

Abstract
Objective:
To determine how sleep-disordered breathing, nocturnal hypoxia, and changes in sleep architecture in the elderly may be related to the development of the neuropathologic correlates of dementia.
Methods:
The Honolulu-Asia Aging Study is a prospective cohort study of Japanese American men in Honolulu, HI. We examined brain lesions at autopsy (Braak stage, neurofibrillary tangle and neuritic plaque counts, microinfarcts, generalized brain atrophy, lacunar infarcts, Lewy bodies [LBs], neuronal loss and gliosis in the locus ceruleus) in 167 participants who underwent polysomnography in 1999–2000 (mean age, 84 years) and died through 2010 (mean 6.4 years to death). Polysomnography measures included the apnea-hypopnea index, duration of apnea or hypopnea, duration of hypoxemia, minimum oxygen saturation (SpO2), duration of slow-wave sleep (SWS, non-REM stage N3), and arousals.
Results:
Sleep duration with SpO2 <95% was associated with higher levels of microinfarcts (adjusted odds ratio [OR] 3.88, 95% confidence interval [CI] 1.10–13.76, comparing the highest to lowest quartiles of %sleep with SpO2 <95%). Greater SWS duration was associated with less generalized atrophy (adjusted OR 0.32, 95% CI 0.10–1.03, comparing highest to lowest quartiles of %sleep in SWS). LBs were less common with greater %sleep with SpO2 <95% (adjusted OR 0.17, 95% CI 0.04–0.78, comparing highest to lowest quartiles). Higher minimum SpO2 during REM sleep was associated with less gliosis and neuronal loss in the locus ceruleus. Cognitive scores declined less among men with greater SWS duration.
Conclusions:
The findings support a role for lower nocturnal oxygenation and SWS in the development of microinfarcts and brain atrophy, but not Alzheimer lesions or LBs.
Sleep-disordered breathing (SDB),1 nocturnal hypoxia,1 and changes in sleep architecture2,3 may lead to dementia in the elderly. A causal association is supported by the finding that continuous positive airway pressure treatment in patients with obstructive sleep apnea (OSA) may improve cognition, even after dementia has developed.4
It remains unclear how SDB, nocturnal hypoxia, and sleep architecture may be related to the development of brain lesions associated with dementia. Hypoxia and altered brain perfusion during sleep may cause neuronal injury, leading to impaired cognitive functioning. Alternatively, abnormalities in regions such as the locus ceruleus may alter sleep architecture or control of breathing. We examined associations between sleep characteristics and neuropathologic lesions at autopsy in 167 men who underwent polysomnography (PSG) in 1999–2000 and died through 2010.
METHODS
Study population.
The Honolulu-Asia Aging Study (HAAS) is a prospective cohort of 3,374 Japanese American men followed since 1965 as part of the Honolulu Heart Program (HHP).5,6 The HAAS was established in 1991 to study aging-related conditions, with a focus on brain diseases (figure). Participants (aged 71–93 years) represented approximately 80% of the surviving HHP cohort.7 The HAAS began at examination 4 of the HHP cohort. Subsequent examinations occurred every 2 to 3 years thereafter, with the last completed in 2012. Brain autopsies were completed on approximately 18% of HAAS decedents through 2010,8,9 including 15% of those without dementia and 39% of those identified as having dementia during the final years of life. Except for the higher rates of dementia, those autopsied were representative of all deaths among cohort members.
Figure. Derivation of the analytic sample.
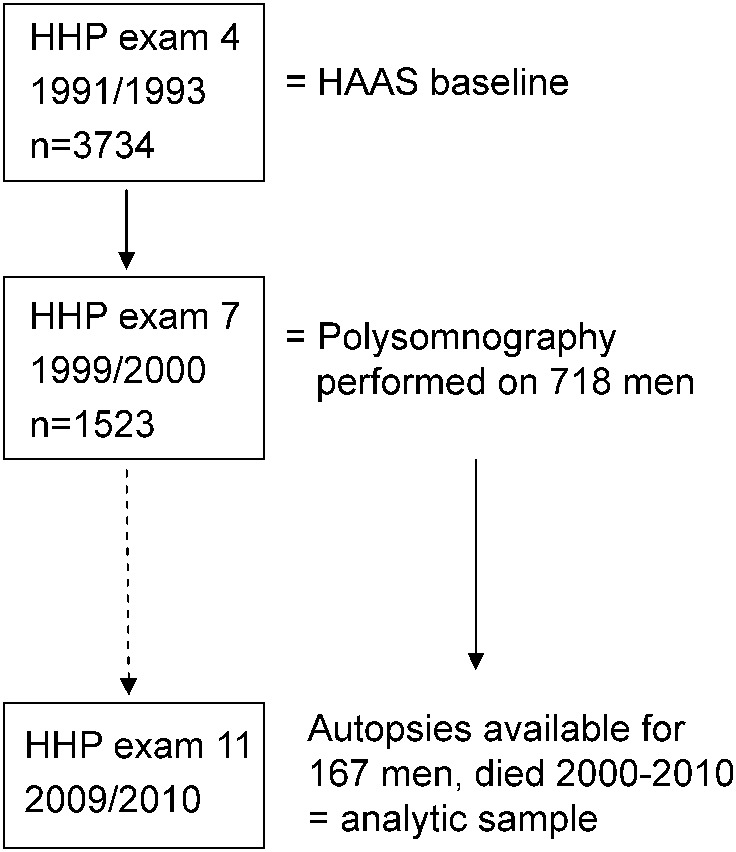
HAAS = Honolulu-Asia Aging Study; HHP = Honolulu Heart Program.
Overnight PSG was performed on 718 of 1,523 men who participated in examination 7 (1999–2000). Excluded from PSG recruitment were men with moderate or severe dementia10 (n = 37) and those receiving continuous positive airway pressure or oxygen during sleep (n = 13). Of the remaining 1,473, 49% completed the PSG.11 Of these 718 men, 502 died through 2010, and, of these, 167 were included in the autopsy substudy and in this analysis. Data were made available through the National Institute on Aging.
Standard protocol approvals, registrations, and patient consents.
The Kuakini Medical Center institutional review board reviewed and approved this study. Informed consents for participation were obtained at each HAAS examination. Consents for autopsy were obtained from next of kin.
Polysomnography.
PSGs were done in the home where sleep typically occurred. Details of the PSG protocol have been described.11 We examined apnea-hypopnea index (AHI), duration of apnea or hypopnea, duration of hypoxemia, minimum oxygen saturation during REM and non-REM sleep, duration of slow-wave (SWS, non-REM stage N3) and REM sleep, and arousals. SWS scoring was based on strict amplitude and frequency criteria.
The AHI (number of apneas plus hypopneas per hour) was examined in quartiles and dichotomously (</≥15 events/h). Apneas were defined as cessation of airflow for ≥10 seconds and hypopneas as a decrease in airflow or thoracoabdominal excursion by ≥30% for ≥10 seconds, both with ≥3% decrease in oxygen saturation by finger pulse oximetry (SpO2). Duration of apnea or hypopnea (%sleep in apnea or hypopnea) was examined in tertiles and predefined categories (<2.3%, 2.3% to <7.0%, ≥7.0%).1 Duration of hypoxemia, defined as %sleep with SpO2 <95% (and alternatively as <90%), and minimum SpO2, were examined in quartiles and as continuous variables.
Percentage of sleep in SWS (SWS%) or REM (REM%) was evaluated in quartiles and continuously. Analyses of SWS and REM excluded studies with unreliable stage information (n = 16 and 13, respectively). An arousal index was calculated as the number of arousals per hour of sleep, excluding 50 studies with unreliable arousal data.
Neuropathologic lesions.
Details of the autopsy protocol have been described.8,9 We examined the following categories of lesions without considering clinical condition: microinfarcts, lesions related to Alzheimer disease (AD) (Braak stage,12 average neocortical neurofibrillary tangle [NFT] and neuritic plaque [NP] counts), generalized brain atrophy, lacunar infarcts, Lewy bodies (LBs), and neuronal loss and gliosis in the locus ceruleus. Microinfarcts were counted from 18 brain regions, amyloid NPs and NFTs from 4 neocortical regions, LBs from the substantia nigra, locus ceruleus, amygdala, medulla, and neocortical regions, and lacunar infarcts from the entire brain. Moderate or high levels of microinfarcts were defined as ≥2 temporally remote microinfarcts across 8 neocortical regions, and/or ≥2 microinfarcts in the left and right caudate, putamen, and thalamus (6 deep gray nuclei). Microscopic cystic lesions with surrounding astrocytic gliosis in the deep gray matter were treated as microinfarcts. Subcortical lacunes observed at gross examination in the deep gray or white matter (estimated volumes, 0.01–0.2 mL) were also viewed as microvascular lesions. A total of 3 such lacunes were counted as equivalent to one subcortical microinfarct. Such subcortical lacunes contributed minimally to the definition of microinfarcts.8 Generalized brain atrophy was based on an index previously described.6 A summary measure of neuronal loss and gliosis in the locus ceruleus bilaterally was used, giving more weight to neuronal loss.
Four-level indices were used for each lesion type, based on previously demonstrated associations with cognitive impairment in the HAAS.6,8 Levels for each lesion were defined for comparability with Braak score frequencies (negligible = 0–3, low = 4, moderate = 5, high = 6), because this index is widely used in studies linking brain lesions to cognitive impairment.6,8,12 For our analyses, each lesion was considered present if moderate or high levels were identified. Given a lower prevalence, LBs were defined as present if any were observed.
Cognitive measures.
Cognitive assessments occurred at each HAAS examination according to a standardized protocol,6,7 using the 100-point Cognitive Abilities Screening Instrument (CASI).13,14 We evaluated decline in CASI score as a continuous variable, defined as the difference between the last available and the examination 7 scores. In prior HAAS analyses, CASI scores <74 had approximately 80% sensitivity and 90% specificity for identifying dementia.15
Covariates.
Covariates included age at death (years) and the following from examination 4: body mass index (BMI, kg/m2), smoking (never, past, current), diabetes (serum glucose ≥126 mg/dL fasting or ≥200 mg/dL 2 hours after a 75-g oral glucose load, or use of antidiabetic medications), hemoglobin (g/dL), and fasting plasma high-density lipoprotein cholesterol level (mg/dL). Midlife systolic blood pressure (SBP, mm Hg) was estimated as the mean from the first 3 HHP examinations.16 Genotyping for APOE ε4 (1 or 2 alleles vs none) was performed at Duke University, Durham, NC, using conventional methods.17
Data analyses.
We compared participant characteristics according to sleep characteristics using analysis of variance for continuous variables and χ2 tests for categorical variables. Correlations among sleep variables were assessed using Spearman correlation coefficients (ρ). We used logistic regression to estimate odds ratios (ORs) for lesions according to sleep features.
Two multivariable-adjusted models were considered for analyses of microinfarcts and atrophy. Model 1 adjusted for age at death, smoking, and BMI. Model 2 additionally adjusted for other possible confounders: hemoglobin, high-density lipoprotein cholesterol, diabetes, and midlife SBP.16
Analyses of Alzheimer lesions adjusted for age at death, APOE ε4, smoking, BMI, hemoglobin, diabetes, and midlife SBP. For analyses of gliosis and neuronal loss in the pons and for LBs, model 1 adjusted for age at death and smoking. Model 2 additionally adjusted for BMI, hemoglobin, diabetes, and midlife SBP. Separate models also adjusted for age at the PSG.
Secondary analyses excluded 39 men who died early in follow-up (≤3 years) or who had baseline (examination 7) CASI scores <74, to address the possibility of preexisting brain lesions accounting for observed sleep features. Models incorporating joint terms examined interactions between SWS and hypoxia. Likelihood ratio tests (LRTs) were used to assess effect modification, adjusting for age at death. In generalized linear models, we examined how CASI score decline varied according to sleep characteristics, adjusting for age at death, baseline CASI score, and time to last available CASI, using the difference between the last available and the examination 7 scores as the dependent variable. Two-sided p values are reported; p < 0.05 was considered statistically significant. Analyses were performed using SAS version 9.3 (SAS Institute, Inc., Cary, NC).
RESULTS
Among 167 men who completed the PSG and underwent autopsy, mean age was 83.7 (79–95) years at PSG, average time to death from PSG was 6.4 (SD, 2.8) years, and 23 (13.8%) died within the first 3 years of follow-up. Mean baseline CASI score was 83.5 (range, 61.6–98.5), and 25 men (15.0%) had scores <74. An AHI ≥15 was observed in 55.1% (n = 92). Higher BMI was associated with nocturnal hypoxemia (table 1). Other participant characteristics, including examination 7 CASI scores, were not significantly associated with hypoxemia, and none were associated with SWS duration (table 2). Self-reported history of chronic lung disease (available from examination 4) was rare (n = 19/167).
Table 1.
Participant characteristics according to sleep time with SpO2 <95%
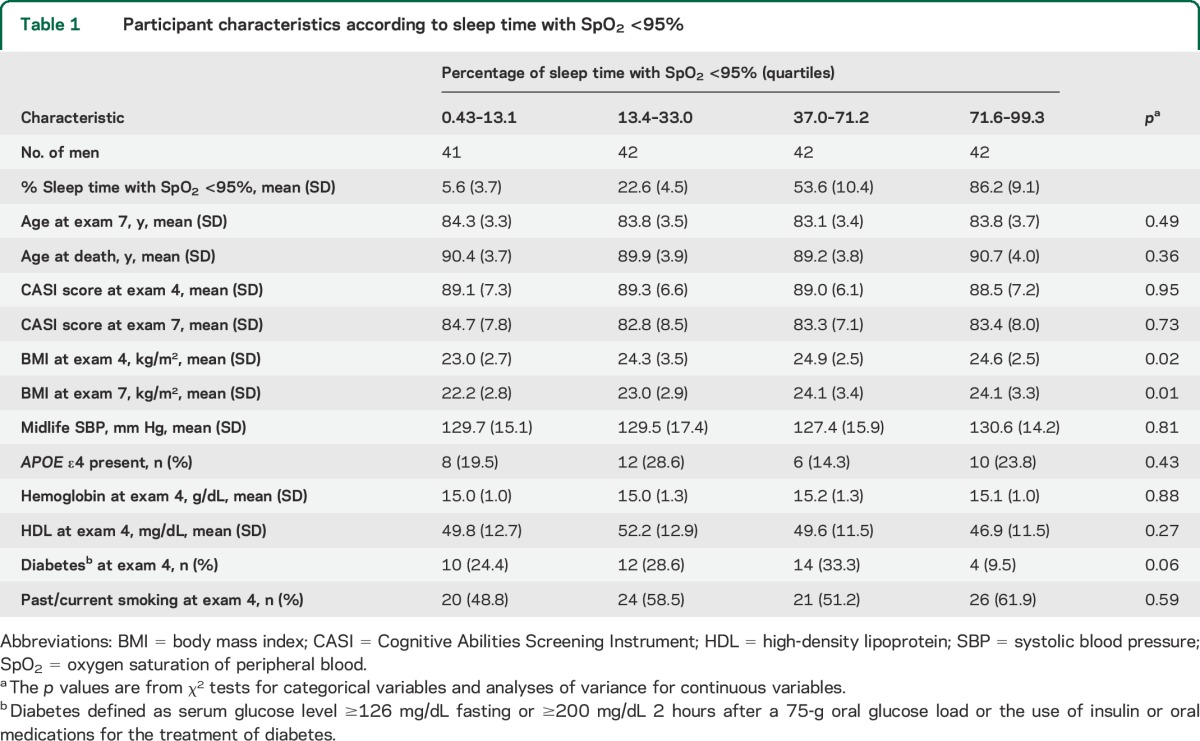
Table 2.
Participant characteristics according to duration of SWS
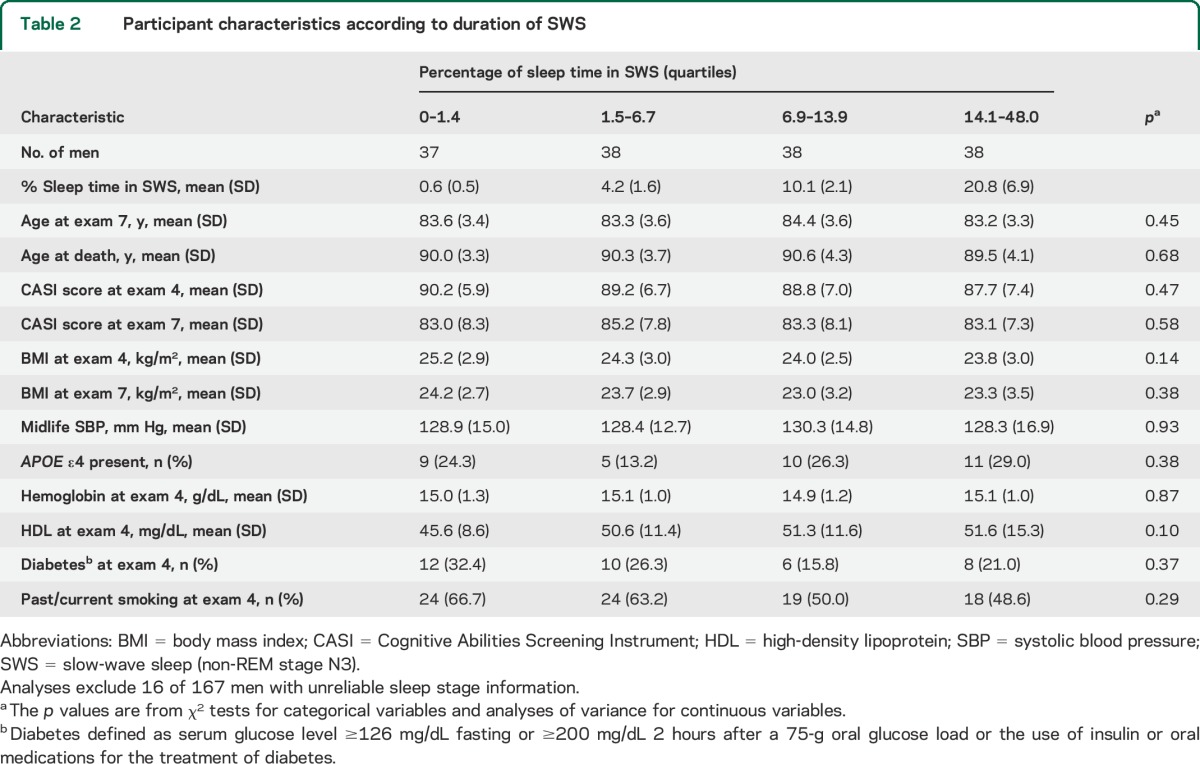
The AHI was correlated with %sleep with SpO2 <95% (ρ = 0.26, p < 0.001) and the arousal index (ρ = 0.52, p < 0.001), but not with SWS% (ρ = −0.12, p = 0.13). SWS% was not correlated with %sleep with SpO2 <95% (ρ = 0.07, p = 0.40).
High/moderate levels of brain lesions were observed as follows: microinfarcts, 19.8% (n = 33); atrophy, 29.9% (n = 50); lacunar infarcts, 21.0% (n = 35); Braak stage 5/6, 18.6% (n = 31); NFTs, 15% (n = 25); NPs, 31.1% (n = 52); and gliosis and neuron loss in the locus ceruleus, 27.5% (n = 46). LBs were present in 13.8% (n = 23). The expected associations of Braak stage with NFTs and NPs were observed. Lacunar infarcts were correlated with microinfarct levels (ρ = 0.22, p = 0.004). Brain atrophy was correlated with neuron loss and gliosis in the locus ceruleus (ρ = 0.26, P ≤ 0.001).
Greater %sleep with SpO2 <95% was associated with microinfarcts at autopsy (table 3). Results were comparable examining absolute sleep time with SpO2 <95% (model 2 OR, 95% confidence interval [CI] was 3.15, 0.89–11.17 comparing >220.0 vs <41.7 minutes). For each increase in %sleep with SpO2 <95% of 31.7% (SD unit), the model 2 OR (95% CI) was 1.53 (1.02–2.31). Percentage of sleep with SpO2 <90% (quartiles) was also associated with microinfarcts, although less consistently. The ORs (95% CIs) were 3.58 (1.03–12.43), 2.87 (0.82–10.03), and 2.34 (0.64–8.52) for 0.1%–0.6%, >0.6%–3.8%, and >3.8%–50.4% of sleep with SpO2 <90%, as compared with <0.1%, adjusting for age at death. Other sleep features were not significantly associated with microinfarcts.
Table 3.
ORs for brain lesions at autopsy, according to sleep time with SpO2 <95%
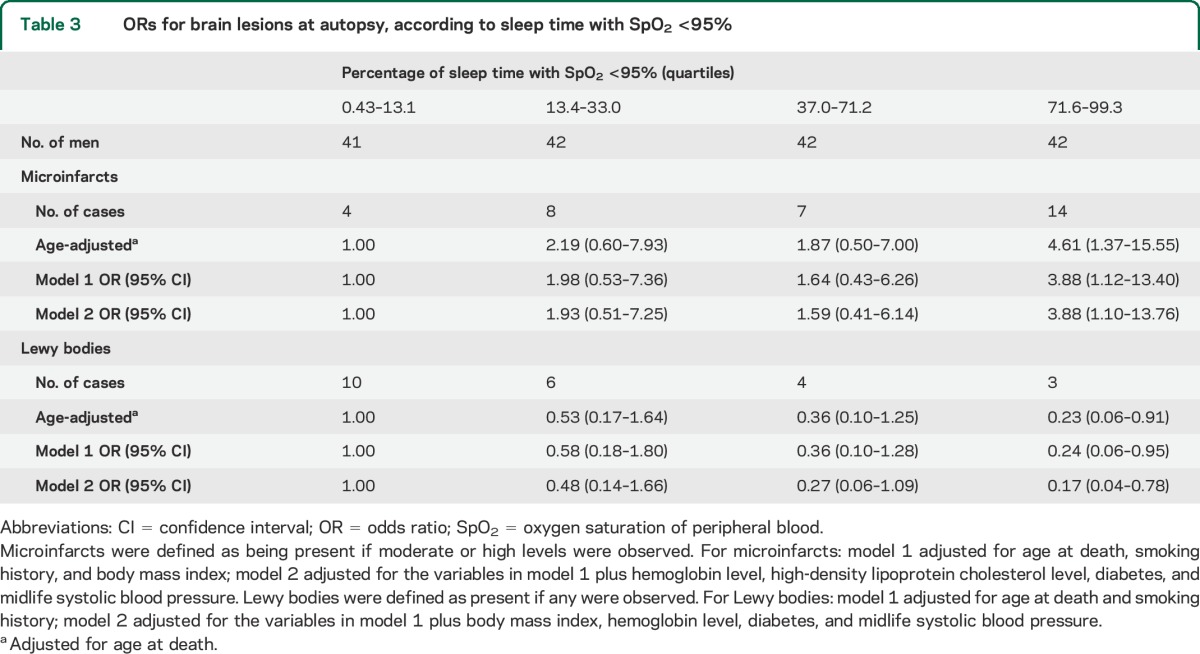
LBs were less common among men with greater %sleep with reduced SpO2 (table 3). Examining absolute sleep time, the model 2 OR (95% CI) was 0.16 (0.03–0.88), comparing >220.0 vs <41.7 minutes with SpO2 <95%. For each increase in %sleep with SpO2 <95% of 31.7% (SD unit), the model 2 OR was 0.45 (95% CI, 0.24–0.82). Using %sleep with SpO2 <90% as a continuous variable (per SD unit, 7.1%), the model 2 OR was 0.08 (95% CI, 0.01–0.97). Other sleep characteristics were not significantly associated with LBs.
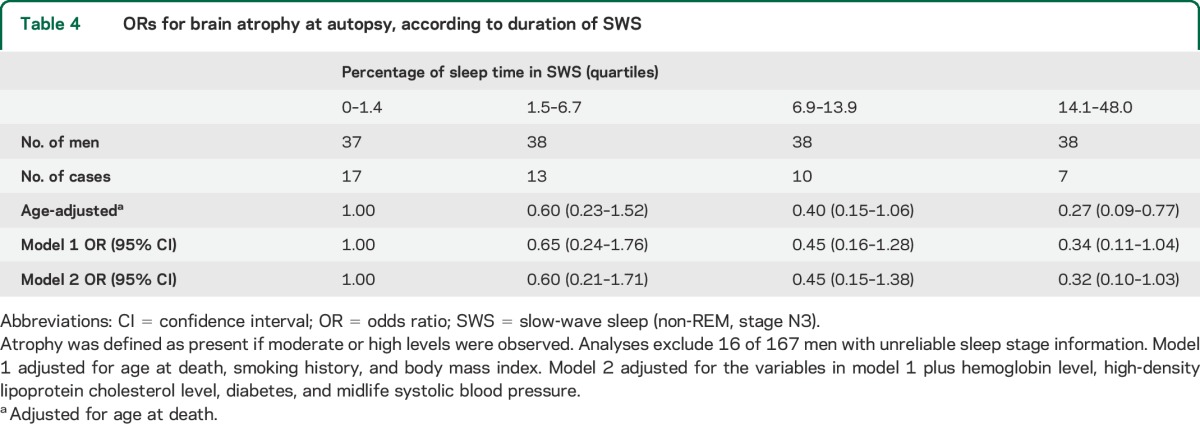
Higher SWS% was associated with less brain atrophy (table 4). Results were similar examining total time in SWS. The model 2 OR (95% CI) was 0.30 (0.10–0.94) for men with >47.0 minutes in SWS vs <4.9 minutes. For each increase in SWS% of 8.5% (SD unit), the model 2 OR was 0.62 (95% CI, 0.39–1.01). Other sleep measures were not significantly associated with atrophy.
Table 4.
ORs for brain atrophy at autopsy, according to duration of SWS
Higher minimum SpO2 during REM was associated with lower levels of gliosis and neuronal loss in the locus ceruleus (not shown). The model 1 OR (95% CI) was 0.21 (0.06–0.66) for men with a minimum SpO2 during REM of 90%–96% vs 42%–81%. Examining minimum SpO2 during REM continuously (per SD unit, 8.1%), the model 1 OR (95% CI) was 0.70 (0.49–1.00). Other sleep variables were not significantly associated with gliosis and neuronal loss in the locus ceruleus.
There were no consistent associations between the sleep features and Braak stage, NPs, NFTs, or lacunar infarcts. AHI, duration of apnea or hypopnea, %sleep in REM, and the arousal index were not consistently associated with any of the neuropathologic endpoints.
Results were similar in secondary analyses excluding 39 participants who died within the first 3 years of follow-up or who had examination 7 CASI scores <74. The model 1 OR was 5.91 (95% CI, 1.14–30.69) for microinfarcts and 0.07 (95% CI, 0.01–0.64) for LBs, comparing the highest to lowest quartiles of %sleep with SpO2 <95%. The model 1 OR was 0.26 (95% CI, 0.07–1.00) for generalized atrophy, comparing the highest to lowest quartiles of SWS duration. Findings were also comparable excluding men with a self-reported history of Parkinson disease, stroke, or TIA at examination 7 (n = 11) or those with a history of chronic lung disease at examination 4 (n = 19). Adjusting for age at examination 7 also did not affect the overall results.
The association of higher SWS duration with less generalized atrophy appeared stronger among men with lower oxygen saturation levels, although there was no evidence for statistical effect modification (LRT, p = 0.37). Among men with ≥37.0% of sleep with SpO2 <95%, the OR for atrophy was 0.12 (95% CI, 0.02–0.67), comparing the highest to lowest SWS quartiles, adjusting for age at death. By contrast, among men with <37.0% of sleep with SpO2 <95%, the corresponding OR was 0.50 (95% CI, 0.13–2.01). Although limited by small numbers, there was no evidence for interaction between SWS (</≥6.9%) and nocturnal hypoxia (quartiles) in analyses of microinfarcts (LRT, p = 0.75).
Cognitive scores declined less during follow-up among men with greater SWS duration. Mean adjusted declines in scores (SE) were 18.5 (3.2), 19.6 (2.8), 15.0 (2.8), and 10.2 (3.0) for the lowest to highest quartiles of SWS duration, although the association was not statistically significant (p = 0.12). Nocturnal hypoxemia, AHI, and duration of sleep in apnea or hypopnea were not associated with cognitive decline.
DISCUSSION
In this prospective cohort, men with lower SpO2 during sleep were more likely to have higher levels of microinfarcts, while those with less SWS had more brain atrophy at autopsy. Those with greater hypoxemia during REM also had more gliosis and neuronal loss in the locus ceruleus. In contrast, none of the sleep measures were significantly associated with the lesions of AD, and LBs were less common in those with lower SpO2 during sleep. The observed associations remained consistent adjusting for potential confounders and excluding participants who died early in follow-up and those with lower baseline cognitive scores, supporting the inference that the sleep features may have preceded the development of the lesions. Furthermore, decline in cognitive scores during follow-up was attenuated among men with higher levels of SWS, suggesting that SWS may protect against cognitive decline, possibly through a reduction in brain atrophy.
The finding of less atrophy with increased SWS appeared stronger among men with longer durations of nocturnal hypoxemia. Hypoxia may lead to microinfarcts, which in turn contribute to the development of brain atrophy and cognitive impairment before the development of dementia,18 but this process may be attenuated if SWS is preserved.
AHI, a measure used in standard definitions of OSA,19 and arousals were not associated with any of the lesions examined, whereas oxygen saturation level was associated with microinfarcts, the major lesion of vascular dementia,6 and a precursor in the development of brain atrophy and cognitive impairment.18 Together with the correlation noted between AHI and SpO2, our findings support the idea that hypoxia may be the main feature of OSA affecting cognition.1,20
Prior studies of SDB have generally shown an increased risk of dementia, but none have examined neuropathologic outcomes. In a study of elderly women, higher AHI and duration of sleep in apnea or hypopnea were associated with cognitive impairment.1 Prior imaging studies further support a role for OSA in the development of brain atrophy21,22 and cerebral white matter disease.23–25
SWS has not been previously examined as a risk factor for the pathologic correlates of dementia. SWS duration tends to decline with age26,27 and may be particularly important in the formation of declarative memory.28–30 Locus ceruleus–mediated increases in cortical norepinephrine during prolonged wakefulness appear to promote SWS, supporting a restorative role for SWS with increased sleep need.31
While we did not find associations between sleep features and Alzheimer lesions, others have suggested a link between sleep quality and AD. The APOE ε4 allele has been associated with OSA,32 and the adverse cognitive effects of OSA may be worse among allele carriers.33,34 Studies in mice have demonstrated increased β-amyloid clearance during sleep.35 In addition, sleep fragmentation appears to increase AD risk,2 and the effect of APOE ε4 on AD risk appears to be reduced with better sleep consolidation.3 While sleep stages were not measured in that study, consolidation may reflect enhanced duration of SWS, which, given our findings, may lead to less brain atrophy and cognitive impairment.
Our finding of an inverse association between oxygen saturation level and LB presence was unexpected and deserves further evaluation. Smoking has been associated with a reduced risk of LBs36 but not clinical dementia with LBs.37 Avoidance of severe hypoxia during REM, by contrast, appeared to protect against neuronal degeneration in the locus ceruleus, an area involved in alertness and control of sleep architecture. In animal studies, hypoxia38 and extended wakefulness39 lead to neuronal degeneration in the locus ceruleus, and damage to this area is seen in Parkinson disease and REM sleep behavior disorder.40 It remains unclear to what extent neuronal changes in this area may either lead to or result from changes in sleep architecture and SDB.
Strengths of our study include its prospective design, adjustment for potential confounders, standardized measurements of sleep variables, and detailed neuropathologic assessments. Certain limitations should be considered. Causes of nocturnal hypoxemia were unclear, and daytime oxygen saturation measures were unavailable. In addition, approximately 49% of participants completed the PSG, which may contribute to selection bias, because these men were more educated and functional at baseline.11 Participants who came to autopsy also may not be comparable to the entire HAAS cohort. Lastly, while analyses excluded those with low baseline cognitive scores and those who died early in follow-up, it remains possible that development of brain lesions preceded the PSG findings.
Reduced oxygen saturation levels may contribute to the development of microinfarcts, the major pathologic correlate of vascular dementia, and attenuate LB formation. This latter finding is unexpected and warrants additional study. Furthermore, SWS appears to protect against brain atrophy, and this may translate into a reduced risk of cognitive decline. Our findings suggest that hypoxia during sleep and reductions in SWS may contribute to the major pathologic processes underlying cognitive decline in the elderly. How SWS may have a restorative role in brain function, and whether prevention of nocturnal hypoxia may reduce the risk of cognitive decline, require additional investigation.
ACKNOWLEDGMENT
The authors are indebted to the participants in the Honolulu-Asia Aging Study and their families, for their outstanding commitment and cooperation, and to the entire Honolulu-Asia Aging Study staff, for their expert, unfailing assistance. The authors also acknowledge the expert support of Susan Surovec and Jean Arnold at the Sleep Reading Center.
GLOSSARY
- AD
Alzheimer disease
- AHI
apnea-hypopnea index
- BMI
body mass index
- CASI
Cognitive Abilities Screening Instrument
- CI
confidence interval
- HAAS
Honolulu-Asia Aging Study
- HHP
Honolulu Heart Program
- LB
Lewy body
- LRT
likelihood ratio test
- NFT
neurofibrillary tangle
- NP
neuritic plaque
- OR
odds ratio
- OSA
obstructive sleep apnea
- PSG
polysomnography
- SBP
systolic blood pressure
- SDB
sleep-disordered breathing
- SpO2
oxygen saturation as measured by pulse oximetry
- SWS
slow-wave sleep
AUTHOR CONTRIBUTIONS
Dr. Gelber drafted the manuscript and contributed to the study concept, study design, and the analysis and interpretation of data. Dr. Gelber conducted the statistical analysis. Drs. Redline, Ross, Petrovitch, Sonnen, Zarow, Uyehara-Lock, Masaki, Launer, and White revised the manuscript for content, and contributed to the interpretation of data. Dr. White also contributed to the study concept and design.
STUDY FUNDING
Supported by the National Institute on Aging (NIA) (R03 AG046614, U01 AG046871, R01 AG017155, U01 AG019349); the Intramural Research Program, NIA; the Alzheimer's Association (ZEN-12-239028); the Hawaii Community Foundation; and the Office of Research and Development, Department of Veterans Affairs Pacific Islands Health Care System. The information contained in this article does not necessarily reflect the position or the policy of the United States Government, and no official endorsement should be inferred.
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Yaffe K, Laffan AM, Harrison SL, et al. Sleep-disordered breathing, hypoxia, and risk of mild cognitive impairment and dementia in older women. JAMA 2011;306:613–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lim AS, Kowgier M, Yu L, Buchman AS, Bennett DA. Sleep fragmentation and the risk of incident Alzheimer's disease and cognitive decline in older persons. Sleep 2013;36:1027–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lim AS, Yu L, Kowgier M, Schneider JA, Buchman AS, Bennett DA. Modification of the relationship of the apolipoprotein E epsilon4 allele to the risk of Alzheimer disease and neurofibrillary tangle density by sleep. JAMA Neurol 2013;70:1544–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ancoli-Israel S, Palmer BW, Cooke JR, et al. Cognitive effects of treating obstructive sleep apnea in Alzheimer's disease: a randomized controlled study. J Am Geriatr Soc 2008;56:2076–2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Syme SL, Marmot MG, Kagan A, Kato H, Rhoads G. Epidemiologic studies of coronary heart disease and stroke in Japanese men living in Japan, Hawaii and California: introduction. Am J Epidemiol 1975;102:477–480. [DOI] [PubMed] [Google Scholar]
- 6.Gelber RP, Launer LJ, White LR. The Honolulu-Asia Aging Study: epidemiologic and neuropathologic research on cognitive impairment. Curr Alzheimer Res 2012;9:664–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.White L, Petrovitch H, Ross GW, et al. Prevalence of dementia in older Japanese-American men in Hawaii: the Honolulu-Asia Aging Study. JAMA 1996;276:955–960. [PubMed] [Google Scholar]
- 8.White L. Brain lesions at autopsy in older Japanese-American men as related to cognitive impairment and dementia in the final years of life: a summary report from the Honolulu-Asia Aging Study. J Alzheimers Dis 2009;18:713–725. [DOI] [PubMed] [Google Scholar]
- 9.Petrovitch H, Ross GW, Steinhorn SC, et al. AD lesions and infarcts in demented and non-demented Japanese-American men. Ann Neurol 2005;57:98–103. [DOI] [PubMed] [Google Scholar]
- 10.Berg L. Mild senile dementia of the Alzheimer's type: diagnostic criteria and natural history. Mt Sinai J Med 1988;55:87–96. [PubMed] [Google Scholar]
- 11.Foley DJ, Masaki K, White L, Larkin EK, Monjan A, Redline S. Sleep-disordered breathing and cognitive impairment in elderly Japanese-American men. Sleep 2003;26:596–599. [DOI] [PubMed] [Google Scholar]
- 12.Braak H, Braak E. Staging of Alzheimer's disease-related neurofibrillary changes. Neurobiol Aging 1995;16:271–278. [DOI] [PubMed] [Google Scholar]
- 13.Teng EL, Hasegawa K, Homma A, et al. The Cognitive Abilities Screening Instrument (CASI): a practical test for cross-cultural epidemiological studies of dementia. Int Psychogeriatr 1994;6:45–58. [DOI] [PubMed] [Google Scholar]
- 14.Gelber RP, Ross GW, Petrovitch H, Masaki KH, Launer LJ, White LR. Antihypertensive medication use and risk of cognitive impairment: the Honolulu-Asia Aging Study. Neurology 2013;81:888–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Masaki KH, Losonczy KG, Izmirlian G, et al. Association of vitamin E and C supplement use with cognitive function and dementia in elderly men. Neurology 2000;54:1265–1272. [DOI] [PubMed] [Google Scholar]
- 16.Freitag MH, Peila R, Masaki K, et al. Midlife pulse pressure and incidence of dementia: the Honolulu-Asia Aging Study. Stroke 2006;37:33–37. [DOI] [PubMed] [Google Scholar]
- 17.Hixson JE, Vernier DT. Restriction isotyping of human apolipoprotein E by gene amplification and cleavage with HhaI. J Lipid Res 1990;31:545–548. [PubMed] [Google Scholar]
- 18.Launer LJ, Hughes TM, White LR. Microinfarcts, brain atrophy, and cognitive function: the Honolulu Asia Aging Study Autopsy Study. Ann Neurol 2011;70:774–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Epstein LJ, Kristo D, Strollo PJ, Jr, et al. Clinical guideline for the evaluation, management and long-term care of obstructive sleep apnea in adults. J Clin Sleep Med 2009;5:263–276. [PMC free article] [PubMed] [Google Scholar]
- 20.Findley LJ, Barth JT, Powers DC, Wilhoit SC, Boyd DG, Suratt PM. Cognitive impairment in patients with obstructive sleep apnea and associated hypoxemia. Chest 1986;90:686–690. [DOI] [PubMed] [Google Scholar]
- 21.Macey PM, Henderson LA, Macey KE, et al. Brain morphology associated with obstructive sleep apnea. Am J Respir Crit Care Med 2002;166:1382–1387. [DOI] [PubMed] [Google Scholar]
- 22.Torelli F, Moscufo N, Garreffa G, et al. Cognitive profile and brain morphological changes in obstructive sleep apnea. Neuroimage 2011;54:787–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Macey PM, Kumar R, Woo MA, Valladares EM, Yan-Go FL, Harper RM. Brain structural changes in obstructive sleep apnea. Sleep 2008;31:967–977. [PMC free article] [PubMed] [Google Scholar]
- 24.Kim H, Yun CH, Thomas RJ, et al. Obstructive sleep apnea as a risk factor for cerebral white matter change in a middle-aged and older general population. Sleep 2013;36:709–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Macey PM, Kumar R, Yan-Go FL, Woo MA, Harper RM. Sex differences in white matter alterations accompanying obstructive sleep apnea. Sleep 2012;35:1603–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pace-Schott EF, Spencer RM. Age-related changes in the cognitive function of sleep. Prog Brain Res 2011;191:75–89. [DOI] [PubMed] [Google Scholar]
- 27.Fung MM, Peters K, Redline S, et al. Decreased slow wave sleep increases risk of developing hypertension in elderly men. Hypertension 2011;58:596–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rauchs G, Desgranges B, Foret J, Eustache F. The relationships between memory systems and sleep stages. J Sleep Res 2005;14:123–140. [DOI] [PubMed] [Google Scholar]
- 29.Daurat A, Terrier P, Foret J, Tiberge M. Slow wave sleep and recollection in recognition memory. Conscious Cogn 2007;16:445–455. [DOI] [PubMed] [Google Scholar]
- 30.Ackermann S, Rasch B. Differential effects of non-REM and REM sleep on memory consolidation? Curr Neurol Neurosci Rep 2014;14:430. [DOI] [PubMed] [Google Scholar]
- 31.Cirelli C, Huber R, Gopalakrishnan A, Southard TL, Tononi G. Locus ceruleus control of slow-wave homeostasis. J Neurosci 2005;25:4503–4511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kadotani H, Kadotani T, Young T, et al. Association between apolipoprotein E epsilon4 and sleep-disordered breathing in adults. JAMA 2001;285:2888–2890. [DOI] [PubMed] [Google Scholar]
- 33.O'Hara R, Schroder CM, Kraemer HC, et al. Nocturnal sleep apnea/hypopnea is associated with lower memory performance in APOE epsilon4 carriers. Neurology 2005;65:642–644. [DOI] [PubMed] [Google Scholar]
- 34.Cosentino FI, Bosco P, Drago V, et al. The APOE epsilon4 allele increases the risk of impaired spatial working memory in obstructive sleep apnea. Sleep Med 2008;9:831–839. [DOI] [PubMed] [Google Scholar]
- 35.Xie L, Kang H, Xu Q, et al. Sleep drives metabolite clearance from the adult brain. Science 2013;342:373–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tsuang D, Larson EB, Li G, et al. Association between lifetime cigarette smoking and Lewy body accumulation. Brain Pathol 2010;20:412–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boot BP, Orr CF, Ahlskog JE, et al. Risk factors for dementia with Lewy bodies: a case-control study. Neurology 2013;81:833–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang JH, Fung SJ, Xi M, Sampogna S, Chase MH. Apnea produces neuronal degeneration in the pons and medulla of guinea pigs. Neurobiol Dis 2010;40:251–264. [DOI] [PubMed] [Google Scholar]
- 39.Zhang J, Zhu Y, Zhan G, et al. Extended wakefulness: compromised metabolics in and degeneration of locus ceruleus neurons. J Neurosci 2014;34:4418–4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Garcia-Lorenzo D, Longo-Dos Santos C, Ewenczyk C, et al. The coeruleus/subcoeruleus complex in rapid eye movement sleep behaviour disorders in Parkinson's disease. Brain 2013;136:2120–2129. [DOI] [PMC free article] [PubMed] [Google Scholar]