

Abstract
Plasmodium falciparum, the most deadly agent of malaria, displays a wide variety of resistance mechanisms in the field. The ability of antimalarial compounds in development to overcome these must therefore be carefully evaluated to ensure uncompromised activity against real-life parasites. We report here on the selection and phenotypic as well as genotypic characterization of a panel of sensitive and multidrug-resistant P. falciparum strains that can be used to optimally identify and deconvolute the cross-resistance signals from an extended panel of investigational antimalarials. As a case study, the effectiveness of the selected panel of strains was demonstrated using the 1,2,4-oxadiazole series, a newly identified antimalarial series of compounds with in vitro activity against P. falciparum at nanomolar concentrations. This series of compounds was to be found inactive against several multidrug-resistant strains, and the deconvolution of this signal implicated pfcrt, the genetic determinant of chloroquine resistance. Targeted mode-of-action studies further suggested that this new chemical series might act as falcipain 2 inhibitors, substantiating the suggestion that these compounds have a site of action similar to that of chloroquine but a distinct mode of action. New antimalarials must overcome existing resistance and, ideally, prevent its de novo appearance. The panel of strains reported here, which includes recently collected as well as standard laboratory-adapted field isolates, is able to efficiently detect and precisely characterize cross-resistance and, as such, can contribute to the faster development of new, effective antimalarial drugs.
INTRODUCTION
Since the beginning of this century, the revived interest of the international community in the control of malaria, together with the 2007 call for an eradication agenda, resulted in a significant decrease in the burden of mortality and morbidity associated with this parasitic disease (2–4). Over the past 12 years, the global malaria mortality rate has decreased by an estimated 45%, averting 3.3 million deaths, 90% of which were among children under 5 years of age in sub-Saharan Africa (7). This has been achieved, at least in part, by the increased availability of safe and efficacious antimalarial chemotherapies. In 2012, 331 million courses of artemisinin-based combination therapies (ACTs), recommended by the WHO for first-line treatment of uncomplicated Plasmodium falciparum malaria, were delivered to the public and private sectors (7). However, in the absence of a fully protective antimalarial vaccine, the development and spread of drug resistance by P. falciparum are likely to constitute serious obstacles to the eradication of malaria.
The ability of P. falciparum to develop resistance mechanisms had a major impact on the efficacy of two former mainstay therapies for uncomplicated malaria, chloroquine and the sulfadoxine-pyrimethamine combination, whose use for the curative treatment of infections caused by this species had to be abandoned (11, 12). Before then, the prolonged use of partially ineffective chloroquine treatments in sub-Saharan Africa is reported to have caused an increase in malaria mortality of up to 3-fold (14).
ACTs are now threatened by the emergence of P. falciparum strains with reduced sensitivity to artesunate, a phenotype that was first reported in Southeast Asia in 2008 and that has spread locally since then (16–18). Failures of artesunate-mefloquine treatment and, more recently, dihydroartemisinin-piperaquine treatment have been reported in areas of artemisinin resistance (19–23). This suggests that the therapeutic efficacy of ACT might be compromised, which is a major concern, considering that no alternative treatments are ready to be deployed (25).
Resistance to essentially all antimalarial drugs previously or currently used to treat P. falciparum malaria have been identified to various degrees and levels of geographical spread (6). Moreover, resistance to new antimalarial compounds currently in clinical development can often be selected for in vitro, demonstrating the genetic ability of P. falciparum to acquire resistance to these compounds and suggesting that none would be intrinsically refractory to resistance if they happened to be deployed as new antimalarial therapies (27–29). Thus, measures need to be taken at all stages of the drug development process to minimize liabilities due to resistance and to maximize the life span over which antimalarial therapies have efficacy.
The Medicines for Malaria Venture (MMV) is a not-for-profit organization with the mission to discover, develop, and facilitate the delivery of new, effective, and affordable antimalarial drugs (31). Recently, a semiquantitative in vitro framework was developed to evaluate the resistance liabilities of new antimalarials and is currently used in MMV's collaborative projects to identify and deprioritize compounds for which the risk of the development of resistance is overt early in development (15). The underlying strategy is 2-fold: first, to evaluate if new antimalarial compounds show cross-resistance with previously or currently marketed antimalarials, as evidenced by a loss of in vitro activity due to mechanisms of resistance known to operate in natural parasite populations, and second, to evaluate the propensity of de novo mechanisms of resistance to new compounds to be selected in vitro. The first goal is achieved by measuring the in vitro activity of compounds against a panel of standard sensitive and multidrug-resistant (MDR) strains which were selected to represent strains with the major resistance mechanisms known to operate in the field (15). In this study, we deployed this selected panel of strains to determine if they accurately displayed the intended resistance phenotypes, effectively identified signs of cross-resistance to new antimalarial compounds, and allowed the deconvolution of the determinants of the cross-resistance signals. We present and discuss the use of this approach for the detailed evaluation of the cross-resistance of a new antimalarial chemical series identified by high-throughput compound screening.
MATERIALS AND METHODS
Plasmodium falciparum strain culture and drug assays.
P. falciparum strains NF54, D6, HB3, FCB, 7G8, K1, Dd2, and V1/S were obtained from the Malaria Research and Reference Reagent Resource Center (MR4; www.mr4.org). Strain TM90C2B was provided by Dennis E. Kyle (University of South Florida). These strains were maintained in vitro using the method of Trager and Jensen (35). Parasite growth after exposure to serial dilutions of antimalarial compounds was assessed using the [3H]hypoxanthine incorporation assay, and the level of inhibition of growth is expressed as the median (50%) inhibitory concentration (IC50) (36, 37). Exposures of 72 h were performed to allow the detection of slow-acting compounds and to enhance data reproducibility (38).
The multiclonal parasite lines HL1210 and HL1212 were isolated from patients presenting to the Hospital for Tropical Diseases in London, United Kingdom, in 2012 after returning from travel to Ghana and Nigeria, respectively, and are described in detail elsewhere (30). Both strains originated from areas of endemicity where ACT has been in use since the mid-2000s and thus likely have prior exposure to lumefantrine, dihydroartemisinin, and potentially other ACT partner drugs. These strains were maintained in vitro, and drug assays were performed as described in reference 30. These lines have been made publicly available at the European Malaria Reagent Repository (http://www.malariaresearch.eu/).
Plasmodium falciparum sequencing.
DNA for sequencing was obtained from 10 ml of nonsynchronized cultures at approximately 5% parasitemia. Cells were spun down, the pellet was lysed with 0.5% saponin, and the parasites were subsequently spun down and lysed in the lysis buffer contained in the Qiagen DNA purification kit. Nested PCRs were set up for the gene domains in question using the primers listed in Table S1 in the supplemental material and the conditions indicated in the Materials and Methods section in the supplemental material. PCR products were cleaned using a Qiagen PCR cleanup kit and sent to Macrogen (Amsterdam, The Netherlands) for bidirectional sequencing using the same primers used for the nested reaction. All sequences were aligned with the strain 3D7 reference sequences, and single nucleotide polymorphisms were called only if they were present on both strands. pfmdr1 copy numbers were determined by real-time PCR as described previously (39).
Synthesis and characterization of compounds 1, 2, and 3.
The synthesis and characterization of compounds 1, 2, and 3 of the 1,2,4-oxadiazole series are presented in the Materials and Methods section in the supplemental material. Compound 3 was synthesized in a limited amount and could not be included in all experiments reported here.
Saccharomyces cerevisiae heterologous system.
DFR1, the gene encoding dihydrofolate reductase (DHFR) from Saccharomyces cerevisiae (ScDHFR), was PCR amplified from genomic DNA templates and cloned into pCM188 to generate pCMScDHFR (5). The DHFR-coding sequence from Plasmodium falciparum (PfDHFR) with a codon usage suitable for expression in yeast was synthesized by Geneart and was subcloned into pCM188 (1) to generate pCMPfDHFR (5). The DHFR mutations N51I, C59R, and S108N, which confer resistance to antifolates in wild Plasmodium falciparum populations, were introduced by site-directed mutagenesis into pCMPfDHFR to generate pCMPfRdhfrN51I, C59R, pCMPfRdhfrS108N, and pCMPfRdhfrN51I, C59R, S108N. pCM constructs were transformed into a BY4743-derived strain (pdr5::HisMX/PDR5 dfr1::KanMX/DFR1 MATa/MATα his3Δ1/his3Δ1 leu2Δ0/leu2Δ0 met15Δ0/MET15 LYS2/lys2Δ0 ura3Δ0/ura3Δ0), and following sporulation and tetrad dissections, spores with the genotype pdr5::HIS3MX dfr1::KanMX MATα his3Δ1 leu2Δ0 MET15 lys2Δ0 ura3Δ0/pCM were selected for use in drug sensitivity assays.
Standard yeast growth conditions and either YPD (2% peptone, 1% yeast extract, 2% glucose), supplemented sporulation medium (1% potassium acetate, 0.1% yeast extract, 0.05% glucose, amino acid supplements, 2% Bacto agar), or YNB-glucose (0.68% yeast nitrogen base, 2% ammonium sulfate, 2% glucose, 0.015% leucine) were used for all assays.
Serial dilutions (5 times) of stationary-phase cultures were prepared in 96-well plates and replicated onto YNB-glucose agar plates with or without drugs. Cells were allowed to grow for 24 h at 30°C and then photographed.
In vitro hemozoin formation and falcipain 2 enzyme activity assays.
Falcipain 2 activity in the presence of compounds 1 and 2 and chloroquine (5 μM) was assessed using a fluorometric assay, where the release of 7-amino-4-methyl coumarin (AMC) was monitored (excitation, 355 nm; emission, 460 nm) over 30 min at room temperature using an LS50B Perkin-Elmer fluorimeter in a buffer containing 100 mM sodium acetate, pH 5.5, 10 mM dithiothreitol, 7 μM fluorogenic substrate Z-Phe-Arg-AMC, and 200 nM enzyme. Falcipain 2 activity in the absence of any drug was taken as a positive control. Details on the enzyme expression and hemozoin formation assays are presented in the Materials and Methods section in the supplemental material.
In vitro abiotic β-hematin formation assay.
The β-hematin formation inhibition assay method described by Carter and colleagues was modified for manual liquid delivery (8, 9). Solutions of the samples in dimethyl sulfoxide were incubated in 96-well plates with NP-40 detergent (30.55 μM) and hematin (100 μM) in an acetate buffer (1 M, pH 4.8) for 5 to 6 h at 37°C. The detection of free heme was based on the pyridine-ferrichrome method developed by Ncokazi and Egan (10). The detailed assay procedure has been reported previously (13).
RESULTS
Genotypic validation of selected P. falciparum strains.
The selected panel of P. falciparum strains included the MDR strains HB3, FCB, 7G8, K1, Dd2, V1/S, and TM90C2B as well as the sensitive strains NF54 and D6, all of which were selected on the basis of genotypic and phenotypic information previously reported in the literature (15).
In order to evaluate the ability of these strains to identify cross-resistance signals, genes involved in drug resistance were sequenced to confirm the presence of specific mutations known to cause resistance to various antimalarial compounds (Table 1). These genes were those for the P. falciparum chloroquine resistance transporter (pfcrt), multidrug resistance 1 (pfmdr1), dihydrofolate reductase (pfdhfr), dihydropteroate synthase (pfdhps), and cytochrome b (pfcytb). Mutations of pfcrt and pfmdr1 have been shown to mediate resistance to chloroquine and amodiaquine, mutations of pfdhfr have been shown to mediate resistance to pyrimethamine and cycloguanil, mutations of pfdhps have been shown to mediate resistance to sulfadoxine, and mutations of pfcytb have been shown to mediate resistance to atovaquone. The expected pfcrt and pfcytb genotypes were observed, and minor differences from the mutations reported in the literature were observed for pfmdr1, pfdhfr, and pfdhps (see the references in reference 15). Regarding pfmdr1, strain HB3 was found to carry the mutation Y184F, while this residue was expected to be wild type; strain Dd2 had the mutation N86F instead of N86Y, and strain 7G8 had the mutation S1034R instead of S1034C. Four copies of the pfmdr1 gene (instead of two) and three copies of the pfmdr1 gene (instead of four) were identified in strains FCB and Dd2, respectively. FCB was also found to express the pfdhfr mutation A16T instead of A16V. Finally, strains NF54, HB3, and 7G8 were found to carry the pfdhps mutation S436F instead of the wild-type residue.
TABLE 1.
Amino acid sequences of selected gene polymorphisms involved in P. falciparum drug resistance
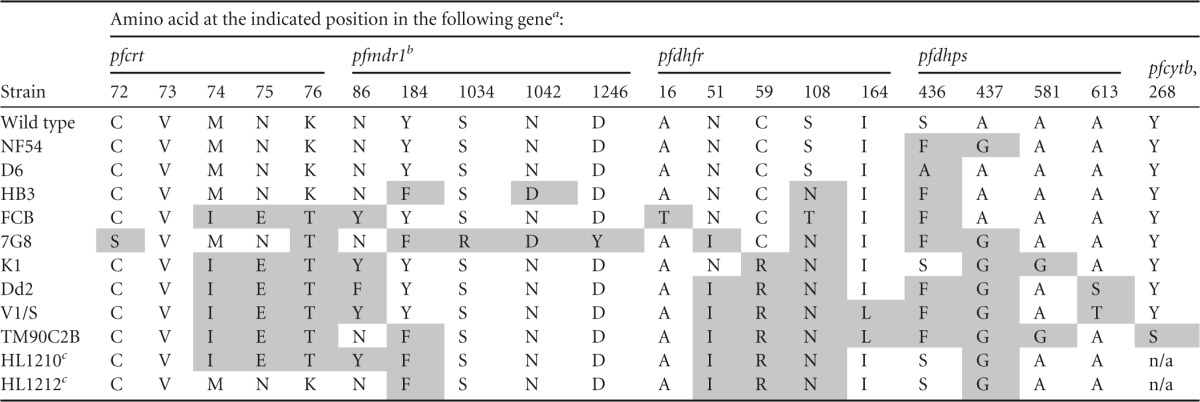
Mutant residues are shaded.
The pfmdr1 copy number was 1 for the wild-type strain and strains NF54, D6, HB3, 7G8, K1, V1/S, TM90C2B, HL1210, and HL1212; 4 for FCB; and 3 for Dd2.
As reported in reference 30.
Phenotypic validation of selected P. falciparum strains.
The presence of the expected resistance phenotypes in the panel of strains was determined by measuring their sensitivity to various standard antimalarial compounds during an in vitro exposure of 72 h. Artesunate, atovaquone, chloroquine, cycloguanil, mefloquine, and pyrimethamine were evaluated, and resistance was considered to occur if the median (50%) inhibitory concentration (IC50) of a given strain was found to be, on average, more than 10-fold greater than that of the sensitive reference strain NF54 (Fig. 1; see Table S2 in the supplemental material). This threshold, which is only of an indicative nature, was selected to be in the range of the smallest IC50 shifts associated with clinical resistance (e.g., for chloroquine [40]), while it simultaneously avoided a false-positive resistance signal due to the intrinsic variation of phenotypic assays.
FIG 1.

Phenotypic validation of the panel of MDR strains. The relative IC50s of artesunate (ART), atovaquone (ATO), chloroquine (CHQ), cycloguanil (CYC), mefloquine (MEF), and pyrimethamine (PYR) against the panel of P. falciparum laboratory strains are shown. The solid line shows a 10-fold increase in the IC50 relative to that for NF54 (n ≥ 2; error bars are SDs).
None of the strains tested showed resistance, as defined here, to artesunate and mefloquine. As expected, TM90C2B was the only strain resistant to atovaquone. NF54, D6, HB3, and 7G8 were found to be sensitive to chloroquine; however, the phenotype of 7G8 was close to the resistance threshold, with 7G8 having an 8.3-fold greater tolerance to the drug than N54. FCB, K1, Dd2, V1/S, and TM90C2B all displayed resistance to chloroquine. All strains except NF54, D6, and HB3 were found to be resistant to cycloguanil, while resistance to pyrimethamine was apparent in all strains except NF54, D6, and FCB. The selected strains effectively displayed various resistance phenotypes and therefore represent an appropriate tool for evaluating the potential cross-resistance of antimalarial compounds with a variety of existing molecular mechanisms of resistance.
Case study with the 1,2,4-oxadiazole series.
To further evaluate the ability of the selected panel of strains to identify and facilitate the characterization of cross-resistance signals from new antimalarial compounds, we studied three analogue compounds (compounds 1, 2, and 3). These three molecules comprise a 1,2,4-oxadiazole moiety, a 1,4-diazepam amide, a tertiary piperidine, and an amide group, and we commonly refer to them as the “1,2,4-oxadiazole” series (Fig. 2A). Compounds 1 and 2 were initially identified as hits on the erythrocytic stage of P. falciparum during the phenotypic screening of a library of more than 250,000 compounds (24). Compound 3 was synthesized while performing the resynthesis of compounds 1 and 2. The IC50s for these three compounds tested against the sensitive strain NF54 ranged from 60 nM to 278 nM (Table 2). This, together with favorable physicochemical properties, suggested that this novel chemical scaffold might be a valuable starting point for a hit-to-lead medicinal chemistry program. As part of a standard resistance risk assessment and as a first filter, the activity of these compounds against multidrug-resistant strain K1 was measured. A strong resistance signal was identified, with the K1 IC50s ranging from 3.8 μM to 7.5 μM, representing, for each compound, an increase in the IC50 of more than 20-fold compared to the values for NF54 (Table 2). The activity of these compounds was further profiled using the complete panel of P. falciparum strains with the objective of identifying the mechanism of resistance responsible for this loss of potency against K1.
FIG 2.

Resistance profile of the 1,2,4-oxadiazole series. (A) Structures of the three compounds from the 1,2,4-oxadiazol series. Ph, phenyl; Bn, benzyl; Me, methyl. (B) Relative IC50s of compounds 1, 2, and 3 against a panel of P. falciparum laboratory strains. The solid line shows a 10-fold increase in the IC50 relative to that for NF54 (n ≥ 2; error bars are SDs).
TABLE 2.
Activity of the 1,2,4-oxadiazole series compounds against the NF54 and K1 P. falciparum strains
| Compound | IC50 (nM)a |
Ratio of IC50 for K1/IC50 for NF54 | |
|---|---|---|---|
| NF54 | K1 | ||
| Chloroquine | 10 | 157 | 16 |
| 1 | 60 | 4,972 | 83 |
| 2 | 163 | 3,813 | 23 |
| 3 | 278 | 7,465 | 27 |
Values are from one representative experiment.
The relative activity of compounds 1, 2, and 3 against the panel of strains is reported in Fig. 2B (absolute values are given in Table S3 in the supplemental material). All three compounds were found to be active against D6 and HB3. Compounds 1 and 2 were inactive against all the other strains tested, that is, FCB, 7G8, K1, Dd2, V1/S, and TM90C2B. Compound 3 displayed a similar lack of activity against these strains, with the exception of 7G8, for which the IC50 increased by only 7-fold. The overall profiles of resistance to the three compounds were very similar. First, the D6 IC50 was slightly elevated; second, a further decrease in activity against 7G8 and Dd2, resulting in resistance, as defined here, except for the activity against 7G8 in the case of compound 3 was detected; and finally, a complete loss of activity against the remaining strains (FCB, K1, V1/S, and TM90C2B) was found.
Compared to the resistance profiles obtained with standard antimalarials, the 1,2,4-oxadiazole series appeared to be cross-resistant with two compounds, chloroquine and cycloguanil, suggesting that genetic determinants of resistance to this series might be mutations in pfcrt and pfdhfr. All the strains fully sensitive to this series of compounds carry a wild-type pfcrt allele, whereas the CVIET or SVMNT mutants displayed an elevated IC50 that was beyond our threshold of resistance in the vast majority of cases. Similarly, all resistant strains contained at least two mutations at pfdhfr codon 16, 51, 59, 108, or 164, while the sensitive ones were either wild type (NF54, D6) or contained only a single S108N mutation (HB3). Conversely, no correlation between specific mutations of pfmdr1, pfdhps, and pfcytb or copy number variations of pfmdr1 and the sensitivity of specific strains to members of the 1,2,4-oxadiazole series was apparent.
In order to further discriminate between the pfcrt and pfdhfr mutations as potential determinants for resistance to compounds 1, 2, and 3, their activity was evaluated in a heterologous system of Saccharomyces cerevisiae strains expressing wild-type or mutated versions of the P. falciparum dhfr gene (1). The growth of yeast strains expressing S. cerevisiae dhfr (ScDHFR), the wild-type P. falciparum dhfr (PfDHFR), or the mutant alleles PfdhfrN51I, C59R, PfdhfrS108N, and PfdhfrN51I, C59R, S108N was determined in the presence of compounds 1, 2, and 3 and cycloguanil (see Fig. S1 in the supplemental material). Cycloguanil specifically inhibited the growth of yeast strains expressing the wild-type PfDHFR enzyme, and this effect was partially lost with the PfdhfrN51I, C59R and PfdhfrS108N alleles and totally lost with the triple mutant, PfdhfrN51I, C59R, S108N. On the other hand, compounds 1, 2, and 3 failed to inhibit the growth of the yeast strain expressing PfDHFR, and no difference between strains expressing wild-type or mutated versions of PfDHFR was noticeable. These data suggest the absence of cross-resistance between cycloguanil and the 1,2,4-oxadiazole series and appear to rule out dhfr mutations as the genetic determinants of resistance to members of this chemical series observed.
We next reasoned that if resistance to the 1,2,4-oxadiazole series was mediated by pfcrt mutations, it might be reversed by verapamil, similar to the findings for chloroquine resistance (26). We exposed the pfcrt mutant and chloroquine-resistant strain K1 to either 0, 50, or 100 ng/ml of verapamil and determined the IC50 of artesunate (negative control), chloroquine (positive control), and compounds 1, 2, and 3 under these conditions (see Fig. S2 in the supplemental material). As expected, the sensitivity of K1 to chloroquine, but not to artesunate, increased in a dose-dependent manner with concomitant incubation with verapamil. The IC50s of compounds 1 and 2 did not change in response to verapamil. The sensitivity of strain K1 to compound 3 increased in response to incubation with verapamil at concentrations of 50 ng/ml and 100 ng/ml, but to an extent lower than that for chloroquine (the IC50s obtained by incubation with 100 ng/ml verapamil decreased by 29% and 51% compared to those with no verapamil for compound 3 and chloroquine, respectively).
The fact that verapamil did not significantly sensitize K1 to the 1,2,4-oxadiazole series suggests that pfcrt might not be the prime genetic determinant of resistance to this series. An alternative hypothesis is that the level of resistance to these compounds is too high to be reversed by verapamil. The IC50s of compounds 1, 2, and 3 for strain K1 ranged from 3.0 μM to 11.0 μM, whereas the IC50 of chloroquine for K1 was 1 order of magnitude lower, at 0.12 μM. The relative shift in the IC50 compared to the NF54 IC50 was also less for chloroquine, at 16-fold, whereas it was 74-, 43-, and 38-fold for compounds 1, 2, and 3, respectively. Alternatively, higher verapamil concentrations might be required to induce a reversion of the resistance of the pfcrt mutant parasites to the 1,2,4-oxadiazole series.
To evaluate the implication of pfcrt mutations in the resistance to this chemical series in a more direct manner, we took advantage of P. falciparum field isolates recently adapted to laboratory culture and fully characterized for key determinants of resistance (30). More particularly, we selected isolates HL1210 and HL1212, which share the same pfdhfr triple mutation haplotype and differ in their pfcrt sequences. HL1210 is a pfcrt mutant (CVIET), and HL1212 is a pfcrt wild-type strain (CVMNK) (Table 1). The chloroquine IC50 observed for these two strains confirmed the resistance and sensitivity status of HL1210 and HL1212, respectively (Table 3). Both strains were equally susceptible to artesunate and resistant to pyrimethamine and cycloguanil, as expected. The activity of the 1,2,4-oxadiazole series mirrored that of chloroquine rather than that of the DHFR inhibitors pyrimethamine and cycloguanil, with a 60-fold or greater increase in activity against pfcrt wild-type strain HL1212 than pfcrt mutant HL1210 (Table 3). These results suggest that mutated PfCRT might play a direct role in P. falciparum resistance to the 1,2,4-oxadiazole series and, by extension, that the mode of action of these compounds could reside in the digestive vacuole, from which mutant forms of the PfCRT transmembrane transporter are postulated to exclude chloroquine in resistant parasites (32, 33).
TABLE 3.
Sensitivity of recently laboratory-adapted P. falciparum isolates to the 1,2,4-oxadiazole series compounds
| Compound | IC50 (nM)a |
Ratio of IC50 for HL1210/IC50 for HL1212 | |
|---|---|---|---|
| HL1210 | HL1212 | ||
| Chloroquine | 150.0 ± 7.0 | 12.0 ± 3.0 | 12.5 |
| Artesunate | 4.2 ± 1.1 | 7.1 ± 1.2 | 0.6 |
| Pyrimethamine | >10,000.0 | >10,000.0 | NAb |
| Cycloguanil | 879.0 ± 218.0 | 1,877.0 ± 156.0 | 0.5 |
| 1 | 2,270.0 ± 548.0 | 33.0 ± 10.0 | 68.8 |
| 2 | 2,694 ± 502.0 | 44.0 ± 11.0 | 61.3 |
| 3 | >10,000.0 | 98.0 ± 26.0 | >100 |
Data are means and standard errors of the means from at least five independent experiments.
NA, not applicable.
The mode of action of chloroquine has been linked with the inhibition of proteases involved in the degradation of hemoglobin as well as with the prevention of hemozoin formation, possibly via heme binding, in the digestive vacuole (see Fig. S3A in the supplemental material) (34). In order to evaluate if compounds from the 1,2,4-oxadiazole series might also interfere with the hemoglobin degradation pathway, we measured the activity of compounds 1 and 2 in an in vitro hemozoin formation assay based on enzymatic or autocatalytic reactions (see Fig. S3B to D and Table S4 in the supplemental material). In vitro, heme can be released from hemoglobin by falcipain 2 and subsequently converted to hemozoin by the heme detoxification protein (HDP) or via an autocatalytic mechanism (34, 41). The autocatalytic formation of hemozoin appears to be slightly inhibited by compounds 1 and 2 (IC50s, 452 μM and 587 μM, respectively, compared to an IC50 of 43.5 μM for amodiaquine; see Table S4 in the supplemental material). Preincubation of HDP with compounds 1 and 2 did not inhibit the formation of hemozoin (see Fig. S3B in the supplemental material). However, the activity of falcipain 2 appeared to be markedly inhibited by compounds 1 and 2 (5 μM) in a fluorometric catalytic assay; in contrast, chloroquine did not have any effect on falcipain 2 activity per se (see Fig. S3C in the supplemental material). In addition, to assess the effect of compounds 1 and 2 on the released heme, they were added to a hemoglobin-to-hemozoin conversion assay. The addition of either compound did not alter the percentage of hemozoin formation, in contrast to chloroquine, which bound free heme and blocked hemozoin formation (see Fig. S3D in the supplemental material). Altogether, these observations suggest that compounds of the 1,2,4-oxadiazole series might be competitive inhibitors of the cysteine protease falcipain 2, a mode of action distinct from that of chloroquine but similarly located in the food vacuole, and thus, the findings are in line with a potential mode of resistance mediated by pfcrt.
A test cascade for resistance deconvolution.
In order to obtain the maximum amount of information about potential in vitro cross-resistance mechanisms, a given set of strains should ideally provide the highest level of resistance specificity. In other words, it is desirable to occupy the maximum possible number of intersections when representing resistance to standard antimalarial compounds as spaces in a Venn diagram (Fig. 3A). Such a graphical representation illustrates that the combination of identical pfdhfr sequences and divergent pfcrt sequences was absent from the selected panel of standard P. falciparum laboratory strains and that this combination is needed to resolve cases of resistance implicating strains with mutations in both these genes, as is the case with compounds 1, 2, and 3. This limitation was circumvented by the addition of HL1210 and HL1212 to the panel.
FIG 3.

Cross-resistance deconvolution. (A) Venn diagram of the profiles of resistance of the P. falciparum strains to atovaquone (ATO), chloroquine (CHQ), cycloguanil (CYC), and pyrimethamine (PYR). The main genetic determinants of resistance for each compound is indicated, and the strains in boxes with bold borders correspond to the ones evaluated in the assay whose results are presented in panel B. (B) Deconvolution cascade of cross-resistance signals determined using the multidrug-resistant P. falciparum strain panel. R, resistant; S, sensitive; CNV, copy number variation.
On the basis of the genotypic and phenotypic data reported here, it is possible to devise an optimal deconvolution scheme that allows acquisition of the maximum amount of information by testing a minimal number of strains (Fig. 3B). At least one strain from each intersection represented in Fig. 3A was included in this scheme. Applying this signal deconvolution to the 1,2,4-oxadiazole series readily identified pfcrt to be a potential genetic determinant of the resistance associated with this series. More generally, compounds active against Dd2 as well as TM90C2B are likely to be pan-active, whereas a resistance signal for Dd2 should trigger the sequential testing of additional strains in order to provide information about the potential genetic determinants of resistance.
DISCUSSION
The genotypic and phenotypic evaluation of a panel of sensitive and MDR P. falciparum strains previously selected to evaluate the cross-resistance potential of antimalarial compounds is reported here. Minor genotypic differences from those reported in the literature were identified for some residues of pfmdr1, pfdhfr, and pfdhps and also for the pfmdr1 copy number. It is unknown whether the sequences reported here are the original haplotypes of these strains or if mutations were acquired during culture passages. Regardless, these are unlikely to significantly alter the respective resistance phenotypes of these strains.
Indeed, the phenotype-based resistance profiles correlated well with expectations on the basis of the findings in the literature as well as with genotypic data. All strains carrying a mutant pfcrt allele at codons 72 to 76 displayed in vitro resistance to chloroquine, according to our definition, with the exception of strain 7G8, the IC50 for which shifted by only 8.3-fold, as expected due to the South American-type (SVMNT) mutant allele of this strain, whereas the other mutant strains carries the Southeast Asian-type allele (CVIET) (42). The SVMNT haplotype is best described as an amodiaquine-resistant genotype, known to impart some survival advantage after chloroquine treatment in vivo, but it does not lead to IC50 estimates as high as those due to the CVIET form of pfcrt in gene replacement studies (32). Strain TM90C2B was resistant to atovaquone and was the only strain with a Y268S mutation in PfCYTB. The responses to the DHFR inhibitors pyrimethamine and cycloguanil were also in line with the observed pfdhfr sequences. The strains that had wild-type sequences at codons 16, 51, 59, 108, and 164 (ANCSI), strains NF54 and D6, were sensitive to both compounds. Strains with the N51I, C59R, and S108N triple mutations, that is, strains K1, Dd2, V1/S, and TM90C2B, showed resistance to both compounds. The mutant with the single S108N mutation, HB3, showed resistance to pyrimethamine and not to cycloguanil, while the converse was true for the mutant with the single S108T mutation, FCB. The pfdhps-mediated resistance to sulfadoxine was not evaluated here because of the specific culture conditions required (43); however, it is expected, on the basis of the pfdhps sequence diversity in specific resistance-mediating residues, that cross-resistance with sulfonamides would be readily and specifically identifiable by using the panel of strains under appropriate culture conditions.
In this study, we defined resistance to be a greater than 10-fold shift in the IC50 compared to the IC50 for strain NF54. This threshold was arbitrary but small enough to be in line with the smallest IC50 shifts documented to be associated with clinical resistance and simultaneously large enough to avoid false-positive signals due to assay variations. An IC50 shift of that magnitude should be a cause for concern, warranting further investigations, such as the ones described here for the 1,2,4-oxadiazole series. The attempt to define more precisely drug-specific in vitro IC50 thresholds directly applicable to clinical situations is a challenging task for reasons described elsewhere (44). We favor a simpler and more conservative approach that is more relevant to antimalarial compounds in development for which no clinical data exist.
We have reported on a new antimalarial chemical series, the 1,2,4-oxadiazole series, to which MDR strain K1 was overtly resistant. Importantly, no analogues of these compounds could be found in the literature, and no hypothesis on their mode of action could be made. The complete profiling of three analogues with the panel of strains suggested that the resistance of P. falciparum to these compounds is likely to be mediated by pfcrt or pfdhfr. Further independent experimental approaches seem to favor a role of pfcrt over a role of pfdhfr but suggest that the mode of action of these compounds is different from that of chloroquine. In particular, the addition of P. falciparum isolates recently adapted for laboratory culture to the panel was necessary to provide further segregation between resistance mediated by pfcrt and that mediated by pfdhfr, revealing a shortcoming in the ability of the original panel of strains to fully deconvolute cross-resistance signals. These two isolates are also evolutionarily closer to the current target parasite populations than the standard laboratory strains, most of which were culture adapted more than 20 years ago (30).
Importantly, the panel of strains was not devised to include all known alleles of each resistance-mediating gene reported here, as it would require a very large number of strains to be tested, rendering screening activities largely ineffective. More than 20 different mutant pfcrt alleles, for instance, have been described to date (32). We favored the selection of a relatively limited number of strains to optimally represent the main causal mutations together with the largest possible diversity in order to facilitate the deconvolution of resistance signals with a manageable number of parasite strains. A direct corollary is that additional specific strains should be screened when a specific mode of resistance might be suspected on the basis of the structure of a compound or a specific activity pattern.
A limitation of our approach is that cross-resistance with artemisinins is not readily detectable with the current panel of P. falciparum strains, first, because none of them has been reported to be resistant to artemisinins and, second, because the standard in vitro IC50 determination assay using 72 h of drug exposure is not suited to the detection of this specific resistance phenotype (45). Consequently, it will be essential to include in the panel one or more of the artemisinin-resistant strains recently made available at MR4 (strains MRA-1236 to MRA-1241) and to profile the activities of new compounds against them using the recently developed ring-stage survival assay, which is able to replicate artemisinin resistance in the laboratory (45, 46). Along the same line, it would be important to determine the genotype of the strains discussed here for K13, a recently described genetic marker of artemisinin resistance (47). These strains were isolated either before the introduction of artemisinins or from areas where resistance has not yet been reported, and consequently, all are expected to be the K13 wild type, but this awaits confirmation. Characterization of the panel could also be reinforced by typing additional minor genetic polymorphisms involved in resistance mechanisms as compensatory mutations or secondary determinants, such as gch1 copy number, pfcrt mutations outside codons 72 to 76, or pfmrp1 polymorphisms (32, 48, 49).
More importantly and in order to stay as representative and effective as possible, the composition of an optimal panel will necessarily need to evolve with time to include strains with new resistance mechanisms emerging in the field. The recent report of apparent clinical resistance to piperaquine might prefigure such a case (23). Once they are available, field-isolated and validated piperaquine-resistant strains should be included in a revised test cascade.
Overall, the multiple sensitive and MDR P. falciparum strains characterized here can be used as a tool to identify cross-resistance with known naturally occurring resistance mechanisms rapidly and effectively. The panel can thus be used to facilitate the identification of pan-active compounds and to determine the genetic determinants of resistance to compounds displaying cross-resistance signals. By testing between 2 strains (for pan-active compounds) and 5 strains (for compounds displaying a pfmdr1-mediated or unresolved resistance), in addition to NF54, the test cascade presented here allows such information to be rapidly gained and facilitates early decision making during drug development (15, 50). An understanding of the mode of resistance to new antimalarial compounds can also potentially inform the design of new analogues to overcome it.
The mechanism of resistance against compounds of the 1,2,4-oxadiazole series, presented here, could successfully be related to pfcrt. This, in turn, guided the identification of the potential mode of action for these compounds to be through the inhibition of falcipain 2 in the food vacuole. Interestingly, the structure of the series does not contain the typical pharmacophore of cysteine protease inhibitors, and the apparent disconnect between falcipain 2 inhibition and whole-cell activity may be explained by the presence of two basic amines that would allow the compounds to be concentrated in the acidic food vacuole by a pH gradient (51). Further studies might investigate the morphological changes to the parasite induced by compounds of the 1,2,4-oxadiazole series, as falcipain inhibitors have previously been associated with impaired hemoglobin digestion, which was visible by light microscopy (52). Analogues potentially able to overcome the pfcrt-mediated resistance might also be synthesized and tested.
Altogether, evaluation of the 1,2,4-oxadiazole case study series illustrates the breadth of information that can be gained by profiling new antimalarial compounds with this set of P. falciparum strains. Ultimately, the careful selection of compounds refractory to already existing resistance mechanisms is an essential step in the development of new antimalarial drugs, which is facilitated by the testing strategy developed here.
Supplementary Material
ACKNOWLEDGMENTS
This work was partially supported by the Medicine for Malaria Venture (MMV). The financial support from MMV donors is gratefully acknowledged. E.B. and S.G.O. are grateful for the award of a Grand Challenges Exploration Grant (OP1087646) from the Bill & Melinda Gates Foundation. P.M.'s laboratory acknowledges financial support from DBT, India.
We thank Jeremy Burrows and Timothy N. C. Wells for their critical reading of the manuscript.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.03265-14.
REFERENCES
- 1.Bilsland E, Pir P, Gutteridge A, Johns A, King RD, Oliver SG. 2011. Functional expression of parasite drug targets and their human orthologs in yeast. PLoS Negl Trop Dis 5:e1320. doi: 10.1371/journal.pntd.0001320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roberts L, Enserink M. 2007. Malaria. Did they really say … eradication? Science 318:1544–1545. doi: 10.1126/science.318.5856.1544. [DOI] [PubMed] [Google Scholar]
- 3.Cotter C, Sturrock HJW, Hsiang MS, Liu J, Phillips AA, Hwang J, Gueye CS, Fullman N, Gosling RD, Feachem RGA. 2013. The changing epidemiology of malaria elimination: new strategies for new challenges. Lancet 382:900–911. doi: 10.1016/S0140-6736(13)60310-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Noor AM, Kinyoki DK, Mundia CW, Kabaria CW, Mutua JW, Alegana VA, Fall IS, Snow RW. 2014. The changing risk of Plasmodium falciparum malaria infection in Africa: 2000-10: a spatial and temporal analysis of transmission intensity. Lancet 383:1739–1747. doi: 10.1016/S0140-6736(13)62566-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garí E, Piedrafita L, Aldea M, Herrero E. 1997. A set of vectors with a tetracycline-regulatable promoter system for modulated gene expression in Saccharomyces cerevisiae. Yeast 13:837–848. doi: 10.1002/(SICI)1097-0061(199707)13:9<837::AID-YEA145>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 6.World Health Organization. 2010. Global report on antimalarial drug efficacy and drug resistance: 2000-2010, p 121 World Health Organization, Geneva, Switzerland. [Google Scholar]
- 7.World Health Organization Global Malaria Programme. 2013. World malaria report 2013, p 1–284 World Health Organization, Geneva, Switzerland. [Google Scholar]
- 8.Carter MD, Phelan VV, Sandlin RD, Bachmann BO, Wright DW. 2010. Lipophilic mediated assays for beta-hematin inhibitors. Comb Chem High Throughput Screen 13:285–292. doi: 10.2174/138620710790980496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sandlin RD, Carter MD, Lee PJ, Auschwitz JM, Leed SE, Johnson JD, Wright DW. 2011. Use of the NP-40 detergent-mediated assay in discovery of inhibitors of β-hematin crystallization. Antimicrob Agents Chemother 55:3363–3369. doi: 10.1128/AAC.00121-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ncokazi KK, Egan TJ. 2005. A colorimetric high-throughput beta-hematin inhibition screening assay for use in the search for antimalarial compounds. Anal Biochem 338:306–319. doi: 10.1016/j.ab.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 11.Wongsrichanalai C, Pickard AL, Wernsdorfer WH, Meshnick SR. 2002. Epidemiology of drug-resistant malaria. Lancet Infect Dis 2:209–218. doi: 10.1016/S1473-3099(02)00239-6. [DOI] [PubMed] [Google Scholar]
- 12.Mita T, Tanabe K, Kita K. 2009. Spread and evolution of Plasmodium falciparum drug resistance. Parasitol Int 58:201–209. doi: 10.1016/j.parint.2009.04.004. [DOI] [PubMed] [Google Scholar]
- 13.Lu W-J, Wicht KJ, Wang L, Imai K, Mei Z-W, Kaiser M, Sayed El IET, Egan TJ, Inokuchi T. 2013. Synthesis and antimalarial testing of neocryptolepine analogues: addition of ester function in SAR study of 2,11-disubstituted indolo[2,3-b]quinolines. Eur J Med Chem 64:498–511. doi: 10.1016/j.ejmech.2013.03.072. [DOI] [PubMed] [Google Scholar]
- 14.Trape JF. 2001. The public health impact of chloroquine resistance in Africa. Am J Trop Med Hyg 64:12–17. [DOI] [PubMed] [Google Scholar]
- 15.Ding XC, Ubben D, Wells TN. 2012. A framework for assessing the risk of resistance for anti-malarials in development. Malar J 11:292. doi: 10.1186/1475-2875-11-292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Noedl H, Se Y, Schaecher K, Smith BL, Socheat D, Fukuda MM, Artemisinin Resistance in Cambodia 1 (ARC1) Study Consortium . 2008. Evidence of artemisinin-resistant malaria in western Cambodia. N Engl J Med 359:2619–2620. doi: 10.1056/NEJMc0805011. [DOI] [PubMed] [Google Scholar]
- 17.Dondorp AM, Nosten F, Yi P, Das D, Phyo AP, Tarning J, Lwin KM, Ariey F, Hanpithakpong W, Lee SJ, Ringwald P, Silamut K, Imwong M, Chotivanich K, Lim P, Herdman T, An SS, Yeung S, Singhasivanon P, Day NPJ, Lindegardh N, Socheat D, White NJ. 2009. Artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med 361:455–467. doi: 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ashley EA, Dhorda M, Fairhurst RM, Amaratunga C, Lim P, Suon S, Sreng S, Anderson JM, Mao S, Sam B, Sopha C, Chuor CM, Nguon C, Sovannaroth S, Pukrittayakamee S, Jittamala P, Chotivanich K, Chutasmit K, Suchatsoonthorn C, Runcharoen R, Hien TT, Thuy-Nhien NT, Thanh NV, Phu NH, Htut Y, Han K-T, Aye KH, Mokuolu OA, Olaosebikan RR, Folaranmi OO, Mayxay M, Khanthavong M, Hongvanthong B, Newton PN, Onyamboko MA, Fanello CI, Tshefu AK, Mishra N, Valecha N, Phyo AP, Nosten F, Yi P, Tripura R, Borrmann S, Bashraheil M, Peshu J, Faiz MA, Ghose A, Hossain MA, Samad R, et al. 2014. Spread of artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med 371:411–423. doi: 10.1056/NEJMoa1314981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rogers WO, Sem R, Tero T, Chim P, Lim P, Muth S, Socheat D, Ariey F, Wongsrichanalai C. 2009. Failure of artesunate-mefloquine combination therapy for uncomplicated Plasmodium falciparum malaria in southern Cambodia. Malar J 8:10. doi: 10.1186/1475-2875-8-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leang R, Barrette A, Bouth DM, Menard D, Abdur R, Duong S, Ringwald P. 2013. Efficacy of dihydroartemisinin-piperaquine for treatment of uncomplicated Plasmodium falciparum and Plasmodium vivax in Cambodia, 2008 to 2010. Antimicrob Agents Chemother 57:818–826. doi: 10.1128/AAC.00686-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carrara VI, Lwin KM, Phyo AP, Ashley E, Wiladphaingern J, Sriprawat K, Rijken M, Boel M, McGready R, Proux S, Chu C, Singhasivanon P, White N, Nosten F. 2013. Malaria burden and artemisinin resistance in the mobile and migrant population on the Thai-Myanmar border, 1999-2011: an observational study. PLoS Med 10:e1001398. doi: 10.1371/journal.pmed.1001398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bustos MD, Wongsrichanalai C, Delacollette C, Burkholder B. 2013. Monitoring antimalarial drug efficacy in the greater Mekong subregion: an overview of in vivo results from 2008 to 2010. Southeast Asian J Trop Med Public Health 44(Suppl 1):S201–S230. [PubMed] [Google Scholar]
- 23.Saunders DL, Vanachayangkul P, Lon C, U.S. Army Military Malaria Research Program, National Center for Parasitology, Entomology, and Malaria Control (CNM), Royal Cambodian Armed Forces . 2014. Dihydroartemisinin-piperaquine failure in Cambodia. N Engl J Med 371:484–485. doi: 10.1056/NEJMc1403007. [DOI] [PubMed] [Google Scholar]
- 24.Avery VM, Bashyam S, Burrows JN, Duffy S, Papadatos G, Puthukkuti S, Sambandan Y, Singh S, Spangenberg T, Waterson D, Willis P. 2014. Screening and hit evaluation of a chemical library against blood-stage Plasmodium falciparum. Malar J 13:190. doi: 10.1186/1475-2875-13-190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dondorp AM, Ringwald P. 2013. Artemisinin resistance is a clear and present danger. Trends Parasitol 29:359–360. doi: 10.1016/j.pt.2013.05.005. [DOI] [PubMed] [Google Scholar]
- 26.Martin SK, Oduola AM, Milhous WK. 1987. Reversal of chloroquine resistance in Plasmodium falciparum by verapamil. Science 235:899–901. doi: 10.1126/science.3544220. [DOI] [PubMed] [Google Scholar]
- 27.Rottmann M, McNamara C, Yeung BKS, Lee MCS, Zou B, Russell B, Seitz P, Plouffe DM, Dharia NV, Tan J, Cohen SB, Spencer KR, Gonzalez-Paez GE, Lakshminarayana SB, Goh A, Suwanarusk R, Jegla T, Schmitt EK, Beck HP, Brun R, Nosten F, Renia L, Dartois V, Keller TH, Fidock DA, Winzeler EA, Diagana TT. 2010. Spiroindolones, a potent compound class for the treatment of malaria. Science 329:1175–1180. doi: 10.1126/science.1193225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McNamara CW, Lee MCS, Lim CS, Lim SH, Roland J, Nagle A, Simon O, Yeung BKS, Chatterjee AK, McCormack SL, Manary MJ, Zeeman A-M, Dechering KJ, Kumar TRS, Henrich PP, Gagaring K, Ibanez M, Kato N, Kuhen KL, Fischli C, Rottmann M, Plouffe DM, Bursulaya B, Meister S, Rameh L, Trappe J, Haasen D, Timmerman M, Sauerwein RW, Suwanarusk R, Russell B, Rénia L, Nosten F, Tully DC, Kocken CHM, Glynne RJ, Bodenreider C, Fidock DA, Diagana TT, Winzeler EA. 2013. Targeting Plasmodium PI(4)K to eliminate malaria. Nature 504:248–253. doi: 10.1038/nature12782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meister S, Plouffe DM, Kuhen KL, Bonamy GMC, Wu T, Barnes SW, Bopp SE, Borboa R, Bright AT, Che J, Cohen S, Dharia NV, Gagaring K, Gettayacamin M, Gordon P, Groessl T, Kato N, Lee MCS, McNamara CW, Fidock DA, Nagle A, Nam T-G, Richmond W, Roland J, Rottmann M, Zhou B, Froissard P, Glynne RJ, Mazier D, Sattabongkot J, Schultz PG, Tuntland T, Walker JR, Zhou Y, Chatterjee A, Diagana TT, Winzeler EA. 2011. Imaging of Plasmodium liver stages to drive next-generation antimalarial drug discovery. Science 334:1372–1377. doi: 10.1126/science.1211936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Schalkwyk DA, Burrow R, Henriques G, Gadalla NB, Beshir KB, Hasford C, Wright SG, Ding XC, Chiodini PL, Sutherland CJ. 2013. Culture-adapted Plasmodium falciparum isolates from UK travellers: in vitro drug sensitivity, clonality and drug resistance markers. Malar J 12:320. doi: 10.1186/1475-2875-12-320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burrows JN, van Huijsduijnen RH, Möhrle JJ, Oeuvray C, Wells TNC. 2013. Designing the next generation of medicines for malaria control and eradication. Malar J 12:187. doi: 10.1186/1475-2875-12-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ecker A, Lehane AM, Clain J, Fidock DA. 2012. PfCRT and its role in antimalarial drug resistance. Trends Parasitol 28:504–514. doi: 10.1016/j.pt.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Summers RL, Nash MN, Martin RE. 2012. Know your enemy: understanding the role of PfCRT in drug resistance could lead to new antimalarial tactics. Cell Mol Life Sci 69:1967–1995. doi: 10.1007/s00018-011-0906-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chugh M, Sundararaman V, Kumar S, Reddy VS, Siddiqui WA, Stuart KD, Malhotra P. 2013. Protein complex directs hemoglobin-to-hemozoin formation in Plasmodium falciparum. Proc Natl Acad Sci U S A 110:5392–5397. doi: 10.1073/pnas.1218412110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Trager W, Jensen JB. 1976. Human malaria parasites in continuous culture. Science 193:673–675. doi: 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- 36.Desjardins RE, Canfield C, Haynes J, Chulay J. 1979. Quantitative assessment of antimalarial activity in vitro by a semiautomated microdilution technique. Antimicrob Agents Chemother 16:710–718. doi: 10.1128/AAC.16.6.710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Snyder C, Chollet J, Santo Tomas J, Scheurer C, Wittlin S. 2007. In vitro and in vivo interaction of synthetic peroxide RBx11160 (OZ277) with piperaquine in Plasmodium models. Exp Parasitol 115:296–300. doi: 10.1016/j.exppara.2006.09.016. [DOI] [PubMed] [Google Scholar]
- 38.Fidock DA, Rosenthal PJ, Croft SL, Brun R, Nwaka S. 2004. Antimalarial drug discovery: efficacy models for compound screening. Nat Rev Drug Discov 3:509–520. doi: 10.1038/nrd1416. [DOI] [PubMed] [Google Scholar]
- 39.Price RN, Uhlemann A-C, Brockman A, McGready R, Ashley E, Phaipun L, Patel R, Laing K, Looareesuwan S, White NJ, Nosten F, Krishna S. 2004. Mefloquine resistance in Plasmodium falciparum and increased pfmdr1 gene copy number. Lancet 364:438–447. doi: 10.1016/S0140-6736(04)16767-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Basco LK, Ringwald P. 2001. Analysis of the key pfcrt point mutation and in vitro and in vivo response to chloroquine in Yaoundé, Cameroon. J Infect Dis 183:1828–1831. doi: 10.1086/320726. [DOI] [PubMed] [Google Scholar]
- 41.Egan TJ, Chen JY-J, de Villiers KA, Mabotha TE, Naidoo KJ, Ncokazi KK, Langford SJ, McNaughton D, Pandiancherri S, Wood BR. 2006. Haemozoin (beta-haematin) biomineralization occurs by self-assembly near the lipid/water interface. FEBS Lett 580:5105–5110. doi: 10.1016/j.febslet.2006.08.043. [DOI] [PubMed] [Google Scholar]
- 42.Sá JM, Twu O, Hayton K, Reyes S, Fay MP, Ringwald P, Wellems TE. 2009. Geographic patterns of Plasmodium falciparum drug resistance distinguished by differential responses to amodiaquine and chloroquine. Proc Natl Acad Sci U S A 106:18883–18889. doi: 10.1073/pnas.0911317106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tan-ariya P, Brockelman CR, Menabandhu C. 1987. Optimal concentration of p-aminobenzoic acid and folic acid in the in vitro assay of antifolates against Plasmodium falciparum. Am J Trop Med Hyg 37:42–48. [DOI] [PubMed] [Google Scholar]
- 44.Ekland EH, Fidock DA. 2008. In vitro evaluations of antimalarial drugs and their relevance to clinical outcomes. Int J Parasitol 38:743–747. doi: 10.1016/j.ijpara.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Witkowski B, Khim N, Chim P, Kim S, Ke S, Kloeung N, Chy S, Duong S, Leang R, Ringwald P, Dondorp AM, Tripura R, Benoit-Vical F, Berry A, Gorgette O, Ariey F, Barale J-C, Mercereau-Puijalon O, Menard D. 2013. Reduced artemisinin susceptibility of Plasmodium falciparum ring stages in western Cambodia. Antimicrob Agents Chemother 57:914–923. doi: 10.1128/AAC.01868-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Witkowski B, Amaratunga C, Khim N, Sreng S, Chim P, Kim S, Lim P, Mao S, Sopha C, Sam B, Anderson JM, Duong S, Chuor CM, Taylor WRJ, Suon S, Mercereau-Puijalon O, Fairhurst RM, Menard D. 2013. Novel phenotypic assays for the detection of artemisinin-resistant Plasmodium falciparum malaria in Cambodia: in-vitro and ex-vivo drug-response studies. Lancet Infect Dis 13:1043–1049. doi: 10.1016/S1473-3099(13)70252-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ariey F, Witkowski B, Amaratunga C, Beghain J, Langlois A-C, Khim N, Kim S, Duru V, Bouchier C, Ma L, Lim P, Leang R, Duong S, Sreng S, Suon S, Chuor CM, Bout DM, Ménard S, Rogers WO, Genton B, Fandeur T, Miotto O, Ringwald P, Le Bras J, Berry A, Barale J-C, Fairhurst RM, Benoit-Vical F, Mercereau-Puijalon O, Menard D. 2014. A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature 505:50–55. doi: 10.1038/nature12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nair S, Miller B, Barends M, Jaidee A, Patel J, Mayxay M, Newton P, Nosten F, Ferdig MT, Anderson TJC. 2008. Adaptive copy number evolution in malaria parasites. PLoS Genet 4:e1000243. doi: 10.1371/journal.pgen.1000243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Koenderink JB, Kavishe RA, Rijpma SR, Russel FGM. 2010. The ABCs of multidrug resistance in malaria. Trends Parasitol 26:440–446. doi: 10.1016/j.pt.2010.05.002. [DOI] [PubMed] [Google Scholar]
- 50.Leroy D, Campo B, Ding XC, Burrows JN, Cherbuin S. 2014. Defining the biology component of the drug discovery strategy for malaria eradication. Trends Parasitol 30:478–490. doi: 10.1016/j.pt.2014.07.004. [DOI] [PubMed] [Google Scholar]
- 51.Ettari R, Bova F, Zappalà M, Grasso S, Micale N. 2010. Falcipain-2 inhibitors. Med Res Rev 30:136–167. doi: 10.1002/med.20163. [DOI] [PubMed] [Google Scholar]
- 52.Rosenthal PJ, McKerrow JH, Aikawa M, Nagasawa H, Leech JH. 1988. A malarial cysteine proteinase is necessary for hemoglobin degradation by Plasmodium falciparum. J Clin Invest 82:1560–1566. doi: 10.1172/JCI113766. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.