

Abstract
Objectives:
We aimed to determine the proportion of individuals in our schwannomatosis cohort whose disease is associated with an LZTR1 mutation.
Methods:
We used exome sequencing, Sanger sequencing, and copy number analysis to screen 65 unrelated individuals with schwannomatosis who were negative for a germline NF2 or SMARCB1 mutation. We also screened samples from 39 patients with a unilateral vestibular schwannoma (UVS), plus at least one other schwannoma, but who did not have an identifiable germline or mosaic NF2 mutation.
Results:
We identified germline LZTR1 mutations in 6 of 16 patients (37.5%) with schwannomatosis who had at least one affected relative, 11 of 49 (22%) sporadic patients, and 2 of 39 patients with UVS in our cohort. Three germline mutation–positive patients in total had developed a UVS. Mosaicism was excluded in 3 patients without germline mutation in NF2, SMARCB1, or LZTR1 by mutation screening in 2 tumors from each.
Conclusions:
Our data confirm the relationship between mutations in LZTR1 and schwannomatosis. They indicate that germline mutations in LZTR1 confer an increased risk of vestibular schwannoma, providing further overlap with NF2, and that further causative genes for schwannomatosis remain to be identified.
Schwannomatosis is a member of the neurofibromatosis family of neurogenetic disorders, which predispose to benign tumors throughout the nervous system. Within this group, neurofibromatosis type 2 (NF2) and schwannomatosis share clinical overlap and can be difficult to distinguish, particularly in cases of mosaic disease.1 The main tumor type seen in both NF2 and schwannomatosis is the schwannoma, although the location of these tumors differs somewhat between these 2 syndromes, with bilateral vestibular schwannomas being almost universal in patients with classic NF2,2,3 and with both intradermal and nonintradermal schwannomas also frequently seen. In contrast, schwannomatosis-associated schwannomas tend to be nonintradermal and nonvestibular, although rare cases of unilateral vestibular schwannomas (UVS) have been observed.4 Both conditions also lead to meningiomas, although there is a much higher incidence in NF2 (>50%) than in schwannomatosis (approximately 5%). In either disease, meningiomas may be the only tumors that develop.5–7 Ependymomas and cataracts are also confined to individuals with NF2.2,3
Genetic characterization is an important tool for distinguishing between diseases, because germline mutation of the NF2 gene is reported in more than 90% of individuals with nonmosaic NF2,8 but is not present in patients with schwannomatosis, although somatically acquired NF2 mutations are usually found in tumors.9 Germline mutation of the SWI/SNF chromatin remodeling complex gene, SMARCB1, is responsible for approximately 20% of patients with schwannomatosis disease, with a much higher detection rate in familial patients (∼50%) than sporadic patients (∼10%),10 indicating genetic heterogeneity. Recently, leucine-zipper-like transcription regulator 1 (LZTR1), which lies approximately 3 Mb centromeric to SMARCB1 on chromosome 22, was identified as a second causative gene for schwannomatosis, with loss of function mutations identified in 80% of the study’s SMARCB1 mutation–negative schwannomatosis cohort, all of whom had somatic loss of chromosome 22 in their tumors.11
We investigated the frequency of LZTR1 mutations in our own cohort of 16 families with multiple affected members with schwannomatosis and 49 individuals with sporadic schwannomatosis and no known germline SMARCB1 or NF2 mutations, with or without known somatic loss of chromosome 22 in their tumors, to determine the proportion of patients with genetically uncharacterized schwannomatosis whose disease is accounted for by this new gene.
METHODS
Patient material.
We analyzed genomic DNA from peripheral lymphocytes from familial and sporadic patients meeting clinical diagnostic criteria for schwannomatosis,12,13 who had previously tested negative for constitutional mutations in both NF2 and SMARCB1 genes using Sanger sequencing and multiplex ligation-dependent probe amplification (MLPA) probesets P258-B1 and P044 (MRC-Holland, Amsterdam, the Netherlands).10 DNA extracted from paraffin-embedded or fresh-frozen tumors was used to confirm mutations and screen for second somatic mutations.
Standard protocol approvals, registrations, and patient consents.
Ethical approval for use of anonymized samples from a historical retrospectively collected archive in this study was obtained from the North West 7–Greater Manchester Central Research Ethics Committee (reference 10/H1008/74). Written consent was obtained from prospectively collected individuals.
Exome sequencing.
Whole-exome targeted enrichment and sequencing were performed on lymphocyte DNA extracted from 7 unrelated individuals with a family history of schwannomatosis. Enrichment was performed using the SureSelect Human All Exon Kit v.1 (Agilent, Santa Clara, CA) for the Illumina HiSeq 2500 system (Illumina, Inc., San Diego, CA). Sequence data were mapped to the human reference sequence hg19 (build GRCh37) with the Burrows-Wheeler aligner (BWA v0.6.2).14 The genome analysis tool kit (GATK v2.4.7)15 was used for base quality score recalibration and indel realignment before variant calling using the unifiedGenotyper. Single nucleotide polymorphisms (SNPs) with ≥5× coverage and indels were annotated to genes using Ensembl v68, and the functional consequences were defined. Between 85% and 98% of the targeted exome was covered at least 20× for each sample. Additional annotation was provided from OMIM and Genomic Evolutionary Rate Profiling (35 species alignment) as well as population frequencies from 1000 Genomes Project (phase 1 release), NHLBI Exome Sequencing Project (v6500), and our own in-house frequencies. PolyPhen-2 and sorting intolerant from tolerant (SIFT) predictions were also included to help determine pathogenicity. All candidate mutations were verified by Sanger sequencing.
Sanger sequencing.
Primers were designed to amplify each of the 21 LZTR1 exons, including all of the coding regions and approximately 50 to 100 bases of flanking intronic sequence per exon. Each fragment was amplified by PCR, using the GoTaq Green Master Mix (Promega, Southampton, UK). PCR products were purified using Agencourt AMPure XP beads (Beckman Coulter Genomics, Danvers, MA). Sequencing was performed using BigDye Terminator v3·1 Cycle Sequencing Kit (ABI, Life Technologies, Foster City, CA). Sequencing PCR products were purified using Agencourt CleanSEQ beads (Beckman Coulter Genomics) and sequence analysis was performed using the ABI 3730xl DNA Analyzer (ABI, Life Technologies).
Design and application of an LZTR1-specific MLPA probeset.
The genomic sequence of the 21-exon human LZTR1 gene (NM_006767) was downloaded from the University of California Santa Cruz genome browser (www.genome.ucsc.edu). A total of 8 MLPA probes were placed such as to target exons 1, 3, 5, 7, 10, 15, 18, and 20. Probes were designed according to criteria provided by MRC-Holland at www.mlpa.com. Seven reference probes targeting physically distinct genomic regions were derived from previously established probesets (unpublished). Oligonucleotides for MLPA probes were from Biolegio (Nijmegen, the Netherlands). MLPA reactions utilized reagents from MRC-Holland, and products were visualized on a LICOR4200 (LICOR Biosciences, Lincoln, NE). Relative MLPA signals were calculated as described previously.16
RESULTS
We undertook exome sequencing analysis on lymphocyte DNA samples from 7 patients with familial schwannomatosis who had previously been found negative for germline SMARCB1 and NF2 mutations. We identified novel heterozygous loss of function mutations in the LZTR1 gene in 3 of these patients (shown in table 1 and figure 1, A–C). The mutations were confirmed by Sanger sequencing and segregated with all affected family members available for testing (table 1). Of note, one clinically unaffected father in family 2 carried a pathogenic mutation (figure 1B). This may indicate nonpenetrance in this individual, although he has not undergone full-body MRI and may therefore harbor undetected tumors. Sanger sequencing of probands from 9 additional schwannomatosis families identified 3 further germline loss-of-function mutations (figure 1, D–F).
Table 1.
LZTR1 mutations identified in individuals with schwannoma disease
Figure 1. Pedigrees of LZTR1 mutation–positive families.

(A) Family 1; (B) family 2; (C) family 3; (D) family 4; (E) family 5; and (F) family 6. Asterisks indicate family members screened for the mutation. Black stripe indicates an asymptomatic mutation carrier.
Tumor DNA for 2 familial patients, in whom no germline mutation had been detected, showed a reduced signal for known SNPs on one allele in comparison to heterozygous signals in germline DNA on sequencing chromatograms, suggestive of loss of heterozygosity (LOH).
We next sequenced LZTR1 in 49 patients with sporadic schwannomatosis and identified 11 (22%) additional germline point mutations (listed in table 1). Matched tumor DNA was available for 4 of the mutation-positive patients and all showed evidence of loss of the wild-type allele (table 1).
To determine whether larger single- or multiexon deletions of LZTR1 were present in the samples negative for point mutations, we developed an MLPA assay containing 8 probes spanning the LZTR1 gene. Lymphocyte DNA of sufficient quantity and quality was available to perform MLPA analysis on 9 familial patients and 30 sporadic patients. The tumor from the familial patient with an exon 7 missense mutation and 4 tumors from mutation-negative sporadic patients were also tested.
No significant copy number changes were seen in any germline DNA samples. Loss of the wild-type allele was seen for the tumor from the patient with an exon 7 mutation. Three of the 4 tumors from sporadic patients with no detected germline mutation showed a reduced copy number of the entire LZTR1 gene at levels indicating loss of one allele with some nontumor cell contamination in the sample. The remaining tumor showed no evidence of LOH. No point mutations were identified by Sanger sequencing in this tumor.
None of the mutations identified in our cohort were seen on dbSNP137 or on the ESP6500. The variant c.2062C>T, identified in one individual in our study, has been previously associated with schwannomatosis disease.11 In silico analysis of all the missense mutations by PolyPhen2, SIFT, Align GVGD, and MutationTaster predicted 5 of 8 different mutations to be damaging to the protein by all 4 algorithms, 2 more were predicted to be damaging by 3 of 4 algorithms, and one was predicted to be damaging by 2 of 4 and likely to be damaging by a third (table e-1 on the Neurology® Web site at Neurology.org). This mutation was also found in 2 unrelated individuals in our cohort and was retained in the matched tumor, available for one of these individuals, in conjunction with loss of the wild-type allele, further suggesting that it is pathogenic. Evolutionary conservation of the affected amino acids (table e-1) shows that the missense mutations occurred at conserved residues.
These results determine an overall detection rate of LZTR1 mutations in 6 of 16 (38%) familial patients and 11 of 49 (22%) sporadic patients in our cohort without germline SMARCB1 mutations.
None of the LZTR1 mutation carriers identified in the schwannomatosis cohort had meningiomas, ependymomas, intracutaneous schwannomas, or cataracts. Two patients had facial nerve schwannomas and one a lower cranial nerve schwannoma, which was initially thought to be a vestibular schwannoma. Schwannomas typically occurred at deep locations, including spinal nerve roots with pain as the predominant symptom.
One individual with an inherited LZTR1 mutation (family 1, table 1) had a schwannoma removed at 37 years of age, which was clearly identified at surgery as emanating from the vestibular nerve during cochlear nerve–preserving surgery in Germany. Because of this result, we screened 39 additional lymphocyte samples from individuals with a UVS and at least one additional schwannoma, but who had no identifiable NF2 mutation in blood or proven mosaic mutation in tumors. Tumors from 7 of these individuals were also sequenced. Most of these patients met Manchester criteria for NF2 with at least 2 nonvestibular schwannomas or meningiomas in addition to a UVS.2 We detected germline mutations in 2 of these individuals and one somatic mutation, not present in blood, in a third individual (table 1). The exon 1 frameshift mutation, c.27delG, p.(Gln10Argfs*15), previously associated with schwannomatosis,11 was identified in one of these individuals. This mutation has subsequently been added to the Exome Variant Server, seen in 59 of 11,848 alleles. It is difficult to surmise that this mutation would not lead to total loss of protein product by nonsense mediated decay, but a mechanism for reinitiation of RNA sequence has been described recently for SMARCB1.17 Two of these last 3 individuals had a definite UVS whereas the third had a cerebellopontine angle tumor identified on MRI with a location consistent with involvement of the eighth nerve (figure 2). In total, LZTR1 mutations were identified in 3 of 39 individuals (8%) with a UVS and at least one other schwannoma, but without a germline NF2 mutation.
Figure 2. Cerebellopontine angle tumor in an LZTR1 mutation carrier.
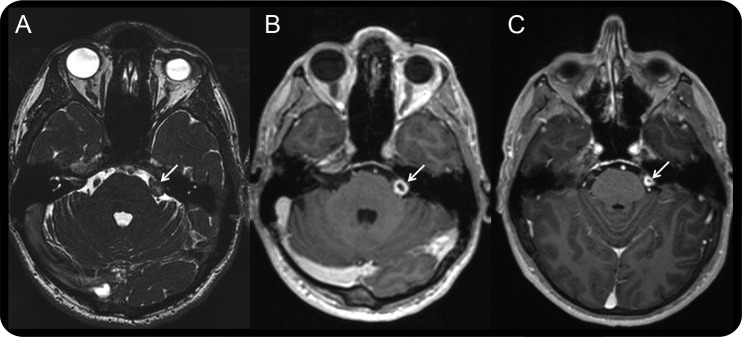
MRIs showing (A) contact image sensors image taken in 2006, (B) postcontrast image taken in 2006, and (C) postcontrast image taken in 2013 showing cystic degeneration around the tumor. White arrows indicate the location of the tumor.
The results of schwannoma tumor analysis from patients with schwannomatosis without known NF2 mosaicism are shown in table 2. All 12 tumors from SMARCB1 mutation carriers had the typical 22q loss including LOH for NF2 and a point mutation. All 11 tumors from LZTR1 carriers had LOH, but in 4 cases this was due to mitotic recombination. A substantial proportion of patients with typical NF2 involvement in their tumors, including 4 with typical different NF2 point mutations in different schwannomas, had no identifiable LZTR1 or SMARCB1 mutation. Three of these patients whose lymphocyte DNA showed no mutation of the LZTR1, SMARCB1, or NF2 gene, had 2 tumors tested for somatic mutations. None of these patients had 2 identical point mutations in both tumors in any of these 3 genes.
Table 2.
NF2 mutational hits in schwannomas from patients with a SMARCB1 germline mutation, an LZTR1 germline mutation, or no identified mutation who have been genetically determined not to have mosaic NF2

DISCUSSION
In contrast to the 80% of individuals identified with a germline LZTR1 mutation in the initial disease gene discovery publication linking LZTR1 to schwannomatosis,11 we identified novel germline mutations in LZTR1 in only 26% of our cohort of 65 patients with SMARCB1 mutation–negative familial and sporadic schwannomatosis. Unlike the initial report, we did not confine our analysis to patients with proven involvement of the chromosome 22q locus. However, even in patients with 22q involvement in tumor, but no proven NF2 or SMARCB1 mosaicism, we found only 28.5% with germline LZTR1 mutations. It is therefore likely that a further schwannomatosis gene exists on chromosome 22q.
Among the mutation-positive cases, we identified both truncating and nontruncating mutations across the length of LZTR1 (8 missense, 3 splice-site, 2 nonsense, and 4 frameshift), consistent with the hypothesis that at least some of these mutations will produce hypomorphic protein products similar to the predicted effect of schwannomatosis-associated SMARCB1 mutations.18
We identified an LZTR1 mutation in 4 individuals with a UVS plus at least one other schwannoma (table 1): one diagnosed clinically to have familial schwannomatosis, one who met Manchester criteria for a diagnosis of NF2, and 2 with a clinically uncertain disease status because mutation analysis in NF2 was negative in DNA extracted from lymphocytes. The identification of LZTR1 mutations in these individuals supports our previous assertion that UVS can occur in the context of schwannomatosis,4 and further suggests that LZTR1 mutations may confer a greater risk of vestibular schwannoma than SMARCB1 mutations, because there have been no reports of SMARCB1 mutation–positive schwannomatosis patients with a proven vestibular schwannoma. Vestibular schwannomas are still considered exclusion criteria for schwannomatosis in published criteria,12 although limiting this restriction to patients with bilateral tumors has been proposed.19 In light of our results, a UVS should not be considered an exclusion criterion for schwannomatosis, and LZTR1 mutation analysis should be considered in patients with a UVS and other painful schwannomas, who do not have other typical NF2 features, or a proven germline or mosaic NF2 mutation.
The LZTR1 gene encodes a member of the BTB-Kelch superfamily of proteins, exclusively localized in the Golgi network. The cellular function of LZTR1 is not clear, although mutations in this gene have recently been implicated in the development of glioblastomas.20 LZTR1 lies on chromosome 22, approximately 3 megabases centromeric to SMARCB1 and 9 megabases centromeric to NF2. Three patients whose lymphocyte DNA showed no mutation of the LZTR1, SMARCB1, or NF2 gene had 2 tumors tested for somatic mutations (a fourth had no DNA left for LZTR1 analysis). One of these patients has been reported previously to have LOH at the NF2 locus in one tumor without a change in copy number, resulting from mitotic recombination and with a breakpoint occurring downstream of both LZTR1 and SMARCB1.21 None of these 3 patients had identical mutations in both tumors in any of the 3 genes. This shows that the schwannomas in these patients are not due to mosaic disease caused by any of the 3 genes and suggests that there are further causative genes still to be identified.
In the context of our analysis, we present a flow diagram of the ideal screening protocol for mutation analysis of schwannoma disease, assuming availability of relevant samples (figure 3). This will evolve as further genes are discovered and sequencing of panels of genes relevant to schwannoma disease become available and in time exome and/or genome sequencing become routine first-line diagnostics. Currently, we would recommend that next-generation sequencing panels for patients with NF2 without bilateral vestibular schwannoma and those with schwannomatosis should include the NF2, SMARCB1, and LZTR1 genes for both blood and tumor analysis and that, where possible, 2 tumors should be analyzed from patients with no known family history.
Figure 3. Flow diagram for schwannomatosis mutation screening.

The diagram indicates an ideal mutation screening strategy for schwannomatosis mutations, assuming availability of all samples.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Thomas Rio Frio and Virginie Bernard for their help with the NGS platform.
GLOSSARY
- LOH
loss of heterozygosity
- LZTR1
leucine-zipper-like transcription regulator 1
- MLPA
multiplex ligation-dependent probe amplification
- NF2
neurofibromatosis type 2
- SIFT
sorting intolerant from tolerant
- SMARCB1
SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily b, member 1
- SNP
single nucleotide polymorphism
- UVS
unilateral vestibular schwannoma
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
Miriam J. Smith: drafting/revising the manuscript, study concept or design, analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval, acquisition of data, statistical analysis. Bertand Isidor: drafting/revising the manuscript, analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval, acquisition of data. Christian Beetz: drafting/revising the manuscript, analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval, acquisition of data. Simon G. Williams: analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval. Sanjeev S. Bhaskar: analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval. Wilfrid Richer: drafting/revising the manuscript, analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval, statistical analysis. James O'Sullivan: study concept or design, analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval. Beverly Anderson: analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval, acquisition of data. Sarah B. Daly: drafting/revising the manuscript, accepts responsibility for conduct of research and will give final approval, acquisition of data. Jill E. Urquhart: drafting/revising the manuscript, analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval, acquisition of data. Alan Fryer: drafting/revising the manuscript, accepts responsibility for conduct of research and will give final approval, contribution of vital reagents/tools/patients, acquisition of data. Cecilie F. Rustad: drafting/revising the manuscript, accepts responsibility for conduct of research and will give final approval, acquisition of data. Samantha J. Mills: drafting/revising the manuscript, analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval, review of imaging and acquisition of appropriate images for the publication. Amir Samii: drafting/revising the manuscript, accepts responsibility for conduct of research and will give final approval. Daniel du Plessis: analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval, acquisition of data. Dorothy Halliday: drafting/revising the manuscript, analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval. Sebastien Barbarot: analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval, acquisition of data. Franck Bourdeaut: drafting/revising the manuscript, analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval. William G. Newman: drafting/revising the manuscript, study concept or design, analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval, acquisition of data, statistical analysis, study supervision, obtaining funding. D. Gareth Evans: drafting/revising the manuscript, study concept or design, analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval, contribution of vital reagents/tools/patients, acquisition of data, statistical analysis, study supervision, obtaining funding.
STUDY FUNDING
The project was funded by a Young Investigator Award from the Children's Tumor Foundation and a project grant from the Association of International Cancer Research. High-throughput sequencing has been performed in part by the NGS platform of the Institut Curie, supported by grants ANR-10-EQPX-03 and ANR10-INBS-09-08 from the Agence Nationale de le Recherche (investissements d'avenir) and by the Canceropôle Ile-de-France.
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Leverkus M, Kluwe L, Roll EM, et al. Multiple unilateral schwannomas: segmental neurofibromatosis type 2 or schwannomatosis? Br J Dermatol 2003;148:804–809. [DOI] [PubMed] [Google Scholar]
- 2.Evans DG, Huson SM, Donnai D, et al. A clinical study of type 2 neurofibromatosis. Q J Med 1992;84:603–618. [PubMed] [Google Scholar]
- 3.Evans DG. Neurofibromatosis type 2 (NF2): a clinical and molecular review. Orphanet J Rare Dis 2009;4:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith MJ, Kulkarni A, Rustad C, et al. Vestibular schwannomas occur in schwannomatosis and should not be considered an exclusion criterion for clinical diagnosis. Am J Med Genet A 2012;158A:215–219. [DOI] [PubMed] [Google Scholar]
- 5.Christiaans I, Kenter SB, Brink HC, et al. Germline SMARCB1 mutation and somatic NF2 mutations in familial multiple meningiomas. J Med Genet 2010;48:93–97. [DOI] [PubMed] [Google Scholar]
- 6.Bacci C, Sestini R, Provenzano A, et al. Schwannomatosis associated with multiple meningiomas due to a familial SMARCB1 mutation. Neurogenetics 2010;11:73–80. [DOI] [PubMed] [Google Scholar]
- 7.Evans DG, Birch JM, Ramsden RT. Paediatric presentation of type 2 neurofibromatosis. Arch Dis Child 1999;81:496–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Evans DG, Ramsden RT, Shenton A, et al. Mosaicism in neurofibromatosis type 2: an update of risk based on uni/bilaterality of vestibular schwannoma at presentation and sensitive mutation analysis including multiple ligation-dependent probe amplification. J Med Genet 2007;44:424–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.MacCollin M, Willett C, Heinrich B, et al. Familial schwannomatosis: exclusion of the NF2 locus as the germline event. Neurology 2003;60:1968–1974. [DOI] [PubMed] [Google Scholar]
- 10.Smith MJ, Wallace AJ, Bowers NL, et al. Frequency of SMARCB1 mutations in familial and sporadic schwannomatosis. Neurogenetics 2012;13:141–145. [DOI] [PubMed] [Google Scholar]
- 11.Piotrowski A, Xie J, Liu YF, et al. Germline loss-of-function mutations in LZTR1 predispose to an inherited disorder of multiple schwannomas. Nat Genet 2014;46:182–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.MacCollin M, Chiocca EA, Evans DG, et al. Diagnostic criteria for schwannomatosis. Neurology 2005;64:1838–1845. [DOI] [PubMed] [Google Scholar]
- 13.Baser ME, Friedman JM, Evans DG. Increasing the specificity of diagnostic criteria for schwannomatosis. Neurology 2006;66:730–732. [DOI] [PubMed] [Google Scholar]
- 14.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010;20:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beetz C, Nygren AO, Schickel J, et al. High frequency of partial SPAST deletions in autosomal dominant hereditary spastic paraplegia. Neurology 2006;67:1926–1930. [DOI] [PubMed] [Google Scholar]
- 17.Hulsebos TJ, Kenter S, Verhagen WI, Baas F, Flucke U, Wesseling P. Premature termination of SMARCB1 translation may be followed by reinitiation in schwannomatosis-associated schwannomas, but results in absence of SMARCB1 expression in rhabdoid tumors. Acta Neuropathol 2014;128:439–448. [DOI] [PubMed] [Google Scholar]
- 18.Smith MJ, Walker JA, Shen Y, Stemmer-Rachamimov A, Gusella JF, Plotkin SR. Expression of SMARCB1 (INI1) mutations in familial schwannomatosis. Hum Mol Genet 2012;21:5239–5245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Plotkin SR, Blakeley JO, Evans DG, et al. Update from the 2011 International Schwannomatosis Workshop: from genetics to diagnostic criteria. Am J Med Genet A 2013;161A:405–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frattini V, Trifonov V, Chan JM, et al. The integrated landscape of driver genomic alterations in glioblastoma. Nat Genet 2013;45:1141–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hadfield KD, Smith MJ, Urquhart JE, et al. Rates of loss of heterozygosity and mitotic recombination in NF2 schwannomas, sporadic vestibular schwannomas and schwannomatosis schwannomas. Oncogene 2010;29:6216–6221. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.