

Abstract
Drug-resistant tuberculosis (TB) has lent urgency to finding new drug leads with novel modes of action. A high-throughput screening campaign of >65,000 actinomycete extracts for inhibition of Mycobacterium tuberculosis viability identified ecumicin, a macrocyclic tridecapeptide that exerts potent, selective bactericidal activity against M. tuberculosis in vitro, including nonreplicating cells. Ecumicin retains activity against isolated multiple-drug-resistant (MDR) and extensively drug-resistant (XDR) strains of M. tuberculosis. The subcutaneous administration to mice of ecumicin in a micellar formulation at 20 mg/kg body weight resulted in plasma and lung exposures exceeding the MIC. Complete inhibition of M. tuberculosis growth in the lungs of mice was achieved following 12 doses at 20 or 32 mg/kg. Genome mining of lab-generated, spontaneous ecumicin-resistant M. tuberculosis strains identified the ClpC1 ATPase complex as the putative target, and this was confirmed by a drug affinity response test. ClpC1 functions in protein breakdown with the ClpP1P2 protease complex. Ecumicin markedly enhanced the ATPase activity of wild-type (WT) ClpC1 but prevented activation of proteolysis by ClpC1. Less stimulation was observed with ClpC1 from ecumicin-resistant mutants. Thus, ClpC1 is a valid drug target against M. tuberculosis, and ecumicin may serve as a lead compound for anti-TB drug development.
INTRODUCTION
Mycobacterium tuberculosis is the cause of one of the most deadly diseases of mankind, and despite the availability of effective treatments, tuberculosis (TB) remains a major public health threat. The difficult challenges in treating multiple-drug-resistant (MDR) and extensively drug-resistant (XDR) TB and the importance of shortening the duration of treatment to improve patients' compliance make the discovery of new anti-TB drugs imperative (1–5). Attempts to discover new TB drugs and targets via large-scale screening against intact mycobacteria have largely been confined to synthetic compound libraries and to date have yielded only one new clinical TB drug, the diarylquinoline bedaquiline (6, 7). Although very potent, to be of maximum benefit, bedaquiline, a diarylquinoline, and nitroimidazoles (8) require new companion drugs to be used in a multidrug regimen.
While the intensive search for antibiotics from soil microorganisms in the mid-20th century yielded several clinically useful TB drugs, the pathogenic nature of M. tuberculosis and its extremely slow growth rate did not allow classical agar diffusion tests and excluded M. tuberculosis from the initial target panel. The discovery of TB drugs of natural origin at that time therefore relied upon the detection of activity against nonmycobacteria in agar diffusion assays followed by bioassay-guided isolation of the active principle, again using nonmycobacteria. Activity against M. tuberculosis was only assessed once the active principle was purified.
Because M. tuberculosis is uniquely susceptible to a number of antimicrobial agents, a high-throughput screening (HTS) of actinomycete extracts directly against the virulent H37Rv strain was conducted, and this campaign revealed selective anti-TB peptides produced by a genetically distinct Nonomuraea species, strain MJM5123. Here, we describe the activity profile of ecumicin, its efficacy in infected mice, the identification of its molecular target, and the elucidation of its unusual mechanism of action.
MATERIALS AND METHODS
High-throughput screening.
Approximately 7,000 actinomycete cultures isolated from Korea, China, Nepal, the Philippines, Vietnam, Antarctica, and the Arctic Circle and maintained at Myongji University, South Korea, were fermented in 20-ml cultures in glucose-soybean starch (GSS) medium (rich medium), Bennett's medium (normal medium), and dextrin-yeast-corn steep liquor (DYC) medium (minimal medium) (see Table S1 in the supplemental material). The mycelia and culture medium supernatants were separated and extracted with methanol and ethyl acetate, respectively. Nine extracts were thus generated from each microbial isolate. Each was dissolved in dimethyl sulfoxide (DMSO) and screened for inhibition of replicating M. tuberculosis (9).
Bacterial strains and chemicals.
M. tuberculosis strain H37Rv (ATCC 27294) was used in all assays except as otherwise stated. All chemicals were purchased from Sigma-Aldrich except as otherwise stated.
MICs versus M. tuberculosis.
All MICs were determined using the microplate alamarBlue assay (MABA) as previously described (9) except that we now use 7H12 medium (instead of 7H9 plus glycerol plus Casitone plus oleic acid-albumin-dextrose-catalase [OADC]) (10, 11) and resazurin (instead of the alamarBlue reagent). The MIC was defined as the lowest concentration effecting a reduction in fluorescence of 90% relative to that of controls.
Cytotoxicity.
Compounds were tested for cytotoxicity using Vero cells (ATCC CRL-1586) (10, 12, 13). After 72 h of exposure, viability was assessed on the basis of cellular conversion of 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) into a soluble formazan product using the Promega CellTiter 96 AQueous nonradioactive cell proliferation assay. Rifampin (RMP) was included as a control.
Cytotoxicity testing was repeated, using the J774.1 macrophage cell line since these cells are used in the macrophage assay and were usually somewhat more sensitive than Vero cells. This was important for interpreting data from the macrophage assay.
MICs against monodrug-resistant isolates.
The MABA was used to assess MICs against isogenic M. tuberculosis H37Rv-derived strains from ATCC with monoresistance to isoniazid (INH) (ATCC 35822), rifampin (RMP) (ATCC 35838), streptomycin (SM) (ATCC 35820), kanamycin (KM) (ATCC 35827), cycloserine (CS) (ATCC 35826), moxifloxacin (MFX), and capreomycin (CAP). The latter two strains were selected in our laboratory and harbored mutations in gyrA and tylA, respectively.
MICs against nonmycobacteria.
MICs against Escherichia coli (ATCC 25922), Staphylococcus aureus (ATCC 29213), Acinetobacter baumannii (ATCC BAA-747), Enterococcus faecalis (ATCC 29212), and Pseudomonas aeruginosa (ATCC 27853) were determined by measuring optical density at 570 nm (OD570) after 16 h of incubation in 2.2% Mueller-Hinton II broth (Becton Dickinson, Sparks, MD, USA), against Streptococcus pneumoniae (ATCC 49619) at OD490 after 20 h in 2.2% Mueller-Hinton II broth supplement with 2% horse blood, and against Candida albicans (ATCC 10231) at OD570 after 48 h in 1% Cellgro RPMI 1640 medium (Mediatech Inc., Manassas, VA, USA) supplemented with 1.8% d-(+)-dextrose (ICN Biomedicals, Aurora, OH, USA) and 3.5% morpholinepropanesulfonic acid (MOPS) (Acros, NJ, USA). The MIC was defined as the lowest concentration resulting in ≥90% reduction in absorption relative to that of untreated control cultures.
MICs against nontuberculous mycobacteria.
MICs against Mycobacterium smegmatis (ATCC 700084) and Mycobacterium abscessus (ATCC 19977) were determined by the MABA in a manner similar to that used for M. tuberculosis except that cultures were incubated for 72 h prior to addition of 0.6 mM resazurin and Tween 80 and then fluorescence was recorded after an additional 4 h of incubation. Mycobacterium chelonae (ATCC 35752) and Mycobacterium marinum (ATCC 927) were cultured in 7H9 medium plus OADC supplement at 30°C for 72 h and 120 h, respectively. Mycobacterium avium (ATCC 15769) and Mycobacterium kansasii (ATCC 12478) were cultured in 7H9 medium plus OADC supplement at 37°C for 6 days and 7 days, respectively. The MIC against Mycobacterium bovis BCG (ATCC 35734) was determined by the MABA.
MICs against clinical isolates (including MDR and XDR M. tuberculosis).
Nine clinical strains were isolated from pulmonary TB patients hospitalized in the National Masan Hospital, Masan, South Korea. M. tuberculosis isolates freshly grown on 7H11 agar medium for 2 weeks were used for determination of the MICs by the MABA.
MICs against genetically diverse M. tuberculosis.
M. tuberculosis isolates collected from throughout the world were grouped into six major single nucleotide polymorphism (SNP) clusters. MICs were determined by the MABA against isolates from each cluster (total of six isolates) to ensure the lack of major heterogeneity with respect to susceptibility. The isolates are X001354 corresponding to the Indo-Oceanic lineage, X004439 and X004244 to the East Asian lineage, X005282 and X005319 to the Euro-American lineage, and X001354 to the East African Indian lineage (14).
MBCs.
Minimal bactericidal concentration (MBCs) for replicating cultures were determined by subculture from the MABA plates onto 7H11 agar just prior to addition of resazurin and Tween 80 to the test wells. The cultures were washed with phosphate-buffered saline (PBS) once to remove test compounds prior to plating. The MBC was defined as the lowest concentration reducing CFU by 99% relative to the zero time inoculum.
MBCs for nonreplicating (NR) cultures were determined by subculture from the low-oxygen recovery assay (LORA) (15) plates onto 7H11 agar. M. tuberculosis isolates had been exposed for 10 days to test compounds in a low-oxygen environment prior to subculture. The MBC was defined as the lowest concentration reducing CFU by 99% relative to the zero time inoculum. The reported MBC results were from one representative experiment of three that were independently performed.
Activity in macrophage culture.
Inhibition of growth of M. tuberculosis Erdman (ATCC 35801) in a macrophage cell culture was assessed as previously described (16) except that cultures were not activated with interferon. Briefly, J774A.1 cells were cultured on 13-mm coverslips in 24-well plates and infected with M. tuberculosis Erdman, extracellular bacilli were removed by washing, and cultures were incubated in Dulbecco's modified Eagle's medium (DMEM) containing ecumicin at 0.012, 0.12, and 1.2 μM and rifampin at 0.024, 0.12, and 0.61 μM. All experimental conditions were set up in triplicate. Before treatment (T0) (for untreated controls) and after 7 days, the incubation medium was removed, and macrophages were lysed with 200 μl of 0.25% SDS. After 10 min of incubation at 37°C, 200 μl of fresh medium was added. The contents of the wells were transferred to a microtube and sonicated (model 1510; Branson Ultrasonics, Danbury, CT) for 15 s, and 1:1, 1:10, 1:100, and 1:1,000 dilutions were plated on 7H11 (Difco) agar plates. Colonies were counted after incubation at 37°C for 2 to 3 weeks.
In vitro absorption, distribution, metabolism, and excretion assays.
Caco-2 cell monolayers were grown in 24-well BD Falcon cell culture insert plates for at least 21 days. The bidirectional permeability of ecumicin was measured in duplicate following a 2-h incubation. The dosing solutions (5 μM) were prepared in transport buffer (TM) containing Hanks balanced salt solution (HBSS) with 10 mM HEPES and 25 mM glucose (HBSSg) (pH 7.4); 4% bovine serum albumin (BSA) in TM buffer (pH 7.4) was used for the receiver side. Digoxin, propranolol, and [14C]mannitol were used as controls. Ecumicin and controls were analyzed using liquid chromatography-tandem mass spectrometry (LC-MS2). For metabolic stability assessment, ecumicin was incubated for 30 min at 1 μM in 100 mM phosphate buffer (pH 7.4), 1 mM NADPH, and 3 mM MgCl2 with 0.5 mg/ml human or mouse CD1 liver microsomes. The final solvent concentration was <0.1%. Ecumicin was analyzed by LC-MS2 using reserpine as an internal control.
Micelle formulation preparation.
PLA(4,000)-b-PEG(5,000)-OCH3 (10 mg) (PLA, polylactic acid; PEG, polyethylene glycol) was dissolved in 0.9 ml dimethylformamide (DMF), to which 1 mg ecumicin dissolved in 0.1 ml DMF was added. The solution was transferred to a dialysis membrane with a molecular weight cutoff (MWCO) of 3,500 and dialyzed against deionized water for 1 day, changing the water frequently. The micelle solution was then lyophilized for further characterization. The particle size was analyzed by dynamic light scattering. Of the total micelle particles by volume, 96.6% had a mean diameter of 51.5 nm, with 9.5 nm (18.5%) deviation; the remaining 3.4% micelle particles had a mean diameter of 691.1 nm, with 105.0 nm (15.2%) deviation. An LC-MS2 method was established to determine the loading of micelles. A 5-μl ecumicin sample was injected onto an XTerra C8 column (2.5 μm, 2.1 by 50 mm). A biphase linear gradient was applied, starting from 50% acetonitrile and 0.1% formic acid water solution at 0 min to 95% acetonitrile at 4 min. The column was then flushed with 95% acetonitrile for 2 more minutes and reequilibrated with 50% acetonitrile for 2 min. The MS2 transition 800.5 to 100.2 (m/z) was used to quantitate ecumicin, and the transition 800.5 to 227.3 (m/z) was used for qualification. The retention time for ecumicin was 5.21 min. Reserpine was used as an internal standard in this process at a final concentration of 0.045 μM; the transition 609.3 to 195.1 (m/z) was monitored to quantitate reserpine. Dried micelles were dissolved in acetonitrile to a final concentration of 10 μg/ml. A standard curve was built with a series of standard ecumicin solutions with concentrations of 0.2, 0.4, 0.6, 0.8, 1.0, and 2.0 μg/ml. To every 100-μl ecumicin solution, 10 μl 0.5 μM reserpine solution was added. The ecumicin loading of this micelle formulation was calculated to be 5.0%.
Mouse pharmacokinetic study.
Healthy male CD-1 mice were administered the micellar ecumicin formulation intravenously (i.v.) or subcutaneously (s.c.) with a single dose or five consecutive daily doses. Blood and lung tissue samples were collected at 0.5, 1, 2, 4, 8, 16, and 24 h posttreatment. Blood was centrifuged at 4,000 × g for 30 min at 4°C. Lung samples were washed in PBS, dried with a paper towel, and weighed. To each lung sample, 3 ml PBS per g tissue was added, and each sample was homogenized by sonication until the homogenate was of an even consistency with no visible chunks. The homogenates were centrifuged at 4,000 × g for 30 min at 4°C, and 50 μl of the resulting supernatants was mixed with 50 μl of cold acetonitrile and centrifuged again at 4,000 × g for 30 min at 4°C. The supernatants were analyzed by LC-MS2 on an AB Sciex QTrap 4000 instrument. To establish a standard curve, ecumicin dissolved in DMSO was spiked into untreated mouse plasma and lung tissue homogenate, followed by the above-mentioned analysis.
In vivo anti-TB activity.
Female BALB/c mice (19 to 20 g) were exposed to Mycobacterium tuberculosis Erdman (ATCC 35801) in an aerosol infection chamber (Glas-Col, Terra Haute, IN). The nebulizer contained a 10-ml suspension of 3 × 106 CFU/ml. On day 3, five mice were sacrificed to determine the baseline bacterial load in lungs and on day 10, seven mice were sacrificed to determine the pretreatment bacterial load. Groups of 7 mice were treated once daily on days 10 to 14, 17 to 21, and 24 to 25 postinfection. Treatments included subcutaneous administration of micellar ecumicin at 20 and 32 mg/kg body weight as well as an empty micelle vehicle control and oral administration of 15 mg/kg rifampin suspended in 0.5% carboxymethylcellulose as well as the latter (vehicle) alone. On day 28 postinfection (72 h after the last dose), all treatment and vehicle control groups were sacrificed. Following euthanasia using carbon dioxide asphyxiation, both lungs were aseptically homogenized in 3 ml of sterile HBSS. Ten-fold serial dilutions of homogenates were prepared in HBSS, and diluted suspensions were plated on Middlebrook 7H11 agar in 6-well plates. CFU were determined following 18 to 21 days incubation at 37°C.
Selection of ecumicin-resistant strains of M. tuberculosis.
7H11 agar plates containing ecumicin at 1×, 2×, 4×, and 8× MIC (determined on 7H11 agar) against M. tuberculosis were prepared by adding an appropriate amount of compound to molten 7H11 agar medium. A total of 109 CFU of M. tuberculosis H37Rv were plated onto 7H11 agar plates containing 1× MIC ecumicin and incubated at 37°C for 3 to 4 weeks. The colonies from the selection plates were phenotypically characterized, and the representatives were restreaked for isolation on 7H11 agar plates containing 2-fold higher concentrations of ecumicin. The process was repeated until no colonies appeared on the selection plates. The resistance frequencies were determined by calculating the number of colonies arising on selective plates divided by the total CFU plated. The colonies that grew on 7H11 agar with the highest ecumicin concentration (4× MIC) were used to inoculate 7H9 liquid medium and incubated at 37°C for 1 to 2 weeks. The harvested cells were washed with PBS and kept at −80°C.
Ecumicin-resistant M. tuberculosis genome sequencing.
Genomic DNAs of ecumicin-resistant M. tuberculosis strains ITR6, ITR7, ITR12, and ITR13 were isolated as described previously (17). Genomic DNA extracted from each of these M. tuberculosis strains and wild-type H37Rv was sequenced on a HiSeq 2000 instrument (Illumina). The raw sequence data were imported into the software package CLC Genomics Workbench (v5.5) as paired reads. Stringent trimming was performed (0.01 quality trimming threshold with no degeneracies allowed, and all sequences shorter than 100 bases after trimming were discarded). After trimming, approximately 5 to 11.5 million reads were recovered from each sample. These reads were mapped to the reference genome of M. tuberculosis H37Rv (GenBank accession no. NC_000962) using default assembly parameters (0.5 length fraction and 0.8 similarity fraction) implemented within CLC Genomics Workbench. The variants relative to the reference genome, including single nucleotide variants as well as insertions and deletions, were detected using the probabilistic variant detection routine within CLC Genomics Workbench. The parameters included a minimum coverage of 50× and a minimum variant probability of 50%. PCR, Sanger DNA sequencing, and Sequencher (Gene Codes) were used to confirm nonsynonymous variants.
IPTG-inducible M. tuberculosis clpC1 and espG3 expression constructs.
The coding sequences of clpC1 (Rv3596c) and espG3 (Rv0289) were PCR amplified from genomic DNA of wild-type and mutant ecumicin-resistant M. tuberculosis strain H37Rv to create recombinant pET-28a(+) plasmids (Novagen EMD Millipore) with C-terminal 6-histidine tags. Both wild-type and mutant clpC1 and espG3 PCR fragments were cloned into the NcoI and HindIII restriction enzyme (RE) sites of pET-28a(+). All recombinant clones were confirmed with Sanger DNA sequencing and Sequencher (GeneCodes). All recombinant plasmids were transformed into BL21-Gold (DE3) expression competent E. coli (Agilent Technologies) for isopropyl-β-d-thiogalactopyranoside (IPTG)-induced expression. The recombinant His-tagged ClpC1 and EspG3 proteins resolved at approximately 95 and 35 kDa, respectively, using NuPAGE Novex 4 to 12% bis-Tris gels and Novex InVision His tag In-gel stain with the Novex sharp protein standard (Life Technologies).
DARTS and protein identification.
The recombinant E. coli whole-cell lysates were prepared by glass bead beating (BioSpec) and sonication as previously described (18). The protein concentrations of cell lysates were determined by a BCA protein assay kit (Thermo Scientific) and adjusted to 2 mg/ml total protein concentration with 1× TNC buffer (50 mM Tris-Cl [pH 8.0], 50 mM NaCl, 10 mM CaCl2) for drug affinity responsive target stability (DARTS) application (19). The lysates were divided into 50-μl aliquots and incubated with DMSO (control) or treated with ecumicin at 4 ng/ml to 40 μg/ml for 2 h at 4°C. Pronase (2 μl at 12.5 μg/ml) (Roche Applied Science) was then added to each sample, and incubation was continued for an additional hour at 37°C. The cell lysate sample proteins were separated on NuPAGE Novex 4 to 12% bis-Tris gels (Life Technologies) and stained with SimplyBlue SafeStain (Life Technologies). According to the supplier's instructions, Western blot transfer onto polyvinylidene difluoride (PVDF) iBlot transfer stacks was completed using an iBlot dry transfer system (Life Technologies). Chromogenic immunodetection was completed with 5-bromo-4-chloro-3-indolylphosphate-nitroblue tetrazolium (BCIP-NBT) substrate and anti-rabbit secondary antibodies coupled to alkaline phosphatase (WesternBreeze WB7105; Life Technologies). The primary anti-His antibody (2365S; Cell Signaling) was diluted 1,000:1. Images of gels and Western blots were obtained using a Canon PowerShot SD400 digital camera. To identify proteins protected by ecumicin, the protein bands of interest were excised from SDS-PAGE gels and in-gel digested with trypsin. The extracted peptides were then analyzed by LC-MS2 with data-dependent acquisition using an Ultimate 3000 nanoscale liquid chromatograph coupled to an Orbitrap Velos Pro (Thermo Scientific) mass spectrometer. The raw data were processed for protein database searching, which was conducted using a Mascot search engine against an NCBI protein database with a mass accuracy of 10 ppm and a 95% confidence cutoff. All recombinant E. coli cultures used in the DARTS assay were plasmid DNA authenticated by Sanger DNA sequencing and Sequencher.
Measurement of ClpC1 ATPase activity.
The ATPase activity was measured using a coupled enzyme assay with pyruvate kinase and lactic dehydrogenase (PK/LDH) (20). Briefly, 2 μg of pure ClpC1 (or other ATPase) was mixed with 100 μl of the assay buffer (50 mM Tris-HCl [pH 7.8], 100 mM KCl, 10% glycerol, 1 mM phosphoenolpyruvate, 1 mM NADH, 2 units of pyruvate kinase/lactic dehydrogenase [Sigma], 4 mM MgCl2, and 1 mM ATP) and the ATPase activity of ClpC1 was measured by following the coupled oxidization of NADH to NAD spectrometrically at 340 nm. Measurements were performed in triplicate, and deviations between them were <5%.
Measurement of casein degradation.
The proteolytic activity was measured at 37°C in 96-well plates using a SpectraMax M5 plate reader (Molecular Devices, USA). Wells contained 3 μg of ClpP1P2, 6 μg of ClpC1, 5 mM Z-Leu-Leu, and 2 to 5 μg of fluorescein isothiocyanate (FITC)-casein in 80 μl of 50 mM phosphate buffer (pH 7.6) with 5% glycerol, 100 mM KCl, 8 mM MgCl2, and 2 mM ATP. The FITC-casein hydrolysis was continuously monitored at 518 nm (excitation at 492 nm). Measurements were performed in triplicate, and deviations between them were <10%.
RESULTS
High-throughput screening.
Of approximately 65,000 extracts, 349 (0.54%) effected a >90% inhibition of fluorescence relative to that of the untreated control cultures. Such high activity was primarily found in the organic solvent extracts of normal or rich medium. Ninety of the initial hits were then profiled for activity against: mammalian cells (Vero cells, 50% inhibitory concentration [IC50]), nonreplicating M. tuberculosis (15), isogenic M. tuberculosis strains that are resistant to SM, RMP, and CS (to ensure that we are not merely again finding these actinomycete-derived antibiotics), and M. smegmatis, S. aureus, E. coli, and C. albicans.
The 22 samples with the best profiles were then retested for these activities. Based on the cumulative data, 18 of these hits were refermented at a 1-liter scale, with subsequent solid-phase extraction on RP-18 cartridges and bioassay profiling for the above activities.
Ecumicin (Fig. 1), a macrocyclic tridecapeptide, was isolated from strain MJM5123, which was prioritized based on the strong activity in the lipophilic fractions of the mycelial methanolic extract against both replicating and nonreplicating M. tuberculosis H37Rv as well as strains that are resistant to the existing actinomycete-derived antibiotics (21–23).
FIG 1.
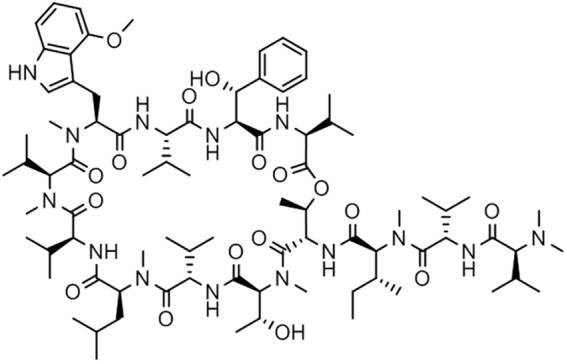
Structure of ecumicin.
Ecumicin has potent and selective in vitro bactericidal activity against M. tuberculosis.
Ecumicin has an in vitro anti-M. tuberculosis activity profile comparable to or superior to that of the current first-line anti-TB drugs (Tables 1 and 2). This level of activity is maintained against M. tuberculosis H37Rv-isogenic strains monoresistant to RMP, INH, SM, CS, KM, and the cyclic peptide CAP, thus suggesting that ecumicin has a molecular target distinct from that of these drugs. Activities were also maintained against nine clinical strains, including MDR and XDR M. tuberculosis isolates, and against M. tuberculosis clinical isolates representing the six major global clades (14) and five of six nontuberculous mycobacterial species. No inhibitory activity was observed against S. aureus, E. coli, or C. albicans at 32 μM. Moreover, ecumicin is selective with respect to mammalian cells (Vero and mouse macrophage J774.1 cell lines) (Table 2), with a selectivity index (IC50/M. tuberculosis MIC) of >640. The high bactericidal activity against M. tuberculosis replicating in culture broth medium (MBC of 0.34 μM) is dependent upon both the concentration and time of exposure and was similar when two inocula were used, differing by an order of magnitude (Fig. 2; see also Fig. S1 in the supplemental material). More modest but still potent activity was observed against nonreplicating cultures (MBC of 1.5 μM), indicating the possibility of shortening the duration of treatment.
TABLE 1.
MICs of ecumicin and anti-TB drugs versus M. tuberculosis
| M. tuberculosis strain | MIC(s) (μM) ofa: |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| ECM | RMP | INH | CAP | MFX | SM | KM | CS | PA-824 | |
| H37Rv in 7H12 medium containingb: | |||||||||
| 0.5% BSA | 0.16 | 0.08 | 0.29 | 1.9 | 0.05 | 1.2 | 2.1 | <0.08 | |
| 4% BSA | 0.58 | 0.10 | 0.44 | 1.0 | 1.8 | 0.96 | 0.22 | ||
| 10% FBS | 0.36 | 0.23 | 0.22 | 4.5 | 0.47 | 3.2 | 0.47 | ||
| H37Rv resistant to: | |||||||||
| RMP | 0.19 | >1 | 0.47 | 3.4 | 0.11 | 0.72 | 0.06 | ||
| INH | <0.12 | 0.01 | >8 | 7.5 | 0.11 | 0.48 | 0.06 | ||
| CAP | 0.29 | 0.06 | 0.46 | 52 | 0.24 | 3.3 | |||
| MFX | 0.31 | 0.06 | 0.24 | 3.6 | >8 | 0.49 | – | ||
| SM | <0.12 | 0.02 | 0.23 | 3.7 | 0.06 | >8 | 0.18 | ||
| CS | <0.12 | 0.01 | 0.42 | 3.8 | 0.12 | 0.89 | >100 | ||
| 6 global clade representatives | 0.13–0.38 | 0.03–0.06 | 0.03–0.06 | 0.24–0.45 | 0.43–1.5 | 0.31–2.0 | |||
| Clinical | |||||||||
| 3 pan-susceptible strains | 0.31–0.62 | 0.30–0.61 | 0.88–1.8 | 0.30–0.62 | 0.86–1.7 | 2.1–4.1 | |||
| 3 MDR strains | 0.31 | ≥1.2 | 15–29 | 0.12–0.25 | 3.4–6.9 | 4.1 | |||
| 3 XDR strains | 0.31–0.62 | >1.2 | >15 | ≥1.9 | ≥14 | >66 | |||
| ECMr strains | |||||||||
| ITR6 | 1.5 | <0.02 | 0.08 | 0.37 | <0.06 | <0.031 | |||
| ITR7 | 1.4 | <0.02 | 0.12 | 0.49 | 0.08 | <0.031 | |||
| ITR12 | 1.5 | <0.02 | 0.12 | 0.91 | 0.89 | <0.031 | |||
| ITR13 | 1.0 | <0.02 | 0.07 | 0.42 | 0.16 | <0.031 | |||
ECM, ecumicin; RMP, rifampin; INH, isoniazid; CAP, capreomycin; MFX, moxifloxacin; SM, streptomycin; KM, kanamycin; CS, cycloserine.
BSA, bovine serum albumin; FBS, fetal bovine serum.
TABLE 2.
Ecumicin spectrum of activities
| Target | MIC (μM) ofa: |
IC50 (μM) of: |
|||||
|---|---|---|---|---|---|---|---|
| ECM | RMP | CAP | MFX | KM | ECM | RMP | |
| E. faecalis | >32 | 12 | |||||
| P. aeruginosa | >32 | 0.10 | |||||
| S. pneumonias | >32 | 0.05 | |||||
| A. baumannii | >32 | ||||||
| E. coli | >32 | >50 | |||||
| S. aureus | >32 | >50 | |||||
| C. albicans | >32 | >50 | |||||
| M. smegmatis | 1.7 | 12 | 49 | 0.48 | 0.48 | ||
| M. chelonae | 0.97 | ||||||
| M. abscessus | >63 | ||||||
| M. kansasii | <0.24 | ||||||
| M. avium | 0.35 | ||||||
| M. marinum | 0.95 | ||||||
| Vero cells | >63 | 86 | |||||
| J774 cells | >32 | ||||||
ECM, ecumicin; RMP, rifampin; CAP, capreomycin; MFX, moxifloxacin; KM, kanamycin.
FIG 2.
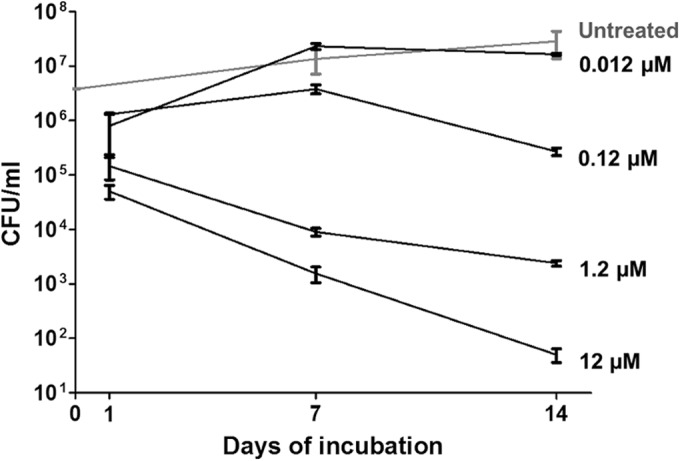
Ecumicin has time- and concentration-dependent bactericidal activity against M. tuberculosis. The inoculum size was 3.8 × 106 CFU/ml. Error bars represent the standard deviation (SD). Each data point indicates the mean of two measurements.
In the presence of 4% bovine serum albumin (physiological concentration) or 10% fetal bovine serum (Table 1), the MIC only increased by 2- to 4-fold, suggesting that protein binding may not cause any significant loss of efficacy.
The cyclic structure and N-methylation pattern of ecumicin likely contribute to the observed resistance to enzymatic degradation. After a 30-min incubation with human or mouse liver microsomes, ecumicin remained substantially unchanged (95% and 91% remaining, respectively).
The permeability coefficients calculated from a Caco-2 cell assay were 16 nm/s apical to basolateral and 28 nm/s basolateral to apical. This suggests poor to modest intestinal absorption, and, therefore, subsequent pharmacokinetic experiments focused on parenteral administration as a prelude to proof-of-concept efficacy studies.
In J774 macrophages, ecumicin clearly demonstrated concentration-dependent killing (Fig. 3). At 0.12 μM, ecumicin and rifampin reduced M. tuberculosis viabilities by 2 × log10 and 1 × log10 CFU, respectively. Thus, ecumicin retains potent bactericidal activity within host cells.
FIG 3.
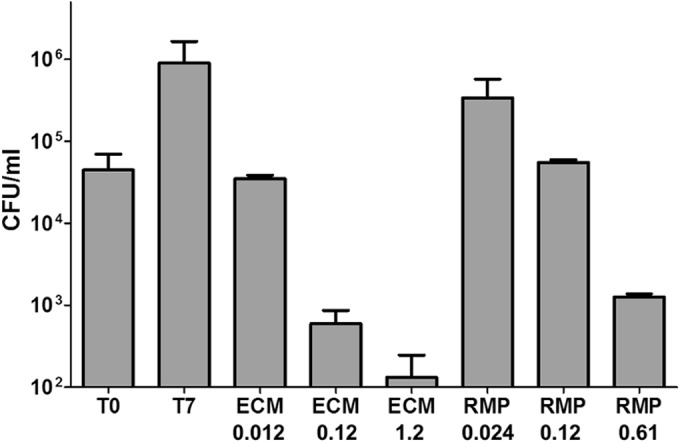
Ecumicin is bactericidal against M. tuberculosis in murine macrophages. Bars represent M. tuberculosis CFU prior to treatment (T0) or after 7 days of incubation without treatment (T7) or with rifampin (RMP) or ecumicin (ECM) at the indicated concentrations (μM). Values are means ± SD. According to Bonferroni's method (43), significant differences (P < 0.01) were observed between the following untreated and ecumicin-treated groups: T0 and ECM 1.2 and T0 and ECM 0.2.
Ecumicin encapsulated in micelles distributes to mouse lung tissue.
In order to overcome the poor water solubility of ecumicin, a polymeric micelle formulation was developed for parenteral administration in mouse pharmacokinetic and efficacy studies.
After a single intravenous (i.v.) administration of 5 mg/kg body weight, ecumicin was eliminated from plasma, with a distribution half-life of approximately 30 min and terminal half-life of 3 h (Fig. 4A). Concentrations in lung ranged from 1.5 to 2.8 μg/g. Areas under the curve for concentration versus time from 0 to 4 h (AUC0→4 h) were 9.6 and 8.9 mg · h/ml for plasma and lung tissue, respectively. After a single subcutaneous administration of 20 mg/kg, maximum plasma concentrations (Cmax) reached 0.30 to 0.40 μg/ml within 2 h, and this level was maintained at 8 h postadministration; in lung tissue, a plateau of 0.60 to 0.73 μg/g was achieved at 8 h postadministration and maintained at 24 h. AUC0→24 h values were 5.5 and 11.7 mg · h/ml for plasma and lung tissue, respectively. Ecumicin in the micelle formulation appeared to accumulate in lung tissue when mice were administered five consecutive daily subcutaneous doses of 20 mg/kg, achieving a range of 1.5 to 3.4 μg/g throughout the 24-h dosing interval following the last dose (Fig. 4B). As this concentration exceeds the MIC, even in the presence of physiological albumin (0.58 μM [0.93 μg/ml]), ecumicin was evaluated for efficacy in M. tuberculosis-infected mice.
FIG 4.
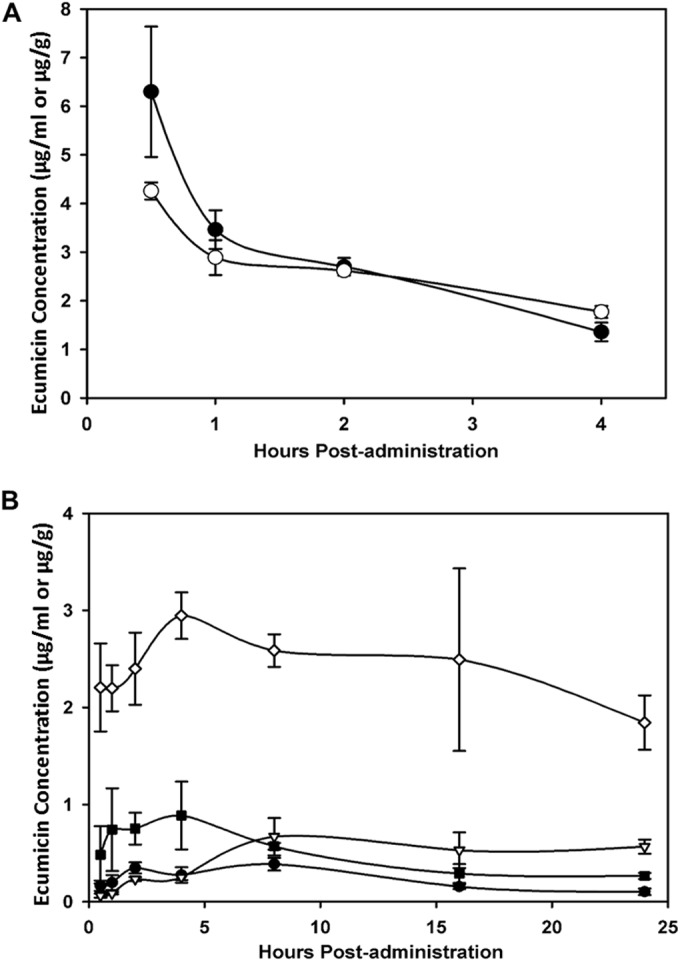
(A) Mean plasma and lung tissue concentrations of ecumicin in mice (n = 3) after a single i.v. dose of ecumicin at 5 mg/kg. ●, plasma; ○, lung tissue. (B) Plasma and lung exposures in mice (n = 3) following single or multiple s.c. doses of ecumicin at 20 mg/kg. ●, plasma after single dose; △, lung tissue after single dose; ■, plasma after 5 daily doses; ◊, lung tissue after 5 daily doses.
Ecumicin inhibits growth of M. tuberculosis in the lungs of mice.
M. tuberculosis CFU in the lungs of mice dosed with either the 0.5% carboxymethyl cellulose vehicle (control for rifampin) or empty micelles increased by 1 to 2 × log10 during the treatment (and posttreatment washout) interval (Fig. 5). Compared with the micellar vehicle control, mice treated with 20 mg/kg and 32 mg/kg ecumicin had reductions in M. tuberculosis lung CFU of 1.3 × log10 and 1.6 × log10, respectively, in the latter case representing essentially complete inhibition of growth. Rifampin demonstrated bactericidal activity by reducing M. tuberculosis CFU in lung relative to the pretreatment tissue load.
FIG 5.
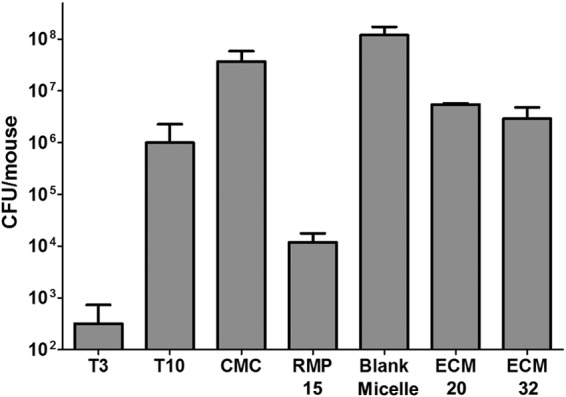
Twelve s.c. doses of micellar ecumicin (ECM) inhibit M. tuberculosis growth in the lungs of mice during acute infection. For all groups, n = 7 except for T3 (n = 5) and RMP 15 (n = 6). T3, untreated and sacrificed at 3 days postinfection; T10, untreated and sacrificed at 10 days postinfection; CMC, treated orally (p.o.) with 0.5% carboxymethyl cellulose; RMP 15, treated p.o. with 15 mg/kg rifampin in 0.5% CMC; blank micelle, treated s.c. with 640 mg/kg blank micelle; ECM 20, treated s.c. with 20 mg/kg ECM; ECM 32, treated s.c. with 32 mg/kg ECM. CMC, RMP 15, blank micelle, ECM 20, and ECM 32 mice were sacrificed 28 days postinfection. Values are means ± SD. Multiple comparisons among pairs were performed by Bonferroni's method (43). There were significant differences between all (P < 0.01) results except between T10 and ECM 32.
Ecumicin-resistant isolates harbor clpC1 mutations.
Spontaneously arising ecumicin-resistant mutants of M. tuberculosis H37Rv were selected in vitro, both to determine the frequency of mutation to resistance and to investigate the mechanism of action. Single-step mutants at 1× MIC were selected at a frequency of 3 × 10−8 and then were inoculated onto 7H11 plates with ecumicin at 2× MIC and finally onto 7H11 plates with ecumicin at 4× MIC.
To determine whether the phenotypes of these resistant mutants of M. tuberculosis were specific for ecumicin, the parent strain and the resistant mutants were tested for sensitivity to a panel of antibiotics with various mechanisms of action. Based on their relatively high level of selective resistance to ecumicin (Table 1), four strains (M. tuberculosis ITR6, ITR7, ITR12, and ITR13) were selected for whole-genome sequencing. Compared with M. tuberculosis H37Rv, the selected mutants all harbored nonsynonymous mutations in clpC1 (Rv3596c), ppsC (Rv2933), and espG3 (Rv0289). All resistant strains had the same mutations to espG3 (GGT→AGT; G191S) and ppsC (CAG→TAG; Q695stop), but harbored one of three mutations in the N-terminal repeat II of ClpC1 (24) that resulted in proline for leucine 96 or serine or phenylalanine for leucine 92 (Table 3). PCR and Sanger DNA sequencing confirmed mutations in all three genes of each resistant strain. Strains ITR5, ITR12, and ITR13, each harboring a different clpC1 mutation, were again cultured in liquid medium. The newly prepared cultures retained clpC1 mutations as confirmed by PCR and Sanger DNA sequencing, and resistance against ecumicin (see Table S2 in the supplemental material), confirming the inherited nature of the resistance.
TABLE 3.
Mutations of clpC1 in ecumicin-resistant strainsa
| Resistant strain | Codon mutation | Amino acid change |
|---|---|---|
| ITR1 | TTG→TCG | L92S |
| ITR2 | CGG→CCG | L96P |
| ITR3 | CGG→CCG | L96P |
| ITR5 | TTG→TCG | L92S |
| ITR6 | CGG→CCG | L96P |
| ITR7 | CGG→CCG | L96P |
| ITR8 | TTG→TCG | L92S |
| ITR9 | TTG→TTT | L92F |
| ITR10 | TTG→TCG | L92S |
| ITR12 | TTG→TCG | L92S |
| ITR13 | TTG→TCG | L92S |
All resistant strains had the same nonsynonymous changes to espG3 (GGT→AGT; G191S) and ppsC (CAG→TAG; Q695stop).
To further examine which of these potential targets actually binds ecumicin, recombinant E. coli BL21 isolates overexpressing M. tuberculosis wild-type and mutant ClpC1 and EspG3 were tested for drug affinity responsive target stability (DARTS) (19). A lysate of each recombinant E. coli was exposed to ecumicin at different concentrations and subjected to pronase digestion. Wild-type M. tuberculosis ClpC1 was protected against pronase digestion by ecumicin, and the protective effect increased with ecumicin concentration (Fig. 6). The protected protein band was excised and identified as ClpC1 by mass spectrometry. Such a protective effect was not observed for any of the three mutant ClpC1s. This result strongly suggests that ClpC1 is at least one of the molecular targets of ecumicin. Previous studies have suggested that clpC1 and espG3 are essential for the viability and infectivity of H37Rv (25–27), while ppsC is nonessential (28–30). As ecumicin failed to protect either the wild-type or mutant EspG3 (see Fig. S2 in the supplemental material) and as the M. tuberculosis CDC1551 ppsC transposon mutant harboring a disrupted ppsC was found to be fully sensitive to ecumicin (see Table S3 in the supplemental material), we conclude that, unlike ClpC1, neither EspG3 nor PpsC directly binds ecumicin.
FIG 6.

Ecumicin protects wild-type (A) but not mutated (B to D) M. tuberculosis ClpC1 from pronase digestion. Analysis was performed by SDS-PAGE (lanes 1 to 7) and Western blotting (lanes 8 to 14). Lanes: M, size marker; 1 and 8, whole-cell lysate without any treatment; 2 and 9, whole-cell lysate digested by pronase; 3 to 7 and 10 to 14, whole-cell lysates treated with ecumicin at increasing concentrations (0.04, 0.4, 4, 40, and 80 μg/ml) and subjected to pronase digestion.
Ecumicin uncouples ClpC1-mediated ATP hydrolysis from ClpP1P2 proteolysis.
ClpC1 is a hexameric ATPase belonging to the large AAA family of ATPases (31, 32), and in M. tuberculosis, unlike in most bacteria, it is an essential gene (30). ClpC1 associates with and supports ATP-dependent protein degradation by ClpP, a compartmentalized protease complex found in many bacteria, mitochondria, and chloroplasts (33, 34). In this process, ClpC1 binds certain cell proteins and unfolds and translocates them into ClpP for degradation. To determine whether ClpC1 is indeed the target of ecumicin in M. tuberculosis and to clarify its mode of action, we tested if ecumicin directly affected the ATPase activity of M. tuberculosis ClpC1, expressed and isolated as described recently (35). Surprisingly, ecumicin did not inhibit this activity but instead stimulated the hydrolysis of ATP by severalfold. The addition of ecumicin did not change the specificity of ClpC1 since it could hydrolyze only ATP but not CTP, GTP, or UTP (data not shown). This stimulation of the ATPase activity by ecumicin occurs at a submicromolar concentration (dissociation constant [Kd] = 0.6 μM) and in a cooperative manner (Hill coefficient = 1.7) (Fig. 7A).
FIG 7.

Ecumicin selectively activates ATPase activity of wild-type ClpC1, but not protein degradation by the ClpC1P1P2 complex. (A) Ecumicin enhances the ATPase activity of wild-type ClpC1 derived from M. tuberculosis. Similar results were obtained when the ATPase activity was measured with the malachite green method. (B) Hill coefficient for activation of ClpC1 by ecumicin. The Kd and Hill coefficient for ecumicin activation of wild-type ClpC1 ATPase were determined using curve fitting with the classic Hill kinetic through a scaled Levenberg-Marquardt algorithm (tolerance 0.0001). (C) Ecumicin stimulates wild-type ClpC1 much more strongly than mutated ClpC1 from ecumicin-resistant strains. The ATPase activities of wild-type and mutant forms of ClpC1 (cloned and expressed in E. coli) are shown. The ATPase activity of each version of ClpC1 in the absence of ecumicin was taken as 100%. (D) ClpC1 (cloned and expressed in M. smegmatis) does not activate degradation of casein by ClpP1P2 in the presence of ecumicin. Each result is the average of three independent experiments. (E) Ecumicin activates M. tuberculosis ClpC1 but not several homologous AAA ATPases. The activities of purified AAA ATPases from several bacteria (M. tuberculosis ClpX; E. coli ClpA and ClpB), archaea (PAN), and mouse (26S proteasome) and the activity of the E. coli molecular chaperone GroEL were measured in the presence and absence of ecumicin (10 μM). The activity of each ATPase in the absence of ecumicin was taken as 100%. The deviations between the measurements were <5% for panels A, B, and C and <10% for panel D.
When mutated forms of ClpC1 from several ecumicin-resistant M. tuberculosis strains were cloned and expressed in E. coli, they showed ATPase activity similar to that of the wild-type enzyme, but reduced the stimulation by ecumicin (Fig. 7B). These observations confirm that ClpC1 is the target of ecumicin in M. tuberculosis and that the changes in ClpC1 function are responsible for ecumicin's bactericidal action. Interestingly, ecumicin did not affect the activity of a variety of purified ATPases, including the E. coli chaperonin GroEL, ClpX from M. tuberculosis, which also functions with ClpP1P2, and homologous AAA ATPases from E. coli (ClpA), archaea (PAN), and mammals (26S proteasome) (Fig. 7C). This specificity and the fact that ClpC1 is essential only in mycobacteria (25) can account for ecumicin's selective toxicity.
In mycobacteria, ClpC1 functions with ClpP1P2, a novel member of the ClpP family found only in mycobacteria, where it is essential for viability (35). Therefore, ClpC1's ability to stimulate ATP-dependent degradation of the model substrate, FITC-casein, by ClpP1P2 (35) was also analyzed in the presence of ecumicin. Surprisingly, even though ecumicin stimulated ATP hydrolysis, it caused a strong inhibition of the ClpC1-dependent degradation of casein (Fig. 7D). Thus, the antibiotic uncoupled ATP hydrolysis from protein degradation. Presumably, this failure to carry out the selective ATP-dependent degradation of key cell proteins accounts for ecumicin's cytotoxic action and because ClpC1 and ClpP1P2 are essential only in mycobacteria, this inhibition of proteolysis can account for the selective killing of these bacteria.
DISCUSSION
This study demonstrates that it is possible to find new potent, narrow-spectrum anti-TB secondary metabolites from actinomycetes by direct whole-cell-based HTS and to both biologically dereplicate and profile them prior to isolation of the active principle. The inclusion of a panel of M. tuberculosis isolates resistant to actinomycete-derived drugs and organisms such as E. coli and S. aureus that have been used in previous screening campaigns minimized the chance of pursuing known compounds. The toxicity of an extract/fraction to a mammalian cell line, while conceivably due to a compound(s) other than that responsible for inhibition of M. tuberculosis, was nonetheless used in the prioritization process and likely significantly reduces the probability of ultimately isolating nonselective compounds at the end of the bioassay-guided isolation process.
Genome sequencing of spontaneous ecumicin-resistant clones and binding assays strengthened by the availability of mutated proteins implicated ClpC1 as the primary molecular target of ecumicin. Whole-genome sequencing of these isolates also revealed common mutations in ppsC and espG3. The gene ppsC encodes the phthiocerol synthesis polyketide synthase type I, PpsC, which is involved in the elongation of C22 to C24 fatty acids by the addition of malonyl-coenzyme (CoA) and methylmalonyl-CoA extender units to yield phthiocerol derivatives (36). That ppsC is a nonessential (28–30) gene and that an M. tuberculosis CDC1551 ppsC transposon mutant was found to be fully sensitive to ecumicin suggest that PpsC is not likely to be the target. The gene espG3 encodes the ESX-3 secretion-associated protein EspG3 and is essential for the viability and infectivity of M. tuberculosis H37Rv (25–27). While its function remains unknown, the analogous protein EspG3 in M. smegmatis is one of the core Esx-3 components that are required for both mycobactin-mediated iron acquisition and EsxG and EsxH export (37). As ecumicin failed to protect either the wild-type or mutant EspG3 (see Fig. S2 in the supplemental material) from nonspecific proteolytic degradation, we conclude that it is unlikely that EspG3 is a target. Ecumicin's actual mode of action, stimulating its ATPase activity but blocking ClpC1-mediated protein breakdown by ClpP1P2, in other words, uncoupling ATP hydrolysis from proteolysis, is quite unusual. ClpP1P2 and its regulatory factors ClpC1 and ClpX are essential in M. tuberculosis, presumably to eliminate the critical proteins whose accumulation is toxic (35). This bactericidal mechanism is opposite to that demonstrated for the acyldepsipeptide (ADEP) antibiotics, which nonspecifically activate degradation of proteins by ClpPs in many bacteria (38). However, in M. tuberculosis, ADEPs appear to act primarily by blocking the interaction of ClpP1P2 with its regulatory ATPases. Enhancement of ClpC1-mediated proteolysis has also been proposed as the mode of action of the antibiotic cyclomarin, which also binds to ClpC1 (39, 40). An uncoupling mechanism similar to that observed here has recently been demonstrated for lassomycin, another M. tuberculosis-specific cyclic peptide antibiotic that also targets ClpC1 (41). Although having similar effects, ecumicin differs markedly from lassomycin in containing many noncanonical amino acids, not being synthesized on ribosomes, and not being extremely basic. So it is very likely that the two agents bind to distinct domains on ClpC1. Our findings imply that certain M. tuberculosis proteins that are normally degraded by the ClpC1P1P2 complex are highly toxic and accumulate in the presence of ecumicin. This unique target and mechanism predict no cross-resistance with existing TB drugs, which is consistent with the observed data (Table 1).
Alignment of the clpC1 sequences of the mycobacteria in Table 2 did not suggest a basis for the relative resistance of M. abscessus, a species that is resistant to many antimycobacterial agents via various mechanisms (42).
Because a bacterial protein appears to be essential in vivo does not necessarily imply that it is a druggable target in a host in vivo, since phenotypes observed in gene knockouts/knockdowns often cannot be replicated with a small molecule inhibitor. Here we have demonstrated that ClpC1 is a druggable target in M. tuberculosis residing in both cellular (macrophage) and mouse hosts. While the former was clearly bactericidal, the static activity observed in the latter may have been due to the limited dosage and treatment duration, which in turn were dictated by the limited solubility/micellar loading and compound availability, respectively. The lung tissue Cmax achieved at 20 mg/kg was only 1.6× to 3.7× the MIC versus M. tuberculosis in the presence of a physiological serum albumin concentration. In consideration of an MBC/MIC ratio of ∼2, this might fall just below an in vivo bactericidal exposure. Two subsequent experiments in mice using a Phosal-based, nonmicellar vehicle demonstrated plasma and lung concentrations exceeding the MIC following either oral or intraperitoneal administration of ecumicin (data not shown).
Future efficacy experiments conducted at higher oral dosages for longer durations will determine in part the potential of ecumicin itself as a therapeutic lead. Closely related analogs of ecumicin isolated from the same Nonomuraea sp. or those produced through genetic engineering of the nonribosomal peptide synthetase (NRPS) coding region or semisynthesis or isolated directly from other species (22) may yet prove to have superior pharmacokinetic and pharmacodynamic profiles. Alternatively, current efforts to cocrystallize ecumicin and ClpC1 may lead to nonpeptidic approaches to exploit this target.
Supplementary Material
ACKNOWLEDGMENTS
We thank Chang-Hwa Hwang and Larry L. Klein for technical assistance and Hyun-Young Jeong and Stefan Green for helpful discussion.
The following reagent was obtained through BEI Resources, NIAID, NIH: Mycobacterium tuberculosis strain CDC1551, transposon mutant 2562 (MT3003, Rv2933), NR-18565.
This work was supported in part by a grant to J.-W. Suh from the Next-Generation BioGreen 21 Program (no. PJ009643), Rural Development Administration, Republic of Korea.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.04054-14.
REFERENCES
- 1.Orenstein EW, Basu S, Shah NS, Andrews JR, Friedland GH, Moll AP, Gandhi NR, Galvani AP. 2009. Treatment outcomes among patients with multidrug-resistant tuberculosis: systematic review and meta-analysis. Lancet Infect Dis 9:153–161. doi: 10.1016/S1473-3099(09)70041-6. [DOI] [PubMed] [Google Scholar]
- 2.Shi W, Zhang X, Jiang X, Yuan H, Lee JS, Barry CE III, Wang H, Zhang W, Zhang Y. 2011. Pyrazinamide inhibits trans-translation in Mycobacterium tuberculosis. Science 333:1630–1632. doi: 10.1126/science.1208813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lienhardt C, Raviglione M, Spigelman M, Hafner R, Jaramillo E, Hoelscher M, Zumla A, Gheuens J. 2012. New drugs for the treatment of tuberculosis: needs, challenges, promise, and prospects for the future. J Infect Dis 205(Suppl 2):S241–S249. doi: 10.1093/infdis/jis034. [DOI] [PubMed] [Google Scholar]
- 4.Keshavjee S, Farmer PE. 2010. Picking up the pace—scale-up of MDR tuberculosis treatment programs. N Engl J Med 363:1781–1784. doi: 10.1056/NEJMp1010023. [DOI] [PubMed] [Google Scholar]
- 5.Dooley KE, Kim PS, Williams SD, Hafner R. 2012. TB and HIV therapeutics: pharmacology research priorities. AIDS Res Treat 2012:874083. doi: 10.1155/2012/874083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Andries K, Verhasselt P, Guillemont J, Gohlmann HW, Neefs JM, Winkler H, Van Gestel J, Timmerman P, Zhu M, Lee E, Williams P, de Chaffoy D, Huitric E, Hoffner S, Cambau E, Truffot-Pernot C, Lounis N, Jarlier V. 2005. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 307:223–227. doi: 10.1126/science.1106753. [DOI] [PubMed] [Google Scholar]
- 7.Diacon AH, Donald PR, Pym A, Grobusch M, Patientia RF, Mahanyele R, Bantubani N, Narasimooloo R, De Marez T, van Heeswijk R, Lounis N, Meyvisch P, Andries K, McNeeley DF. 2012. Randomized pilot trial of eight weeks of bedaquiline (TMC207) treatment for multidrug-resistant tuberculosis: long-term outcome, tolerability, and effect on emergence of drug resistance. Antimicrob Agents Chemother 56:3271–3276. doi: 10.1128/AAC.06126-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mukherjee T, Boshoff H. 2011. Nitroimidazoles for the treatment of TB: past, present and future. Future Med Chem 3:1427–1454. doi: 10.4155/fmc.11.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Collins L, Franzblau S. 1997. Microplate Alamar blue assay versus BACTEC 460 system for high-throughput screening of compounds against Mycobacterium tuberculosis and Mycobacterium avium. Antimicrob Agents Chemother 41:1004–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Falzari K, Zhu Z, Pan D, Liu H, Hongmanee P, Franzblau SG. 2005. In vitro and in vivo activities of macrolide derivatives against Mycobacterium tuberculosis. Antimicrob Agents Chemother 49:1447–1454. doi: 10.1128/AAC.49.4.1447-1454.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Franzblau SG, Degroote MA, Cho SH, Andries K, Nuermberger E, Orme IM, Mdluli K, Angulo-Barturen I, Dick T, Dartois V, Lenaerts AJ. 2012. Comprehensive analysis of methods used for the evaluation of compounds against Mycobacterium tuberculosis. Tuberculosis 92:453–488. doi: 10.1016/j.tube.2012.07.003. [DOI] [PubMed] [Google Scholar]
- 12.Cantrell CL, Lu T, Fronczek FR, Fischer NH, Adams LB, Franzblau SG. 1996. Antimycobacterial cycloartanes from Borrichia frutescens. J Nat Prod 59:1131–1136. doi: 10.1021/np960551w. [DOI] [PubMed] [Google Scholar]
- 13.Mangalindan GC, Talaue MT, Cruz LJ, Franzblau SG, Adams LB, Richardson AD, Ireland CM, Concepcion GP. 2000. Agelasine F from a Philippine Agelas sp. sponge exhibits in vitro antituberculosis activity. Planta Med 66:364–365. doi: 10.1055/s-2000-8554. [DOI] [PubMed] [Google Scholar]
- 14.Filliol I, Motiwala AS, Cavatore M, Qi W, Hazbon MH, Bobadilla del Valle M, Fyfe J, Garcia-Garcia L, Rastogi N, Sola C, Zozio T, Guerrero MI, Leon CI, Crabtree J, Angiuoli S, Eisenach KD, Durmaz R, Joloba ML, Rendon A, Sifuentes-Osornio J, Ponce de Leon A, Cave MD, Fleischmann R, Whittam TS, Alland D. 2006. Global phylogeny of Mycobacterium tuberculosis based on single nucleotide polymorphism (SNP) analysis: insights into tuberculosis evolution, phylogenetic accuracy of other DNA fingerprinting systems, and recommendations for a minimal standard SNP set. J Bacteriol 188:759–772. doi: 10.1128/JB.188.2.759-772.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cho SH, Warit S, Wan B, Hwang CH, Pauli GF, Franzblau SG. 2007. Low-oxygen-recovery assay for high-throughput screening of compounds against nonreplicating Mycobacterium tuberculosis. Antimicrob Agents Chemother 51:1380–1385. doi: 10.1128/AAC.00055-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou B, He Y, Zhang X, Xu J, Luo Y, Wang Y, Franzblau SG, Yang Z, Chan RJ, Liu Y, Zheng J, Zhang ZY. 2010. Targeting mycobacterium protein tyrosine phosphatase B for antituberculosis agents. Proc Natl Acad Sci U S A 107:4573–4578. doi: 10.1073/pnas.0909133107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Belisle J, Mahaffey S, Hill P. 2009. Isolation of Mycobacterium species genomic DNA, p 1–12. In Parish T, Brown AC (ed), Mycobacteria protocols, vol 465 Humana Press, New York, NY. [DOI] [PubMed] [Google Scholar]
- 18.Jaki BU, Franzblau SG, Cho SH, Pauli GF. 2006. Development of an extraction method for mycobacterial metabolome analysis. J Pharm Biomed Anal 41:196–200. doi: 10.1016/j.jpba.2005.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lomenick B, Hao R, Jonai N, Chin RM, Aghajan M, Warburton S, Wang J, Wu RP, Gomez F, Loo JA, Wohlschlegel JA, Vondriska TM, Pelletier J, Herschman HR, Clardy J, Clarke CF, Huang J. 2009. Target identification using drug affinity responsive target stability (DARTS). Proc Natl Acad Sci U S A 106:21984–21989. doi: 10.1073/pnas.0910040106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anderson KW, Murphy AJ. 1983. Alterations in the structure of the ribose moiety of ATP reduce its effectiveness as a substrate for the sarcoplasmic reticulum ATPase. J Biol Chem 258:14276–14278. [PubMed] [Google Scholar]
- 21.Gao W, Napolitano JG, Kim JY, Lee IA, Lee JE, Choi J, Rodríguez-Brasco MF, Jaki BU, Cho S, McAlpine J, Pauli GF, Kim J, Suh JW, Franzblau SG. 2012. New insights on the structure of the anti-TB peptide H14: crystal structure, 1H NMR full spin analysis, and biosynthetic pathway. Planta Med 78:PJ134. doi: 10.1055/s-0032-1321294. [DOI] [Google Scholar]
- 22.Cho S, Choi JK, Gao Franzblau S. Friesen B, Gao W, Jaki B, Jin YY, Kim JY, Kim JW, Lankin D, Lee IA, Lee JE, Lee SW, Mcalpine J, Napolitano J, Park SJ, Pauli G, Rodriguez BMF, Suh JW, Yoon TM. October 2012. Cyclic peptide from nonomuraea sp., process for the production thereof, and pharmaceutical composition for the prevention or treatment of mycobacteria related disease comprising the same. International patent WO/2012/144790 A1. [Google Scholar]
- 23.Gao W, Kim J-Y, Chen S-N, Cho S-H, Choi J, Jaki BU, Jin Y-Y, Lankin DC, Lee J-E, Lee S-Y, McAlpine JB, Napolitano JG, Franzblau SG, Suh J-W, Pauli GF. 19 November 2014. Discovery and characterization of the TB drug lead ecumicin. Org Lett doi: 10.1021/ol5026603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kar NP, Sikriwal D, Rath P, Choudhary RK, Batra JK. 2008. Mycobacterium tuberculosis ClpC1: characterization and role of the N-terminal domain in its function. FEBS J 275:6149–6158. doi: 10.1111/j.1742-4658.2008.06738.x. [DOI] [PubMed] [Google Scholar]
- 25.Raju RM, Goldberg AL, Rubin EJ. 2012. Bacterial proteolytic complexes as therapeutic targets. Nat Rev Drug Discov 11:777–789. doi: 10.1038/nrd3846. [DOI] [PubMed] [Google Scholar]
- 26.Raju RM, Unnikrishnan M, Rubin DH, Krishnamoorthy V, Kandror O, Akopian TN, Goldberg AL, Rubin EJ. 2012. Mycobacterium tuberculosis ClpP1 and ClpP2 function together in protein degradation and are required for viability in vitro and during infection. PLoS Pathog 8:e1002511. doi: 10.1371/journal.ppat.1002511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brodin P, Majlessi L, Marsollier L, de Jonge MI, Bottai D, Demangel C, Hinds J, Neyrolles O, Butcher PD, Leclerc C, Cole ST, Brosch R. 2006. Dissection of ESAT-6 system 1 of Mycobacterium tuberculosis and impact on immunogenicity and virulence. Infect Immun 74:88–98. doi: 10.1128/IAI.74.1.88-98.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Griffin JE, Gawronski JD, Dejesus MA, Ioerger TR, Akerley BJ, Sassetti CM. 2011. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog 7:e1002251. doi: 10.1371/journal.ppat.1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rengarajan J, Bloom BR, Rubin EJ. 2005. Genome-wide requirements for Mycobacterium tuberculosis adaptation and survival in macrophages. Proc Natl Acad Sci U S A 102:8327–8332. doi: 10.1073/pnas.0503272102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lamichhane G, Zignol M, Blades NJ, Geiman DE, Dougherty A, Grosset J, Broman KW, Bishai WR. 2003. A postgenomic method for predicting essential genes at subsaturation levels of mutagenesis: application to Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 100:7213–7218. doi: 10.1073/pnas.1231432100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hanson PI, Whiteheart SW. 2005. AAA+ proteins: have engine, will work. Nat Rev Mol Cell Biol 6:519–529. doi: 10.1038/nrm1684. [DOI] [PubMed] [Google Scholar]
- 32.Erzberger JP, Berger JM. 2006. Evolutionary relationships and structural mechanisms of AAA+ proteins. Annu Rev Biophys Biomol Struct 35:93–114. doi: 10.1146/annurev.biophys.35.040405.101933. [DOI] [PubMed] [Google Scholar]
- 33.Lupas A, Flanagan JM, Tamura T, Baumeister W. 1997. Self-compartmentalizing proteases. Trends Biochem Sci 22:399–404. doi: 10.1016/S0968-0004(97)01117-1. [DOI] [PubMed] [Google Scholar]
- 34.Yu AY, Houry WA. 2007. ClpP: a distinctive family of cylindrical energy-dependent serine proteases. FEBS Lett 581:3749–3757. doi: 10.1016/j.febslet.2007.04.076. [DOI] [PubMed] [Google Scholar]
- 35.Akopian T, Kandror O, Raju RM, Unnikrishnan M, Rubin EJ, Goldberg AL. 2012. The active ClpP protease from M. tuberculosis is a complex composed of a heptameric ClpP1 and a ClpP2 ring. EMBO J 31:1529–1541. doi: 10.1038/emboj.2012.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Camacho LR, Constant P, Raynaud C, Laneelle MA, Triccas JA, Gicquel B, Daffe M, Guilhot C. 2001. Analysis of the phthiocerol dimycocerosate locus of Mycobacterium tuberculosis. Evidence that this lipid is involved in the cell wall permeability barrier. J Biol Chem 276:19845–19854. doi: 10.1074/jbc.M100662200. [DOI] [PubMed] [Google Scholar]
- 37.Siegrist MS, Steigedal M, Ahmad R, Mehra A, Dragset MS, Schuster BM, Philips JA, Carr SA, Rubin EJ. 2014. Mycobacterial Esx-3 requires multiple components for iron acquisition. mBio 5:e01073-14. doi: 10.1128/mBio.01073-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kirstein J, Hoffmann A, Lilie H, Schmidt R, Rubsamen-Waigmann H, Brotz-Oesterhelt H, Mogk A, Turgay K. 2009. The antibiotic ADEP reprogrammes ClpP, switching it from a regulated to an uncontrolled protease. EMBO Mol Med 1:37–49. doi: 10.1002/emmm.200900002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schmitt EK, Riwanto M, Sambandamurthy V, Roggo S, Miault C, Zwingelstein C, Krastel P, Noble C, Beer D, Rao SP, Au M, Niyomrattanakit P, Lim V, Zheng J, Jeffery D, Pethe K, Camacho LR. 2011. The natural product cyclomarin kills Mycobacterium tuberculosis by targeting the ClpC1 subunit of the caseinolytic protease. Angew Chem Int Ed Engl 50:5889–5891. doi: 10.1002/anie.201101740. [DOI] [PubMed] [Google Scholar]
- 40.Vasudevan D, Rao SP, Noble CG. 2013. Structural basis of mycobacterial inhibition by cyclomarin A. J Biol Chem 288:30883–30891. doi: 10.1074/jbc.M113.493767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gavrish E, Sit CS, Cao S, Kandror O, Spoering A, Peoples A, Ling L, Fetterman A, Hughes D, Bissell A, Torrey H, Akopian T, Mueller A, Epstein S, Goldberg A, Clardy J, Lewis K. 2014. Lassomycin, a ribosomally synthesized cyclic peptide, kills Mycobacterium tuberculosis by targeting the ATP-dependent protease ClpC1P1P2. Chem Biol 21:509–518. doi: 10.1016/j.chembiol.2014.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nessar R, Cambau E, Reyrat JM, Murray A, Gicquel B. 2012. Mycobacterium abscessus: a new antibiotic nightmare. J Antimicrob Chemother 67:810–818. doi: 10.1093/jac/dkr578. [DOI] [PubMed] [Google Scholar]
- 43.Godfrey K. 1985. Statistics in practice. Comparing the means of several groups. N Engl J Med 313:1450–1456. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.