperativity of binding expected if the two Fab regions of a single molecule of bezlotoxumab bind to two separate epitopes within one toxin molecule (8). Conversely, the low-affinity binding site was not observed for TcdB of ribotypes 027 and 078, reflecting the possibility that bezlotoxumab binds to a single epitope in toxins of these ribotypes or that the affinity of binding at a single site is too low to be accurately measured using this approach.
FIG 2.
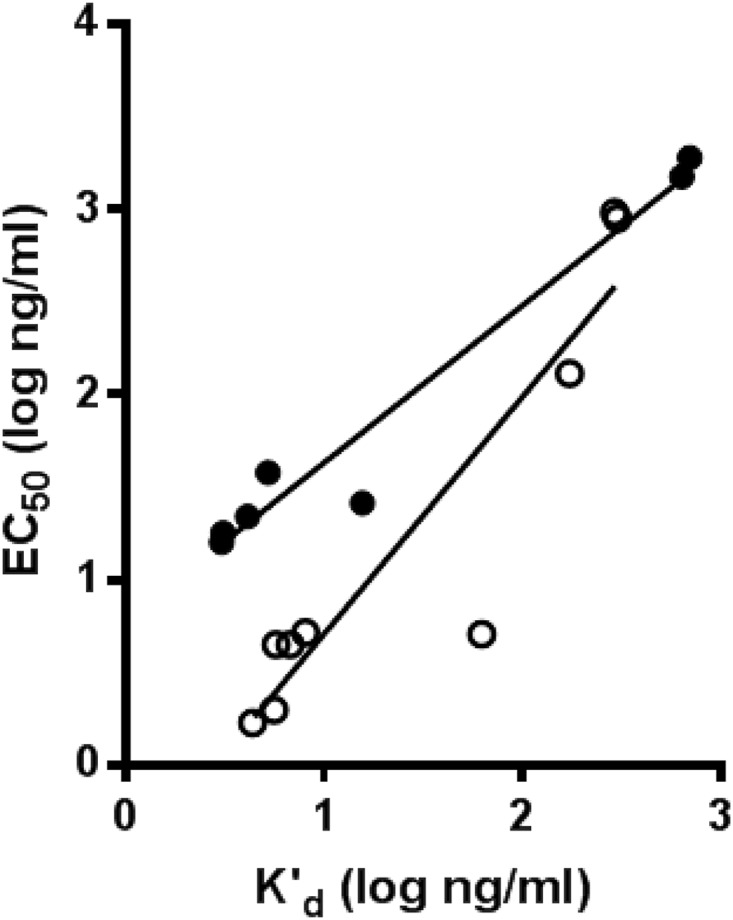
Correlation of neutralization potencies (EC50) and apparent affinities (K′d) of actoxumab (black circles) and bezlotoxumab (white circles) against TcdA and TcdB, respectively, purified from strains of various ribotypes. R2 = 0.96 for TcdA/actoxumab, and R2 = 0.85 for TcdB/bezlotoxumab.
TABLE 2.
Association and dissociation rates and affinities of actoxumab and bezlotoxumab binding to purified toxins, as determined by surface plasmon resonance
| Antibody/toxin | Strain | High-affinity binding site |
Low-affinity binding site |
||||
|---|---|---|---|---|---|---|---|
| kon (M−1 s−1)a | koff (s−1) | K′d (nM) | kon (M−1 s−1) | koff (s−1) | K′d (nM) | ||
| Actoxumab/TcdA | 087b | 1.9 × 105 ± 0.1 × 105 | 1.1 × 10−4 ± 0.1 × 10−4 | 0.61 ± 0.09 | NDc | ND | ND |
| 027 | 1.7 × 104 ± 0.2 × 104 | 1.5 × 10−4 ± 0.2 × 10−4 | 8.7 ± 1.5 | ND | ND | ND | |
| 078 | 5.0 × 104 ± 3.4 × 104 | 2.2 × 10−4 ± 1.0 × 10−4 | 5.2 ± 2.2 | ND | ND | ND | |
| Bezlotoxumab/TcdB | 087 | 1.0 × 106 ± 0.1 × 106 | 5.2 × 10−5 ± 2.0 × 10−5 | 0.050 ± 0.014 | 4.8 × 106 ± 1.4 × 106 | 1.6 × 10−2 ± 0.8 × 10−2 | 3.9 ± 2.9 |
| 027 | 3.9 × 105 ± 0.8 × 105 | 2.8 × 10−4 ± 0.3 × 10−4 | 0.75 ± 0.16 | ND | ND | ND | |
| 078 | 6.7 × 105 ± 0.3 × 105 | 4.4 × 10−4 ± 1.9 × 10−4 | 0.65 ± 0.27 | ND | ND | ND | |
Values are means ± standard deviations of two separate determinations.
Strain 087 is VPI 10463.
ND, not determined because the data best fit a single-site model.
Neutralization of toxins from clinical isolates by actoxumab and bezlotoxumab.
To expand our analysis of toxin neutralization to different clinical strains of C. difficile for which purified toxins are not available, we assessed toxin-containing supernatants of strains grown in culture. We characterized the abilities of actoxumab and bezlotoxumab to neutralize toxins from 81 clinical isolates from the United States, Canada, the United Kingdom, western Europe, and Japan, representing 18 distinct ribotypes and 7 of the known toxinotypes (Table 3; also see Table S1 in the supplemental material). Toxin concentrations in culture medium were first quantified by ELISA for each strain. Since isolates within the same toxinotype tended to produce similar amounts of toxin, the data were grouped by toxinotype rather than by ribotype except for ribotypes trf, smz, 081, and 198, for which the corresponding toxinotypes are unknown (Fig. 3). Similar levels of TcdA expression were observed for toxinotypes 0, I, III, and V (as well as ribotypes smz, 081, and 198), with lower levels of expression for toxinotypes IV and VII. TcdA levels were undetectable for toxinotype VIII and ribotype trf strains, which are known to express TcdB but not TcdA (17, 18). In the case of TcdB expression, toxinotype III strains produced consistently higher levels of toxin, compared to other toxinotypes, and strains of toxinotype IV and VII produced little to no TcdB under these culture conditions.
TABLE 3.
Summary of C. difficile clinical isolates characterized in this study
| Ribotype | Toxinotype | No. of isolates |
|---|---|---|
| 001 | 0 | 9 |
| 002 | 3 | |
| 014 | 6 | |
| 053 | 1 | |
| 077 | 2 | |
| 087 | 2 | |
| 106 | 5 | |
| 003 | I | 3 |
| 012 | 4 | |
| 027 | III | 18 |
| 023 | IV | 2 |
| 078 | V | 7 |
| 063 | VII | 2 |
| 017 | VIII | 4 |
| 081 | ?a | 1 |
| 198 | ? | 2 |
| smz | ? | 5 |
| trf | ? | 5 |
?, toxinotype unknown.
FIG 3.

TcdA (A) and TcdB (B) expression levels in culture supernatants of the various clinical isolates described in Table 3 and Table S1 in the supplemental material, grouped by toxinotype. LOQ, limit of quantitation. *, P < 0.05; **, P < 0.01; ***, P < 0.0001, compared to toxinotype 0.
For determination of neutralization potencies, culture supernatant dilutions leading to 90% reduction in cell number (LC90) were first determined for each isolate in the SRB assay, as described above for purified toxins. Since the supernatants contained a mixture of TcdA and TcdB, we utilized two different cell lines, with different sensitivities to the two toxins, for these assays. To measure TcdB activity, we used Vero cells, which are >100-fold more sensitive to TcdB than to TcdA (Fig. 1A and B). In these cells, TcdA present in the culture supernatants did not measurably contribute to cytotoxicity at LC90 dilutions (data not shown). For assessment of TcdA neutralization, we used T84 cells, which are more sensitive to TcdA than to TcdB. In this case, however, TcdB in the culture supernatant had a significant impact on cell numbers at LC90 dilutions. Therefore, to more accurately determine specific TcdA activity without interference from TcdB, we first removed TcdB from the culture supernatants using an immunodepletion approach (see Materials and Methods), before determining LC90 concentrations with T84 cells.
We evaluated the ability of actoxumab and bezlotoxumab to neutralize TcdA and TcdB at LC90 dilutions for each clinical isolate. Representative neutralization curves are shown in Fig. 4A and B for strains of ribotypes 087 (VPI 10463), 027, and 078, while the EC50s obtained against all individual clinical isolates tested are plotted in Fig. 4C and D, grouped by toxinotype (and listed individually in Table S1 in the supplemental material). Neutralization of TcdA by actoxumab was not evaluated for strains of ribotype 023 (toxinotype IV), since these strains produced insufficient amounts of TcdA and did not reach LC90 even at very low dilutions (Fig. 3). These data are consistent with a recent report by Walker et al. showing that ribotype 023 strains are associated with low mortality rates clinically despite expressing binary toxin (19). Strains of ribotype 063 (toxinotype VII) produced low levels of both TcdA and TcdB and therefore were not evaluated for antibody neutralization. Finally, strains of ribotypes 017 (toxinotype VIII) and trf are known to be TcdA−/TcdB+ strains (17, 18) and therefore were not tested in the actoxumab-TcdA assay. As was observed with purified toxins, actoxumab was significantly less potent against TcdA from the ribotype 027/toxinotype III and ribotype 078/toxinotype V strains (Fig. 4A and C), and the same pattern was observed with bezlotoxumab neutralization of TcdB (Fig. 4B and D). Also similar to data obtained with purified toxins (Fig. 1D), the potencies of bezlotoxumab against TcdB of ribotype 078/toxinotype V strains were intermediate between those against TcdB of ribotype 027/toxinotype III strains and toxinotype 0 strains. Interestingly, bezlotoxumab was significantly more potent against TcdB from ribotype 017/toxinotype VIII and trf strains than against TcdB from other strains, again confirming a trend observed with purified toxins from ribotype 017/toxinotype VIII (Fig. 1D and Table 1). While the EC50s for both actoxumab and bezlotoxumab were higher against ribotype 027 and 078 toxins, these values were still well below plasma antibody levels measured in clinical trials, even up to 84 days following antibody administration (Fig. 4C and D) (7).
FIG 4.

Neutralization of TcdA and TcdB from culture supernatants of various clinical isolates by actoxumab and bezlotoxumab. (A and B) Actoxumab-mediated (A) and bezlotoxumab-mediated (B) neutralization of TcdA (T84 cells) and TcdB (Vero cells), respectively, from culture supernatants of strains of ribotypes 087 (strain VPI 10463) (red circles), 027 (strain 89 from tgcBIOMICS) (see Table S1 in the supplemental material) (green squares), and 078 (strain 73 from tgcBIOMICS) (see Table S1 in the supplemental material) (blue diamonds). (C and D) Actoxumab (C) and bezlotoxumab (D) EC50s against toxins in culture supernatants of all clinical isolates tested (Table 3; also see Table S1 in the supplemental material), grouped by toxinotype. The approximate range of serum antibody concentrations measured in patients participating in phase II clinical studies (7) is also shown. X, EC50 of purified toxins corresponding to each toxinotype (Table 1). *, P < 0.0001, compared to toxinotype 0.
Molecular modeling of bezlotoxumab epitopes across different strains.
Actoxumab and bezlotoxumab bind within the CROP domains of TcdA and TcdB, respectively (6). We recently showed that bezlotoxumab binds to two homologous but not identical epitopes that partially overlap 2 of the 4 putative receptor binding pockets of TcdB, preventing toxin binding to host cells (8). Since toxin neutralization correlates strongly with antibody binding (Fig. 2), we hypothesized that strain-specific sequence differences within the epitopes of bezlotoxumab could be predictive of neutralization potencies across these different strains. To validate this hypothesis, we compared the amino acid sequences of the two bezlotoxumab epitopes (8) across publically available TcdB sequences. As shown in Fig. 5, the known TcdB sequences can be categorized into 7 distinct bezlotoxumab epitope sequences, typified by specific ribotypes/toxinotypes. The residues within each epitope type are compared with those of the VPI 10463/ribotype 087/toxinotype 0 epitopes, and differences are highlighted in red. The numbers of ribotype/toxinotype-specific differences within epitopes 1 and 2 range from a single substitution for ribotype 017/toxinotype VIII up to 9 substitutions for ribotype 019/toxinotype IX and ribotype 036/toxinotype X.
FIG 5.

Alignment of bezlotoxumab epitopes across known TcdB sequences. Residues within TcdB that are known to interact with bezlotoxumab in epitopes 1 and 2 are shown for the control strain (VPI 10463/ribotype 087) against which bezlotoxumab was raised and for other known TcdB sequences. Bezlotoxumab epitopes fall into seven distinct categories represented by specific strains, i.e., (i) ribotype 087/toxinotype 0 (other strains with identical bezlotoxumab epitopes include ribotypes 001, 012, 053, and 056), (ii) ribotype 027/toxinotype III (other strains with identical bezlotoxumab epitopes include ribotypes 111 and 122 and toxinotypes IIIc, XVII, and XXII), (iii) ribotype 019/toxinotype IX, (iv) ribotype 036/toxinotype X, (v) ribotype 078/toxinotype V (other strains with identical bezlotoxumab epitopes include toxinotype XVI), (vi) ribotype 063/toxinotype VII, and (vii) ribotype 017/toxinotype VIII (other strains with identical bezlotoxumab epitopes include ribotype 023 and toxinotype XXI). Positions at which differences exist versus the VPI 10463 epitopes are highlighted in red. Interacting residue(s), residues within bezlotoxumab that interact at each position in the epitopes.
Since bezlotoxumab is significantly less potent against (and binds with lower affinity to) TcdB from the ribotype 027, 078, and 036 strains (Table 1 and Fig. 1 and 5), we surmised that amino acid substitutions within the bezlotoxumab epitopes of these ribotypes would have detrimental effects on antibody binding and neutralization. Based on the crystal structure of bezlotoxumab bound to VPI 10463 TcdB (8), we carried out molecular modeling of TcdB bound to bezlotoxumab and calculated the binding free energy (ΔG) between TcdB and bezlotoxumab for each of the 7 epitope types shown in Fig. 5. The differences in binding free energy (ΔΔG) between the VPI 10463/ribotype 087 epitope and the other epitope types are shown in Table 4. The ΔΔG values for ribotype 027, 036, 078, and 017 TcdB bound to bezlotoxumab are consistent with the data on toxin binding and neutralization. Indeed, TcdB from ribotypes 027, 078, and 036 demonstrated positive ΔΔG values, indicating weaker binding to bezlotoxumab and correlating with the lower antibody potency and toxin affinity observed experimentally, while TcdB from ribotype 017 exhibited a negative ΔΔG value, indicating stronger binding to bezlotoxumab and correlating with the higher antibody potency and toxin affinity observed experimentally.
TABLE 4.
Differences in calculated free energies (ΔΔG) for bezlotoxumab binding to TcdB of different ribotypes and corresponding affinities and neutralization potencies
| Ribotype | ΔΔG (kcal/mol) | K′d ratioa | EC50 ratioa |
|---|---|---|---|
| 087 | 0 | 1 | 1 |
| 027 | 3.2 | 26 | 185 |
| 019 | 3.1 | NDb | ND |
| 036 | 2.3 | 27 | 171 |
| 078 | 2.1 | 15 | 25 |
| 063 | 2.0 | ND | ND |
| 017 | −0.6 | 0.4 | 0.33 |
From values determined by competition ELISA and neutralization studies (Table 1), normalized to values obtained for VPI 10463/ribotype 087.
ND, not determined.
The differences in binding free energies calculated for different epitope types are associated with disrupting or stabilizing effects of distinct amino acid residues at key bezlotoxumab-interacting positions within the epitopes (see Table S2 in the supplemental material). For example, the bezlotoxumab epitopes of ribotype 027/toxinotype III are dissimilar from those of ribotype 087 at 8 distinct positions. Two of those residues, Ile1876 and Val2007, are located relatively distant from the toxin/antibody interface and make minimal contact with antibody residues. The remaining six residues, however, have important interactions with bezlotoxumab and either introduce van der Waals clashes, reduce favorable hydrogen bond and hydrophobic interactions, or remove a favorable salt bridge interaction (see Table S2 in the supplemental material). A favorable van der Waals (and perhaps hydrogen bond) interaction between Asp1939 of TcdB and Trp102 of bezlotoxumab is lost in ribotype 027, as well as in ribotypes 019 and 036, all of which have a Gly residue at position 1939 (Fig. 6A). Modeling experiments predict that the Glu2033Ala substitution in epitope 2 of ribotypes 027, 019, and 036 has destabilizing effects on antibody binding, since it removes a favorable salt bridge interaction and a hydrophobic interaction with residues Arg59 and Trp33, respectively, of the bezlotoxumab heavy chain (Fig. 6B). In the case of ribotype 078, the Phe1905Leu substitution in epitope 1 of the ribotype 078 strain is predicted to have the greatest effect on antibody binding, because of the loss of a favorable van der Waals interaction with Asn101 of bezlotoxumab (Fig. 6A). In addition to providing insight into the lower potencies/affinities of bezlotoxumab against TcdB toxins of ribotypes 027, 078, and 036, our analysis of the bezlotoxumab epitopes reveals why the potency of bezlotoxumab against TcdB of ribotype 017/toxinotype VIII is improved, compared to toxinotype 0 strains (Fig. 1D and 4D). The only sequence difference between ribotypes 017 and 087 within the bezlotoxumab epitopes is a substitution of Asp to Glu at position 1939 of epitope 1, which is predicted to strengthen the interaction between bezlotoxumab and TcdB by forming more-favorable polar interactions between the glutamate side chain and Trp102 of the bezlotoxumab heavy chain (Fig. 6A). TcdB from ribotype 078 also contains a glutamate residue at position 1939, but the nearby Phe1905Leu substitution may introduce a van der Waals clash that changes the orientation of Trp102, thereby preventing formation of the favorable polar interactions with Glu1939.
FIG 6.

Structural basis of strain-specific differences in bezlotoxumab potency and affinity. (A) Favorable van der Waals (and perhaps H-bond) interactions between D1939 of TcdB and W102 of bezlotoxumab are lost in ribotype 027/036 (G1939), and a steric clash (*) is introduced in ribotype 078 (E1939). Favorable van der Waals interactions between F1905 of TcdB and N101 of bezlotoxumab are reduced in ribotype 078 (L1905). (B) Electrostatic interactions (dashed lines) between E2033 and R59 are lost in ribotypes 027/036 (A2033) and 078 (S2033).
DISCUSSION
Current treatments for infection with C. difficile include the antibiotics vancomycin, metronidazole, and fidaxomicin, which target the bacterium. Because the toxins TcdA and TcdB are the main virulence factors responsible for the pathogenic effects of C. difficile, more recent therapies under development for primary and recurrent CDIs are focused on targeting the toxins. The most advanced of these is the combination of actoxumab and bezlotoxumab, which has been shown to be effective in preventing CDI recurrence in a phase II clinical trial. Because hundreds of genetically distinct strains of C. difficile exist, with potentially minor and major differences in the tcdA and tcdB genes (13), it is important to address the question of whether actoxumab and bezlotoxumab can neutralize the toxins of clinically important strains and, by extension, whether these antibodies will prove to be an effective therapy for patients infected with current and emerging strains of C. difficile. In this study, we characterize the neutralizing potency of actoxumab and bezlotoxumab against TcdA and TcdB from a broad range of C. difficile strains. We show that the antibodies have comparable (or superior) potency against toxins of all strains of toxinotypes 0 (including ribotypes 001, 002, 014/020, 053, 077, 087, and 106), I (ribotypes 003 and 012), IV (ribotype 023), and VIII (ribotype 017) and ribotypes 081, trf, and smz (unknown toxinotypes), compared to strain VPI 10463 (against which these antibodies were raised). In contrast, both actoxumab and bezlotoxumab are significantly less potent against strains of ribotypes 027, 036, 078 (toxinotypes III, X, and V, respectively), and 198 (unknown toxinotype). Shifts in potency for ribotypes 027 and 078 were reported by two other groups utilizing their own versions of actoxumab and bezlotoxumab (referred to in those studies as CDA1 and CDB1/MDX-1388, respectively) (20, 21). In the study by Davies et al., the authors reported that CDA1 was “largely impotent” against TcdA from strains of ribotype 027 (20). However, the highest concentration of CDA1 tested in those assays was 500 ng/ml, ∼20-fold lower than plasma concentrations of actoxumab measured in patients a full 84 days after administration of the antibody (and ∼600-fold lower than plasma concentrations at 48 h after infusion) (7). Furthermore, while these neutralization experiments were purportedly carried out at LC80 levels of toxin, the percent neutralization observed at very low concentrations of antibody was not 20%, as expected, but rather closer to 0% (corresponding to 100% cell death) for the CDA1 titrations. In the study by Marozsan et al., higher maximal concentrations of antibody were used (up to 0.1 μM, or approximately 15 μg/ml), and EC50s reported for antibodies having the same amino acid sequence as actoxumab and bezlotoxumab were largely comparable to the results described here (21). The main discrepancies are for neutralization of TcdA, and we surmise that this may be due to the fact that TcdB immunodepletion of the supernatants was not carried out in that study. Indeed, the maximal inhibition observed for many of the strains was at or below 80% for both CDA1 and antibody PA-50, suggesting that residual effects due to TcdB were observed even with full neutralization of TcdA.
While actoxumab and bezlotoxumab have lower neutralization potencies against toxins of the two hypervirulent strains 027 and 078, nearly complete neutralization of these toxins was achieved at concentrations of antibody that are still well below plasma concentrations measured in CDI patients, even up to 84 days after antibody administration (7) (Fig. 1 and 4). However, since the concentration of antibody at the site of infection (i.e., the gut lumen) is difficult to establish experimentally, efficacy against a particular strain cannot be predicted based on plasma exposures alone. To better understand how in vitro potency can predict in vivo clinical efficacy, it is relevant to correlate in vitro neutralization of specific strains with in vivo clinical efficacy against these same strains. In this regard, actoxumab-bezlotoxumab was efficacious against ribotype 027 strains in a gnotobiotic piglet infection model (22) and in mouse models of primary and recurrent CDIs (23). Furthermore, the antibodies reduced the rate of recurrent CDIs in patients infected with ribotype 027 strains to the same extent as in the overall patient population (although the reduction did not reach statistical significance; P = 0.06) (7). These data support the notion that the actoxumab-bezlotoxumab combination will be efficacious against existing strains of C. difficile, since the antibodies neutralized toxins of all strains tested in this study at least as efficiently as those of ribotype 027 (Fig. 1 and 4). Importantly, the strains tested in this study include the most clinically prevalent strains found not only in the United States and Canada but also in Europe and Japan (9–12).
Since binding affinities and neutralization potencies correlate well across different C. difficile strains (Fig. 2), an understanding of the structural basis of antibody binding to toxins should also inform us on potency and, by extension, on predicted clinical efficacy. An analysis of the bezlotoxumab epitopes across known TcdB sequences (Fig. 5), in combination with the neutralization (Fig. 4) and energetics modeling data (Table 4), suggests that substitutions at key residues within the epitopes can negatively affect antibody binding in ribotype 019, 027, 036, 063, and 078 strains. On the other hand, the Asp1939Glu substitution present in ribotype 017 appears to have a beneficial effect on binding, since the neutralization potency of bezlotoxumab against TcdB from this ribotype is improved, compared to other ribotypes. Although the exact epitopes for actoxumab on TcdA have not been identified, the antibody binds within the CROP domain of TcdA (6). As demonstrated for bezlotoxumab, we expect sequence differences within the TcdA CROP domains of ribotypes 027 and 078 to account for the lower binding affinities and neutralization potencies measured with actoxumab against TcdA of these ribotypes. At the amino acid level, the sequence identity between TcdA of the VPI 10463 strain (ribotype 087) and that of ribotype 027 strains is 98%, with a slightly lower identity of 96% within the CROP domain itself. The sequence of ribotype 078 TcdA is unknown.
Epitope modeling not only can allow us to understand why antibodies show superior or inferior binding/neutralization against certain strains but also may help us make predictions regarding the expected affinities and potencies of actoxumab and bezlotoxumab against toxins of emerging strains, as their sequences become available. For example, the bezlotoxumab epitopes of ribotypes 019 and 063 (Fig. 5) are similar to those of ribotypes 027 and 078, respectively, suggesting that bezlotoxumab should have lower potencies against TcdB of these strains versus toxinotype 0 strains. The predictive power of epitope modeling can also be applied in reverse, to predict the TcdB sequences of ribotypes given their neutralization potencies. Thus, the potencies of bezlotoxumab against TcdB of the uncommon ribotypes 081 and 198 (Fig. 4) suggest that their bezlotoxumab epitopes should bear some similarity to those of ribotype 087/toxinotype 0 and ribotype 027/toxinotype III, respectively. Upon sequencing of the tcdB genes of the three clinical isolates corresponding to these ribotypes, we found that (i) the ribotype 081 TcdB sequence differs from the ribotype 087 TcdB sequence at only two positions (Asn to Ser at position 1574 and Tyr to Asp at position 1975), neither of which lies within the bezlotoxumab epitope, and (ii) the ribotype 198 TcdB sequence is 100% identical to known TcdB sequences of ribotype 027 (data not shown).
The importance of the analysis described above is underscored by the fact that the efficacy of actoxumab-bezlotoxumab is unlikely to be demonstrated directly for specific strains in ongoing phase III clinical trials, due to low statistical power (with the likely exception of ribotype 027 and perhaps a few other common strain types). The combination of in vitro neutralization assays and molecular modeling described herein can therefore provide important insights into the efficacy of this novel CDI therapy in patients infected with current and emerging strains of C. difficile. The data described in this study suggest that the actoxumab-bezlotoxumab combination will be efficacious against a broad range of clinically relevant C. difficile strains.
Supplementary Material
ACKNOWLEDGMENTS
We thank Mark Miller (Jewish General Hospital, Montreal, Canada), Mark Wilcox (University of Leeds, Leeds, United Kingdom), Dale Gerding (Veteran's Affairs Hospital, Hines, IL), and Haru Kato (National Institute of Infectious Diseases, Tokyo, Japan) for their generous gifts of C. difficile clinical isolates. We also thank Rumin Zhang for help with interpretation of the surface plasmon resonance data.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.04433-14.
REFERENCES
- 1.Bassetti M, Villa G, Pecori D, Arzese A, Wilcox M. 2012. Epidemiology, diagnosis and treatment of Clostridium difficile infection. Expert Rev Anti Infect Ther 10:1405–1423. doi: 10.1586/eri.12.135. [DOI] [PubMed] [Google Scholar]
- 2.Rupnik M, Wilcox MH, Gerding DN. 2009. Clostridium difficile infection: new developments in epidemiology and pathogenesis. Nat Rev Microbiol 7:526–536. doi: 10.1038/nrmicro2164. [DOI] [PubMed] [Google Scholar]
- 3.Carter GP, Rood JI, Lyras D. 2012. The role of toxin A and toxin B in the virulence of Clostridium difficile. Trends Microbiol 20:21–29. doi: 10.1016/j.tim.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 4.Pruitt RN, Lacy DB. 2012. Toward a structural understanding of Clostridium difficile toxins A and B. Front Cell Infect Microbiol 2:28. doi: 10.3389/fcimb.2012.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shen A. 2012. Clostridium difficile toxins: mediators of inflammation. J Innate Immun 4:149–158. doi: 10.1159/000332946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Babcock GJ, Broering TJ, Hernandez HJ, Mandell RB, Donahue K, Boatright N, Stack AM, Lowy I, Graziano R, Molrine D, Ambrosino DM, Thomas WD Jr. 2006. Human monoclonal antibodies directed against toxins A and B prevent Clostridium difficile-induced mortality in hamsters. Infect Immun 74:6339–6347. doi: 10.1128/IAI.00982-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lowy I, Molrine DC, Leav BA, Blair BM, Baxter R, Gerding DN, Nichol G, Thomas WD Jr, Leney M, Sloan S, Hay CA, Ambrosino DM. 2010. Treatment with monoclonal antibodies against Clostridium difficile toxins. N Engl J Med 362:197–205. doi: 10.1056/NEJMoa0907635. [DOI] [PubMed] [Google Scholar]
- 8.Orth P, Xiao L, Hernandez LD, Reichert P, Sheth PR, Beaumont M, Yang X, Murgolo N, Ermakov G, DiNunzio E, Racine F, Karczewski J, Secore S, Ingram RN, Mayhood T, Strickland C, Therien AG. 2014. Mechanism of action and epitopes of Clostridium difficile toxin B-neutralizing antibody bezlotoxumab revealed by X-ray crystallography. J Biol Chem 289:18008–18021. doi: 10.1074/jbc.M114.560748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McDonald LC, Killgore GE, Thompson A, Owens RC Jr, Kazakova SV, Sambol SP, Johnson S, Gerding DN. 2005. An epidemic, toxin gene-variant strain of Clostridium difficile. N Engl J Med 353:2433–2441. doi: 10.1056/NEJMoa051590. [DOI] [PubMed] [Google Scholar]
- 10.O'Connor JR, Johnson S, Gerding DN. 2009. Clostridium difficile infection caused by the epidemic BI/NAP1/027 strain. Gastroenterology 136:1913–1924. doi: 10.1053/j.gastro.2009.02.073. [DOI] [PubMed] [Google Scholar]
- 11.Bauer MP, Notermans DW, van Benthem BH, Brazier JS, Wilcox MH, Rupnik M, Monnet DL, van Dissel JT, Kuijper EJ. 2011. Clostridium difficile infection in Europe: a hospital-based survey. Lancet 377:63–73. doi: 10.1016/S0140-6736(10)61266-4. [DOI] [PubMed] [Google Scholar]
- 12.Manzo CE, Merrigan MM, Johnson S, Gerding DN, Riley TV, Silva J Jr, Brazier JS. 2014. International typing study of Clostridium difficile. Anaerobe 28:4–7. doi: 10.1016/j.anaerobe.2014.04.005. [DOI] [PubMed] [Google Scholar]
- 13.Rupnik M. 2008. Heterogeneity of large clostridial toxins: importance of Clostridium difficile toxinotypes. FEMS Microbiol Rev 32:541–555. doi: 10.1111/j.1574-6976.2008.00110.x. [DOI] [PubMed] [Google Scholar]
- 14.Friguet B, Chaffotte AF, Djavadi-Ohaniance L, Goldberg ME. 1985. Measurements of the true affinity constant in solution of antigen-antibody complexes by enzyme-linked immunosorbent assay. J Immunol Methods 77:305–319. doi: 10.1016/0022-1759(85)90044-4. [DOI] [PubMed] [Google Scholar]
- 15.Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, Warren JT, Bokesch H, Kenney S, Boyd MR. 1990. New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst 82:1107–1112. doi: 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- 16.Torres J, Camorlinga-Ponce M, Munoz O. 1992. Sensitivity in culture of epithelial cells from rhesus monkey kidney and human colon carcinoma to toxins A and B from Clostridium difficile. Toxicon 30:419–426. doi: 10.1016/0041-0101(92)90538-G. [DOI] [PubMed] [Google Scholar]
- 17.Borriello SP, Wren BW, Hyde S, Seddon SV, Sibbons P, Krishna MM, Tabaqchali S, Manek S, Price AB. 1992. Molecular, immunological, and biological characterization of a toxin A-negative, toxin B-positive strain of Clostridium difficile. Infect Immun 60:4192–4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kato H, Kato H, Ito Y, Akahane T, Izumida S, Yokoyama T, Kaji C, Arakawa Y. 2010. Typing of Clostridium difficile isolates endemic in Japan by sequencing of slpA and its application to direct typing. J Med Microbiol 59:556–562. doi: 10.1099/jmm.0.016147-0. [DOI] [PubMed] [Google Scholar]
- 19.Walker AS, Eyre DW, Wyllie DH, Dingle KE, Griffiths D, Shine B, Oakley S, O'Connor L, Finney J, Vaughan A, Crook DW, Wilcox MH, Peto TE. 2013. Relationship between bacterial strain type, host biomarkers, and mortality in Clostridium difficile infection. Clin Infect Dis 56:1589–1600. doi: 10.1093/cid/cit127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Davies NL, Compson JE, Mackenzie B, O'Dowd VL, Oxbrow AK, Heads JT, Turner A, Sarkar K, Dugdale SL, Jairaj M, Christodoulou L, Knight DE, Cross AS, Herve KJ, Tyson KL, Hailu H, Doyle CB, Ellis M, Kriek M, Cox M, Page MJ, Moore AR, Lightwood DJ, Humphreys DP. 2013. A mixture of functionally oligoclonal humanized monoclonal antibodies that neutralize Clostridium difficile TcdA and TcdB with high levels of in vitro potency shows in vivo protection in a hamster infection model. Clin Vaccine Immunol 20:377–390. doi: 10.1128/CVI.00625-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marozsan AJ, Ma D, Nagashima KA, Kennedy BJ, Kang YK, Arrigale RR, Donovan GP, Magargal WW, Maddon PJ, Olson WC. 2012. Protection against Clostridium difficile infection with broadly neutralizing antitoxin monoclonal antibodies. J Infect Dis 206:706–713. doi: 10.1093/infdis/jis416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Steele J, Mukherjee J, Parry N, Tzipori S. 2013. Antibody against TcdB, but not TcdA, prevents development of gastrointestinal and systemic Clostridium difficile disease. J Infect Dis 207:323–330. doi: 10.1093/infdis/jis669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang Z, Ramsey J, Hamza T, Zhang Y, Li S, Yfantis HG, Lee D, Hernandez LD, Seghezzi W, Furneisen JM, Davis NM, Therien AG, Feng H. 2015. Mechanisms of protection against Clostridium difficile infection by the monoclonal antitoxin antibodies actoxumab and bezlotoxumab. Infect Immun 83:822–831. doi: 10.1128/IAI.02897-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.