

Abstract
Objective:
To determine whether immunoglobulin G (IgG) from patients with Lambert-Eaton myasthenic syndrome (LEMS) decreases action potential–evoked synaptic vesicle exocytosis, and whether the effect is mediated by P/Q-type voltage-gated calcium channels (VGCCs).
Methods:
IgG was obtained from 4 patients with LEMS (3 males, 1 female), including 2 patients with lung malignancy. Antibodies against P/Q-type VGCCs were detected in all 4 patients, and against N-type VGCCs in 2. We incubated neuronal cultures with LEMS IgG and determined the size of the total recycling pool of synaptic vesicles and the rate of action potential–evoked exocytosis using fluorescence imaging of the amphiphilic dye SynaptoRed C1. Pooled IgG from healthy volunteers was used as a control. We repeated the experiments on synapses lacking P/Q-type calcium channels from a Cacna1a knockout mouse to determine whether these channels account for the pathogenic effect of LEMS IgG.
Results:
LEMS IgG had no effect on the total recycling pool size but significantly reduced the rate of action potential–evoked synaptic exocytosis in wild-type neurons when compared with neurons treated with control IgG. In contrast, LEMS IgG had no effect on the rate of synaptic vesicle exocytosis in neurons lacking P/Q-type channels.
Conclusions:
These data provide direct evidence that LEMS IgG inhibits neurotransmitter release by acting on P/Q-type VGCCs.
Lambert-Eaton myasthenic syndrome (LEMS) is an important cause of skeletal muscle weakness. Antibodies against P/Q-type voltage-gated calcium channels (VGCCs) are found in 90% of patients.1,2 Because P/Q-type VGCCs have an important role in triggering acetylcholine release at the neuromuscular junction,3 it has been proposed that muscle weakness is causally related to antibody binding to these channels.4 Passive transfer experiments show that LEMS immunoglobulin G (IgG) leads to a reduction in postsynaptic endplate potentials.5 However, endplate potentials are an indirect readout of presynaptic neurotransmitter release. Moreover, it is not known whether all the effects of LEMS IgG are mediated by a specific effect on P/Q-type channels. It remains possible that different antibodies act on VGCCs and on neurotransmitter release. Approximately 30% of patients with LEMS also have antibodies against N-type channels2 but the significance of these antibodies is unknown. LEMS IgG has been shown to reduce current through HEK cells stably transfected with P/Q-type but not N-type VGCCs.6 However, LEMS IgG has also been reported to decrease N-type currents in small cell lung cancer cells.7
To obtain a direct insight into the mechanism by which neurotransmission is altered, we examined the effect of LEMS IgG on synaptic vesicle exocytosis in neuronal cultures from rats and wild-type mice, as well as from mice lacking P/Q-type channels. We measured exocytosis using a fluorescent amphiphilic dye, which partitions into cell membranes and becomes trapped in synaptic vesicles. The rate of fluorescence loss from synaptic boutons upon stimulation provides a sensitive and specific readout of vesicle exocytosis.8
METHODS
Standard protocol approvals, registrations, and patient consents.
LEMS sample collection was approved by the Oxfordshire Regional Ethical Committee A (07/Q1604/28). Each patient provided written informed consent. Animal experiments were performed in accordance with the UK Animals (Scientific Procedures) Act 1986.
IgG samples were obtained from 4 patients with LEMS (3 males, 1 female; table e-1 on the Neurology® Web site at Neurology.org), and compared with pooled IgG from healthy human controls. Two LEMS patients had lung malignancy and all had antibodies that immunoprecipitated P/Q-type VGCCs (range of titers: 128–10,755 pM, considered positive if >50 pM; table e-1). Two samples additionally immunoprecipitated N-type VGCCs.
Cell culture and imaging solutions.
Hippocampal neurons were isolated from P0–P2 rat pups or Cacna1a−/− mice and their wild-type littermates, and cultured in Neurobasal-based medium.8 Experiments were performed at room temperature 15 to 19 days after plating. The imaging solution contained (in mM) 125 NaCl, 2.5 KCl, 2 MgCl2, 2 CaCl2, 30 glucose, and 25 HEPES (pH 7.4), supplemented with 10 μM 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo[f]quinoxaline-2,3-dione and 50 μM dl-2-amino-5-phosphonopentanoic acid to block glutamate receptors.
Incubation with LEMS IgG.
Neuronal cultures were incubated with LEMS or pooled healthy control IgG (1 mg/mL) at 37°C for 16 to 20 hours before imaging. Experiments were performed blinded to the disease status of each IgG sample.
Fluorescence imaging of synaptic vesicle release in wild-type neurons.
Recycling synaptic vesicles were labeled with the fluorescent dye SynaptoRed C1 (SRC1, 200 μM) using saturating stimulation (4 trains of 120 action potentials at 30 Hz) delivered via platinum bath electrodes. After dye washout, the SRC1 fluorescence decay was monitored, initially at rest for 10 minutes, and then during 0.5-Hz field stimulation for 15 minutes. This was followed by 1,000 stimuli at 10 Hz to evoke exocytosis of all recycling vesicles. Images were acquired every 40 seconds with a QuantEM 512SC EM CCD camera (Photometrics, Tucson, AZ) (figure 1A).
Figure 1. Fluorescence measurements of evoked vesicular release in rat cultures after incubation in control or LEMS IgG.

(A) Experimental protocol showing the sequence of SRC1 incubation, stimulation to load boutons, to evoke exocytosis, and to achieve complete destaining, and fluorescence imaging. (B, C) Representative SRC1 imaging experiments in cultures treated with control (B) or with LEMS IgG (C). Fluorescence microscopy images (top) show gradual decrease of fluorescence at successive time points as indicated during the experiment. Fluorescence time courses in 2 pairs of representative boutons (arrows) are shown below in relative fluorescence units (RFU). Spontaneous and evoked destaining rates were fitted with mono-exponential curves. The specific action potential–dependent rate of destaining kAP was calculated as kAP = kEV − kSP. Scale bars: 5 μm. AP = action potential; IgG = immunoglobulin G; LEMS = Lambert-Eaton myasthenic syndrome; SRC1 = SynaptoRed C1.
Images were analyzed using ImageJ (NIH). Fluorescence was assessed by taking the integrated intensity of individual presynaptic boutons (approximately 1- to 2-μm diameter). The nonspecific residual fluorescence was measured after depleting all labeled recycling vesicles.8 The total recycling pool (TRP) size was calculated by subtracting the residual fluorescence from the initial bouton fluorescence immediately after SRC1 washout. In each bouton, the spontaneous destaining rate in the absence of stimulation (kSP) and the evoked rate during low-frequency 0.5-Hz stimulation (kEV) were calculated by fitting mono-exponential functions to the fluorescence time course. The rate of action potential–evoked fluorescence loss, kAP, which is proportional to the rate of evoked vesicle exocytosis, was calculated as kAP = kEV – kSP. Data are given as mean ± SEM and analyzed with Student 2-tailed t test.
RESULTS
Pretreatment of neuronal cultures with LEMS IgG led to a decrease in action potential–evoked synaptic vesicle exocytosis, as estimated from the rate of destaining of the amphiphilic fluorescent dye SRC1 (figures 1 and 2). Both the action potential–specific SRC1 destaining rate kAP (which is proportional to the average release probability of release-ready vesicles pv [reference 8]) and the overall SRC1 destaining rate during 0.5-Hz action potential stimulation (kEV) were reduced by approximately 23% in LEMS IgG–treated cultures, compared with neurons treated with control IgG (figures 2A and e-1A). LEMS IgG samples from all 4 patients resulted in a lower rate of exocytosis than the pooled control sample (range 16%–39%; table e-2). LEMS IgG also reduced the spontaneous SRC1 destaining rate kSP by approximately 24% (figure e-1B, table e-2). This is consistent with our recent finding that spontaneous exocytosis in the absence of action potentials is in part triggered by stochastic opening of presynaptic VGCCs.9 In contrast, LEMS IgG did not affect the relative TRP size as estimated from the magnitude of the initial SRC1 fluorescence (figure 2B, table e-2). This implies that the effect of LEMS IgG on transmitter release is mainly mediated by a reduction of vesicular release probability pv, as a direct consequence of inhibition of presynaptic VGCC function.
Figure 2. LEMS IgG reduces evoked exocytosis but not the total recycling pool size.
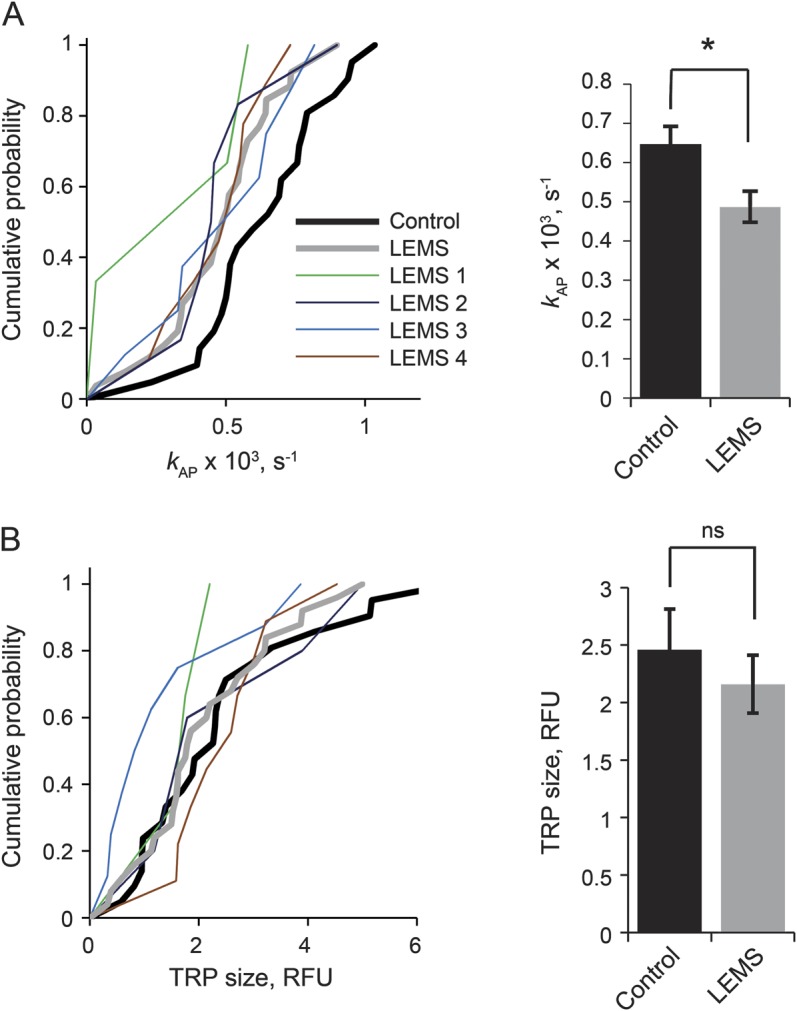
Effects of LEMS and control IgG on kAP (A) and relative TRP size (B). Left panels show cumulative distributions of mean kAP and TRP size values obtained in individual experiments (average of 10–50 boutons in each experiment). Data derived with samples obtained from each of 4 patients with LEMS are shown as thin colored lines. LEMS samples 1–4 are color-coded as in the legend, and were used in 3, 9, 8, and 6 experiments, respectively. Thick red line, pooled LEMS IgG data; thick black line, control IgG data. Right panels show the mean (±SEM) values for the pooled data (LEMS IgG n = 26 experiments, control IgG n = 21 experiments, *p < 0.05). IgG = immunoglobulin G; LEMS = Lambert-Eaton myasthenic syndrome; ns = nonsignificant; RFU = relative fluorescence units; TRP = total recycling pool.
We repeated the experiment in cultures from Cacna1a−/− mice and their wild-type littermates. We first verified that P/Q-type channels were lost in Cacna1a−/− neurons by estimating the contribution of different VGCCs to neurotransmitter release, as measured by the amplitude of evoked excitatory postsynaptic currents (EPSCs). The P/Q-type specific blocker ω-Agatoxin IVA attenuated the EPSC amplitude by approximately 70% in wild-type neurons, but did not affect EPSCs in Cacna1a−/− neurons (figure e-2), consistent with data obtained in another Cacna1a−/− strain.10 Synaptic transmission in Cacna1a−/− neurons was dependent on N-type VGCCs with a contribution from R-type VGCCs. We then tested the effect of LEMS IgG that immunoprecipitated both P/Q- and N-type VGCCs, and control IgG, on the rate of action potential–evoked synaptic vesicle exocytosis in wild-type and Cacna1a−/− neurons (figure e-3). In wild-type cultures, the rate of action potential–evoked exocytosis was decreased by approximately 60% by LEMS IgG when compared with control IgG, qualitatively consistent with the data obtained in rat cultures. In striking contrast, there was no significant effect of LEMS IgG on synaptic vesicle release in Cacna1a−/− cultures (figure 3A). Consistent with the data from rat cultures, TRP was not affected by LEMS IgG in either wild-type or Cacna1a−/− cultures (figure 3B). The LEMS IgG tested here thus required P/Q-type channels to exert an effect on exocytosis.
Figure 3. P/Q-type channels are required for inhibition of vesicular release by LEMS IgG.
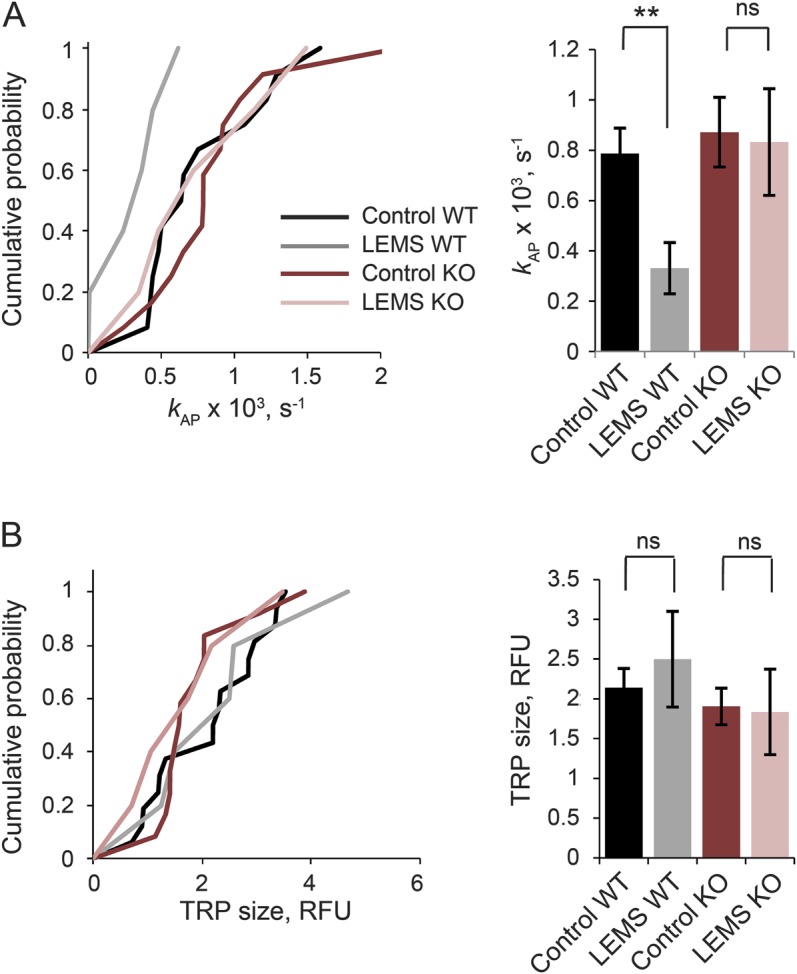
Effects of LEMS and control IgG on kAP (A) and relative TRP size (B) in WT and Cacna1a+/− (KO) neurons. Left panels show cumulative distributions of mean kAP and TRP size values obtained in individual experiments. Right panels show the mean (±SEM) values for the pooled data. Data are from 602 boutons in 15 experiments (WT, control, black line), 138 boutons in 5 experiments (WT, LEMS, gray), 303 boutons in 15 experiments (KO, control, red), and 175 boutons in 6 experiments (KO, LEMS, pink). **p < 0.01. IgG = immunoglobulin G; KO = knockout; LEMS = Lambert-Eaton myasthenic syndrome; ns = nonsignificant; RFU = relative fluorescence units; TRP = total recycling pool; WT = wild-type.
DISCUSSION
The present study demonstrates a direct effect of LEMS IgG on vesicular exocytosis via P/Q-type channels. Although a presynaptic mechanism of action of LEMS IgG has long been assumed, the available evidence to date has been indirect. Our presynaptic imaging data directly show that LEMS IgG decreases action potential–dependent synaptic vesicle release in rat and wild-type mouse neurons. We used a well-characterized model of neurotransmission, in which VGCCs have a similar role as at the neuromuscular junction. We therefore infer that LEMS IgG impairs neuromuscular transmission by reducing the rate of acetylcholine release as a direct consequence of binding to P/Q-type channels.
LEMS IgG had no effect on synaptic vesicle exocytosis when P/Q-type channels were deleted genetically despite the presence of antibodies against N-type VGCCs. We thus found no evidence for an effect of LEMS IgG on synaptic function mediated by N-type VGCCs. However, a more systematic study focusing on samples with high titers of N-type IgG is required to clarify the pathophysiologic role of antibodies directed against this channel subtype.
Supplementary Material
ACKNOWLEDGMENT
The authors are grateful to A.M. van den Maagdenberg for the gift of Cacna1a+/− mice.
GLOSSARY
- EPSC
excitatory postsynaptic current
- IgG
immunoglobulin G
- LEMS
Lambert-Eaton myasthenic syndrome
- SRC1
SynaptoRed C1
- TRP
total recycling pool
- VGCC
voltage-gated calcium channel
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
J. Spillane: design and conceptualization of study, performed experiments, analysis and interpretation of data, drafting and revising of manuscript. Y. Ermolyuk: design of experiments, analysis of data, revising manuscript. M. Cano-Jaimez: preparation of neuronal cultures, revising manuscript. B. Lang: preparation of IgG samples, design of experiments, revising manuscript. A. Vincent: design of experiments, revising manuscript. K.E. Volynski: design and conceptualization of study, analysis and interpretation of data, drafting and revising of manuscript. D.M. Kullmann: design and conceptualization of study, analysis and interpretation of data, drafting and revising manuscript.
STUDY FUNDING
Supported by the Myasthenia Gravis Association, the Medical Research Council, the Wellcome Trust, Epilepsy Research UK, and the Oxford NIHR Biomedical Research Centre.
DISCLOSURE
J. Spillane received a research grant from the Myasthenia Gravis Association UK during the course of this study, has received funding for travel to scientific conferences from the Guarantors of Brain and has received honoraria from educational institutions for teaching. Y. Ermolyuk and M. Cano-Jaimez report no disclosures relevant to the manuscript. B. Lang holds a grant from Epilepsy Research UK (ERUK) and holds a patent with Oxford University for VGKC antibodies, licensed to Euroimmun AG. A. Vincent holds a grant from the NIHR, holds a patent with Oxford University for VGKC antibodies, and receives royalties from Euroimmun AG for LG1 and CASPR2 assays and from Athena Diagnostics for MuSK antibody assays. K. Volynski and D. Kullmann report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Lennon VA, Kryzer TJ, Griesmann GE, et al. Calcium-channel antibodies in the Lambert-Eaton syndrome and other paraneoplastic syndromes. N Engl J Med 1995;332:1467–1474. 10.1056/NEJM199506013322203. [DOI] [PubMed] [Google Scholar]
- 2.Motomura M, Lang B, Johnston I, Palace J, Vincent A, Newsom-Davis J. Incidence of serum anti-P/O-type and anti-N-type calcium channel autoantibodies in the Lambert-Eaton myasthenic syndrome. J Neurol Sci 1997;147:35–42. [DOI] [PubMed] [Google Scholar]
- 3.Protti DA, Uchitel OD. Transmitter release and presynaptic Ca2+ currents blocked by the spider toxin omega-Aga-IVA. Neuroreport 1993;5:333–336. [DOI] [PubMed] [Google Scholar]
- 4.Titulaer MJ, Lang B, Verschuuren JJ. Lambert-Eaton myasthenic syndrome: from clinical characteristics to therapeutic strategies. Lancet Neurol 2011;10:1098–1107. 10.1016/S1474-4422(11)70245-9. [DOI] [PubMed] [Google Scholar]
- 5.Lang B, Newsom-Davis J, Wray D, Vincent A, Murray N. Autoimmune aetiology for myasthenic (Eaton-Lambert) syndrome. Lancet 1981;2:224–226. [DOI] [PubMed] [Google Scholar]
- 6.Pinto A, Iwasa K, Newland C, Newsom-Davis J, Lang B. The action of Lambert-Eaton myasthenic syndrome immunoglobulin G on cloned human voltage-gated calcium channels. Muscle Nerve 2002;25:715–724. 10.1002/mus.10087. [DOI] [PubMed] [Google Scholar]
- 7.Meriney SD, Hulsizer SC, Lennon VA, Grinnell AD. Lambert-Eaton myasthenic syndrome immunoglobulins react with multiple types of calcium channels in small-cell lung carcinoma. Ann Neurol 1996;40:739–749. 10.1002/ana.410400510. [DOI] [PubMed] [Google Scholar]
- 8.Ermolyuk YS, Alder FG, Henneberger C, Rusakov DA, Kullmann DM, Volynski KE. Independent regulation of Basal neurotransmitter release efficacy by variable Ca2+ influx and bouton size at small central synapses. PLoS Biol 2012;10:e1001396. 10.1371/journal.pbio.1001396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ermolyuk YS, Alder FG, Surges R, et al. Differential triggering of spontaneous glutamate release by P/Q-, N- and R-type Ca(2+) channels. Nat Neurosci 2013;16:1754–1763. 10.1038/nn.3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jun K, Piedras-Rentería ES, Smith SM, et al. Ablation of P/Q-type Ca(2+) channel currents, altered synaptic transmission, and progressive ataxia in mice lacking the alpha(1A)-subunit. Proc Natl Acad Sci USA 1999;96:15245–15250. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.