

Abstract
Objective:
To assess the natural history of congenital myopathies (CMs) due to different genotypes.
Methods:
Retrospective cross-sectional study based on case-note review of 125 patients affected by CM, followed at a single pediatric neuromuscular center, between 1984 and 2012.
Results:
Genetic characterization was achieved in 99 of 125 cases (79.2%), with RYR1 most frequently implicated (44/125). Neonatal/infantile onset was observed in 76%. At birth, 30.4% required respiratory support, and 25.2% nasogastric feeding. Twelve percent died, mainly within the first year, associated with mutations in ACTA1, MTM1, or KLHL40. All RYR1-mutated cases survived and did not require long-term ventilator support including those with severe neonatal onset; however, recessive cases were more likely to require gastrostomy insertion (p = 0.0028) compared with dominant cases. Independent ambulation was achieved in 74.1% of all patients; 62.9% were late walkers. Among ambulant patients, 9% eventually became wheelchair-dependent. Scoliosis of variable severity was reported in 40%, with 1/3 of (both ambulant and nonambulant) patients requiring surgery. Bulbar involvement was present in 46.4% and required gastrostomy placement in 28.8% (at a mean age of 2.7 years). Respiratory impairment of variable severity was a feature in 64.1%; approximately half of these patients required nocturnal noninvasive ventilation due to respiratory failure (at a mean age of 8.5 years).
Conclusions:
We describe the long-term outcome of a large cohort of patients with CMs. While overall course is stable, we demonstrate a wide clinical spectrum with motor deterioration in a subset of cases. Severity in the neonatal/infantile period is critical for survival, with clear genotype-phenotype correlations that may inform future counseling.
Congenital myopathies (CMs) are a heterogeneous group of inherited muscle disorders, divided into subtypes based on the predominant histopathologic findings, namely, nemaline rods, cores, central nuclei, or fiber type disproportion (FTD).1–4 Hypotonia and muscle weakness, with neonatal/childhood onset, are the most typical presentations,1,3 but onset and functional deterioration in adult age have been reported.5–9 CMs are rare disorders, with a prevalence estimated between 1:22,480 in Sweden10 and 1:135,000 in Northern England.11 To date, 20 genes have been associated with CMs,12 but approximately one-third of patients remain genetically unresolved.13 Mutations in the same gene may give rise to a wide range of clinicopathologic phenotypes, whereas mutations in different genes may cause the same CM, often due to the similar function of the defective gene products. The need for comprehensive data regarding functional abilities, cardiorespiratory involvement, orthopedic complications, and survival, as a basis for anticipatory patient management and planning of future therapeutic trials, has been recently highlighted.4 The natural history of specific subgroups has been described in only a few cohorts, not all of which had been fully genetically characterized.5,13–19
Herein, we report the clinical course of 125 patients with CM, reviewed at a single neuromuscular center. Furthermore, genotype-phenotype correlations in a subgroup of 99 genetically characterized cases are illustrated.
METHODS
Patients.
This was a retrospective cross-sectional study of cases with a diagnosis of CM. All patients had an evocative clinical picture (characterized by congenital/early childhood onset with hypotonia, static/progressive weakness, affecting predominantly proximal/axial muscles, normal/mildly elevated serum creatine kinase), at least one histopathologic feature suggestive of CM, and/or a genetic diagnosis. Data were obtained from the case notes of patients followed between 1984 and 2012 at the Dubowitz Neuromuscular Centre, London. We collected the following information: family history (consanguinity, miscarriages, premature deaths, ethnic origin), pregnancy (decreased fetal movements, oligohydramnios/polyhydramnios), birth (gestational age, delivery) and neonatal history (respiratory support, nasogastric tube [NGT] feeding), age at onset, motor performance (walking age, maximal motor ability, age when wheelchair use commenced and at loss of ambulation), respiratory involvement (nocturnal/daily noninvasive ventilation [NNIV/DNIV], invasive ventilation), cardiac and bulbar involvement (gastrostomy/jejunostomy [G/J] insertion), orthopedic complications (hip dysplasia, ankle contractures, scoliosis), other features (cognitive impairment, undescended testis/orchidopexy, malignant hyperthermia reactions), and survival.
Standard protocol approvals, registrations, and patient consents.
Parents/guardians provided written informed consent for the diagnostic genetic analysis, according to the Great Ormond Street Hospital guidance. This study was approved by the West London & GTAC Ethics Committee (08/H0707/119).
Genetic analysis.
Genomic DNA was extracted from peripheral blood leukocytes according to standard procedures. Genetic analyses were performed according to the histologic/clinical phenotype, depending on the tests available at the time. Most genes were screened by PCR amplification and Sanger sequencing of coding exons: skeletal muscle α-actin (ACTA1), myotubularin (MTM1), selenoprotein N (SEPN1), β-tropomyosin (TPM2), tropomyosin 3 (TPM3), dynamin 2 (DNM2), ryanodine receptor type 1 (RYR1), cofilin 2 (CFL2), troponin T (TNNT1), amphiphysin 2 (BIN1), and kelch-like family member 40 (KLHL40). In 3 patients, only mutational hotspots of RYR1 were sequenced. Intragenic and flanking microsatellite markers were used to indicate linkage to the nebulin (NEB) gene. A breakpoint PCR assay was used to detect the common exon 55 deletion mutation of NEB.20 In 2 cases, next-generation sequencing analysis has subsequently allowed comprehensive analysis of NEB.21
Histologic analysis.
Muscle biopsies were reviewed and classified according to criteria suggested by Dubowitz et al.22: (1) nemaline myopathy (NM); (2) centronuclear myopathy (CNM); (3) “core myopathies,” including central core disease and multi-minicore disease; (4) FTD; (5) type 1 fiber predominance/uniformity; and (6) nonspecific myopathic changes (NSMC).
Statistical analysis.
Descriptive statistics used were mean, median, and SD. Kaplan-Meier curve was performed using GraphPad Prism 5 software (GraphPad Software, La Jolla, CA).
RESULTS
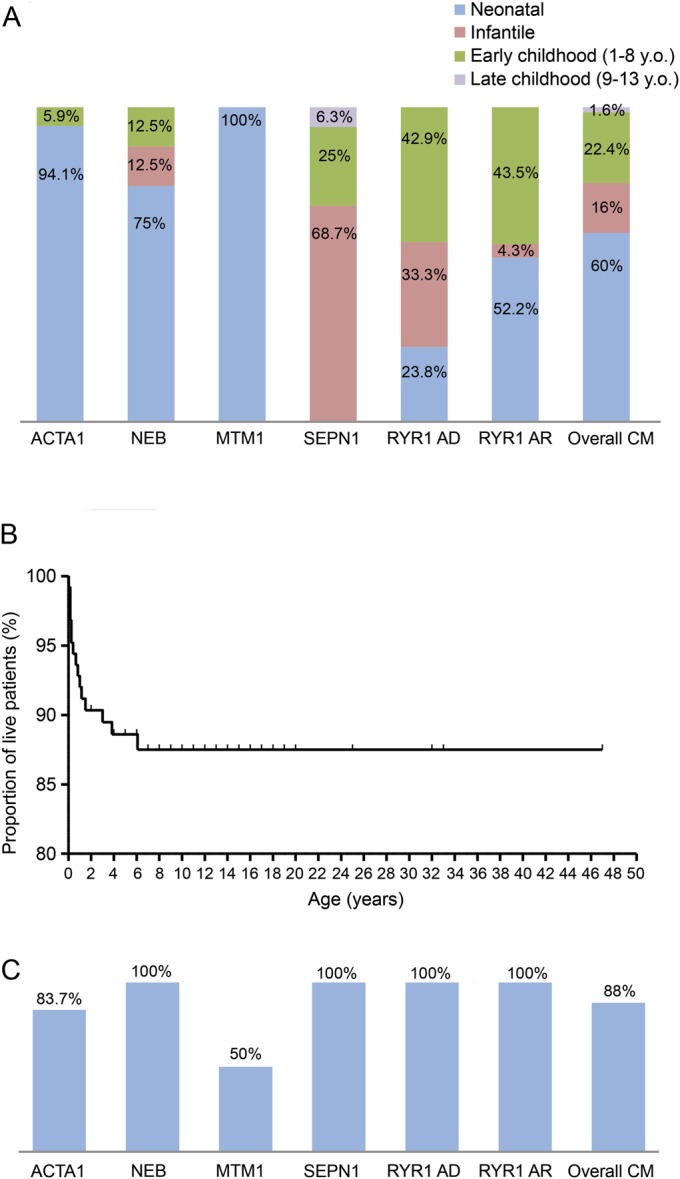
One hundred twenty-five patients according to the above criteria were included; 64 of the 125 were females (51.2%) and 61 were males (48.8%). Mean age at last follow-up was 10.5 years (median 10 years, SD 7.72), ranging from the neonatal period to 47 years, because a few selected cases were followed into adulthood (18/125, 14.4%). A genetic diagnosis was established in 99 of 125 (79.2%), with the following genetic backgrounds identified: RYR1 in 44 of 97 (44.4%), of whom 23 (23.2%) had autosomal recessive (AR) and 21 (21.2%) autosomal dominant (AD) inheritance; ACTA1 in 17 of 99 (17.2%); SEPN1 in 16 of 99 (16.2%); NEB in 8 of 99 (8.1%); MTM1 in 8 of 99 (8.1%); TPM3 and KLHL40 both in 2 of 99 (2.1%); and TPM2 and DNM2 both in 1 of 99 (1%). A muscle biopsy was performed in 104 of 125 (83.2%): the most common diagnosis was core myopathy (39/104, 37.5%), followed by NM (33/104, 31.8%), CNM (18/104, 17.3%), FTD and type 1 fiber predominance/uniformity (both with 5/104, 4.8%), and NSMC (4/104, 3.8%) (figure e-1 on the Neurology® Web site at Neurology.org). All cases presenting with NSMC were genetically characterized and due to SEPN1 mutations. Figure e-2 illustrates histologic features associated with different genetic backgrounds. Clinical information is summarized below with more detail provided in e-Results. Tables e-1 and e-2 summarize clinical information and genetic background, respectively, of each case.
Family history.
Consanguinity was present in 20 of 125 families (16%). There was a history of spontaneous miscarriages in 9 of 125 (7.2%), mainly in families with AD RYR1 mutations (4/9). Premature death in family members was reported in 8 of 125 (6.4%), mainly in the subset with MTM1 mutations (3/8). Thirty-nine of 125 (31.2%) had at least one affected living relative. Among patients with confirmed genetic diagnosis, the most common family ancestry was Caucasian-British (58/99, 58.5%), followed by families of Pakistani origin (14/99, 14.1%). Only 2 cases originated from Arab countries. Within the group of CMs with AR inheritance (50 patients), 22 of 50 patients (44%) had Caucasian-British ancestors and were mainly compound heterozygous for RYR1 mutations (12 cases), whereas 12 of 50 patients (24%) had Pakistani parents and carried mainly homozygous mutations in SEPN1 (5 cases) or NEB (4 cases). Consanguinity was reported in 2 of 50 Caucasian-British families (4%) and in 8 of 50 Pakistani families (16%), a likely explanation for the highest prevalence of homozygous mutations in the latter ethnic subgroup. Within the group of CMs with AD inheritance (49 patients), 36 of 49 (73%) were of Caucasian-British descent and carried mainly heterozygous dominant RYR1 (17 cases) or ACTA1 (11 cases) mutations. Within the subgroup of Pakistani descent, there was no case with dominantly inherited and only one case with recessively inherited RYR1-related myopathy.
Perinatal history and age at onset.
Antenatal onset was evidenced by frequently reduced fetal movements (47/125, 37.6%) and/or polyhydramnios (29/125, 23.2%) (figures 1 and 2A). The prematurity rate was increased (23/110, 21.1%).23 Neonatal bulbar and respiratory involvement requiring tube feeding (32/125, 25.6%) and ventilatory support (38/125, 30.4%) were common.
Figure 1. Perinatal features.
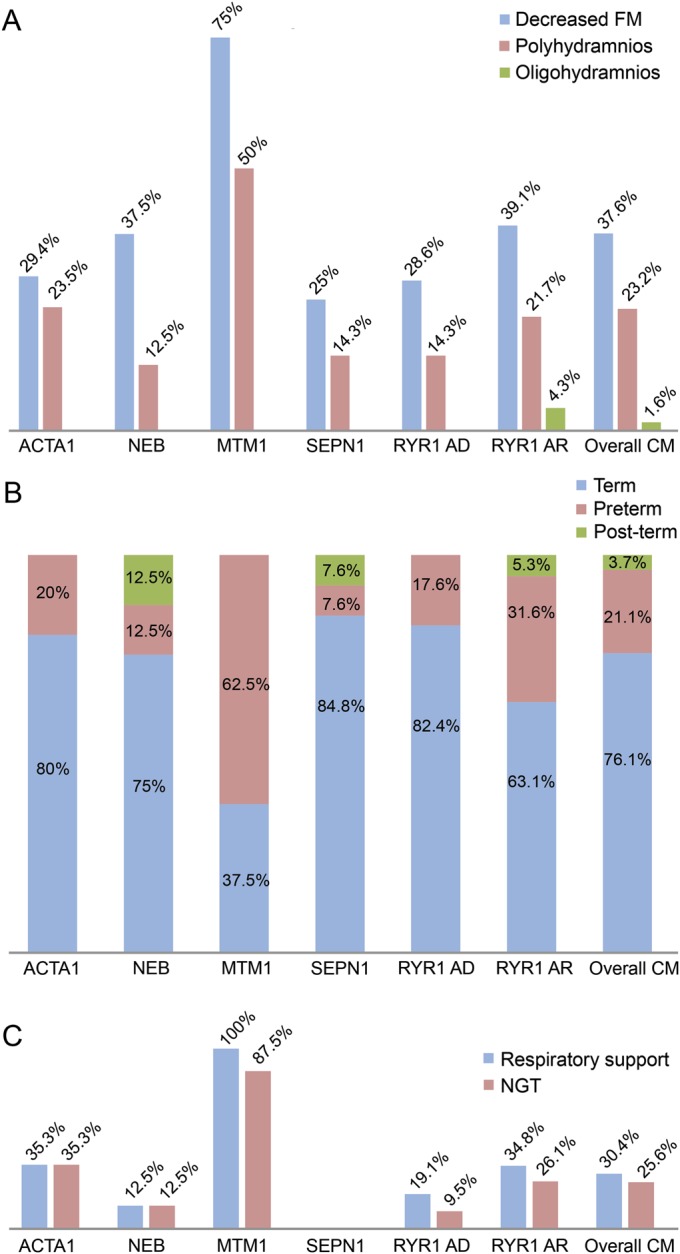
(A) Antenatal history: the highest percentage of decreased FM and polyhydramnios was found in MTM1-related myopathy. (B) Age at birth: the overall prematurity rate in our cohort of CMs (21.1%) was higher than in the general population of England and Wales (7.4% in 2008).23 (C) In the neonatal period, approximately one-third of patients required NGT feeding and respiratory support, with variability according to the genetic background. AD = autosomal dominant; AR = autosomal recessive; CM = congenital myopathy; FM = fetal movements; NGT = nasogastric tube.
Figure 2. Age at onset and survival.
(A) Age at onset according to the genetic background: most cases, presented within the first year of life, with neonatal presentations more common than infantile presentations. (B) Kaplan-Meier survival curve: approximately 2 of 3 deaths occurred within the first year of life. (C) Survival according to genotype: no death was registered in patients with RYR1, SEPN1, and NEB mutations. Of note, 8 of 15 of those who died were affected by NM, 7 of 15 by CNM, with MTM1, ACTA1, and KLHL40 the most common identifiable causes. AD = autosomal dominant; AR = autosomal recessive; CM = congenital myopathy; CNM = centronuclear myopathy; NM = nemaline myopathy.
Mobility and best motor abilities.
Eighty-nine of 120 patients (74.1%), where this information was available, achieved independent ambulation (see figure 3). Eight of 89 patients (9%), who initially acquired independent ambulation, became wheelchair-dependent at a mean age of 10.1 years (median 9.5 years, SD 2.69), irrespective of the genetic background: all were late to walk and no one achieved a full range of gross motor abilities.
Figure 3. Motor abilities.

(A) Maximal motor ability: all patients with SEPN1 and NEB mutations walked independently, whereas motor ability was more variable with other genetic backgrounds. (B) Walking age: the majority of ambulant patients walked late (39.3%) or at the upper limit of normal at 18 months (23.6%). At last follow-up, 3.2% were younger than 18 months. (C) Kaplan-Meier curve showing wheelchair use in patients who achieved independent ambulation: 20 of 89 (22.5%) started manual wheelchair for long distances, whereas a further deterioration of motor performances was observed in 8 of 20, who became wheelchair-bound.
Respiratory involvement.
Eighty of 125 (64.1%) had highly variable respiratory involvement, ranging from patients needing neonatal respiratory support (38/125, 30.4%) to those having recurrent chest infections (51/128, 40%) and/or decreased forced vital capacity (<60%) (51/125, 40.8%). Figure 4 shows NNIV provision due to nocturnal hypoventilation. Permanent tracheostomy and also diurnal ventilation (DNIV) were required, respectively, in 4 of 125 (3.2%) and 6 of 125 patients (4.8%), without consistent genotype-phenotype correlations.
Figure 4. Respiratory, feeding, and orthopedic procedures.

(A) Prevalence of NNIV, G/J, and SS according to genetic background: overall about one-third of cases required NNIV and G/J insertion. Only a minority of cases required scoliosis surgery. (B) Kaplan-Meier curves showing ventilation, G/J, and SS-free patients: NNIV was started at a mean age of 8.53 years, whereas G/J was placed earlier, at a mean age of 2.74 years, usually within the first year. SS was performed at a mean age of 12.0 years. AD = autosomal dominant; AR = autosomal recessive; CM = congenital myopathy; G/J = gastrostomy/jejunostomy; NNIV = nocturnal noninvasive ventilation; SS = scoliosis surgery.
Bulbar impairment.
Fifty-eight of 125 (46.4%) had bulbar impairment, and figure 4 illustrates the proportion requiring G/J insertion. Seven patients in whom neonatal NGT was required died in their first months, without undergoing any feeding tube placement because of marked respiratory impairment. Among the surviving patients requiring NGT at birth, all underwent subsequent G/J placement, but 4 patients affected by RYR1-related myopathy (2 with AR, 2 with AD inheritance) improved to an extent that this became unnecessary. In 11 patients who did not require neonatal NGT, G/J was placed because of subsequent failure to thrive (height/weight less than third percentile).
Orthopedic complications.
There were orthopedic complications in mainly 50 of 125 patients (40%) who developed scoliosis, at a mean age of 7.2 years (median 7.0 years, SD 4.33, age range birth to 16 years). Only 3 cases had congenital scoliosis. Scoliosis surgery was performed in 17 of 125 (13.6%) (figure 4). Patients requiring scoliosis surgery were equally distributed between ambulant (8/17) and nonambulant (9/17) groups. Almost all patients had respiratory impairment at the time of surgery. There were no perioperative deaths. All patients maintained previous motor abilities after scoliosis surgery. In 71 of 125 (56.8%), tightness of the tendon Achilles was observed throughout different genetic backgrounds; 5 of 125 (4%) underwent tendon Achilles lengthening. Nine of 125 (7.2%) had congenital hip dysplasia, mainly in the AD RYR1-mutated subset (4/9). Three of 125 (2.4%) required hip surgery. Congenital talipes equinovarus was present in 11 of 125 (8.8%) (4/11 NEB-mutated, 3/11 currently genetically unsolved NM).
Other complications.
Complications included a dilated cardiomyopathy with onset at 14 years of age in patient 108 with genetically unresolved CNM. Mild learning difficulties were reported in 5 of 125 (4%) (4/5 born from consanguineous parents), without correlation to specific genotypes. Undescended testis was reported in 15 of 61 males (24.5%), mainly with MTM1 and recessive RYR1 mutations (4/7, 57.1%, and 6/9, 66.7%, respectively). Seven of 15 underwent orchidopexy. Malignant hyperthermia was reported in families with dominant RYR1 mutations (patients 59–60 and 67), and in patients 77 and 79 with recessive RYR1; the latter patients had both facial dysmorphisms and contractures, compatible with King-Denborough syndrome.24
Mortality.
Mortality was higher in the group with early-onset severe presentations: 15 of 125 (12%) died, at a mean age of 1.29 years (median 0.66 years, SD 1.72, range birth to 6 years), mainly secondary to respiratory infections (figure 2, B and C) and on the background of universal respiratory impairment, with 8 of 15 ventilated at the time of death.
Distinct clinical presentations and clear genotype-phenotype correlations were identified in the most common genetic subtypes (figures 1–4).
MTM1 patients universally had severe neonatal onset requiring respiratory (8/8) and feeding (7/8) support. Neonatal complications were anticipated in a high proportion of pregnancies with decreased fetal movements (6/8, 75%) and polyhydramnios (4/8, 50%); the latter feature probably also responsible for the high percentage of preterm deliveries (5/8, 62.5%), contributing to a more severe course. All 3 long-term survivors were wheelchair-dependent and ventilated: patients 30 and 33 on NNIV and patient 32 on DNIV; the latter was also the only patient with severe learning difficulties and focal epilepsy in our cohort.
Within the RYR1 group, neonatal onset was more frequent in AR compared with AD cases (12/23, 52.2%, and 5/21, 23.8%, respectively), with an increased occurrence of neonatal complications in the AR subset. Bulbar and respiratory involvement was more frequently observed in the AR subgroup, where 8 of 23 (34.8%) underwent G/J (p = 0.0028, χ2 test) and 3 of 23 (13.4%) required NNIV; this was not a feature in AD cases. Motor abilities were comparable in both subgroups: 17 of 21 AD (81%) and 18 of 22 AR patients (82%) achieved independent ambulation; 10 of 17 AD (58.8%) and 11 of 18 AR patients (67.1%) had walking delay. Six of 44 ambulant patients (13.6%) became wheelchair-dependent (4 with AR inheritance, 2 with AD). No patient required DNIV and tracheostomy in either subset. Lethal cases were not observed.
ACTA1 and NEB were the genes most frequently implicated in NM. All ACTA1 mutations were de novo dominant with the exception of patient 14 with a homozygous recessive mutation inherited from asymptomatic parents, and patient 13 who carried the same dominant mutation as her mildly affected mother. Neonatal onset was the main presentation in both subgroups, but ACTA1 patients had a higher rate of neonatal complications compared with NEB. All NEB cases acquired independent ambulation, while motor abilities of patients with the ACTA1 mutation were more variable, with 10 of 15 (66.74%) achieving independent ambulation. Patient 8 was the only patient with the ACTA1 mutation who became wheelchair-dependent, whereas patient 25, with the NEB mutation, was using a wheelchair outdoors at 14 years of age. Both G/J and NNIV were required in about half of cases and at a similar age: NNIV was started in patients with ACTA1 mutation at a mean age of 8.5 years (median 2.5 years, SD 11.36) and in patients with NEB mutation at a mean age of 7.3 years (median 6.5 years, SD 4.3); G/J was placed in patients with ACTA1 mutation at a mean age of 2.8 years (median 1 year, SD 4.14) and in patients with NEB mutation at a mean age of 5.4 years (median 2.65 years, SD 7.03). In NEB cases, G/J was never preceded by NGT placement, because failure to thrive rather than feeding difficulties was the main feature, whereas in most ACTA1 patients, neonatal swallowing dysfunction was observed.
All SEPN1 patients presented beyond the neonatal period; the only 2 cases with onset in the second decade both had SEPN1 mutations. All SEPN1 cases achieved independent ambulation, mainly before 18 months. Patients with the SEPN1 mutation had the highest prevalence of scoliosis and NNIV requirement. Four of 16 cases (25%), all with restrictive respiratory impairment requiring NNIV, showed right ventricle impairment and pulmonary hypertension.
DISCUSSION
CMs are rare neuromuscular conditions with distinct histologic and clinical features, and multiple genetic backgrounds.1,3,12 Longitudinal studies outlining frequency of complications and progression over time are scarce. Here, we delineate the natural history of a large cohort of CM cases. Most patients had a genetic diagnosis, with core myopathies and RYR1 mutations, respectively, the most common histopathologic and genetic diagnoses, as reported.11,13,25 Bearing in mind that the defining features of the CMs (such as FTD, nemaline rods, cores, and central nuclei) are not specific and may occur in other clinical situations,26 including CNS developmental malformations27 and recently recognized multisystem disorders with primary autophagy defects,28,29 we took great care that only patients with clinicopathologic features of a CM without evidence of other underlying conditions were included in this study.
The onset of clinical signs was observed predominantly within the first year of life: approximately half of the patients presented in the neonatal period with severe feeding and respiratory complications, as described.1–3,13–15,19 Neonatal presentation was more frequently preceded by pregnancy complications, including polyhydramnios and preterm deliveries, compared with a previous study in which only a few neonates were included.30
Although lethal outcome for a few specific conditions has been reported,15,17,19,26,27 the relative mortality rate of different CM subgroups has not been investigated. In our cohort, 12% of patients died, predominantly within the first year of life. The high mortality of X-linked myotubular myopathy18 was confirmed, together with the lethality of severe congenital NM.19 All deceased patients had profound respiratory impairment, with respiratory infections often the precipitating event in neonates/infants with respiratory involvement, highlighting the need for early recognition of infections, timely institution of treatment, and consideration of ventilatory support as part of a proactive management plan.31
Although CMs are considered essentially static conditions, the clinical course may be progressive. In particular, failure to thrive and insidious respiratory insufficiency beyond the first year were observed in a substantial proportion, as reported in smaller series.5,13–15,19 It is often thought that children with CMs who achieve independent ambulation usually do not lose this ability later.4 However, intermittent wheelchair use was needed in 22% of our ambulant patients, whereas 9% became completely wheelchair-dependent within a few years, supporting observations of deterioration in muscle strength after the achievement of ambulation.32 All patients who lost ambulation were late walkers; this may suggest that ambulatory loss reflects the additional effects of increased height/weight on already substantially compromised muscle power, rather than a progressive loss of muscle strength.
The prevalence of orthopedic complications, in particular scoliosis, has not been extensively investigated in CMs.5,33 We demonstrated that early-onset scoliosis is common, but usually mild and needing surgery only in 13.6% of all CMs. Half of all patients with scoliosis were ambulant at the time of surgery, suggesting that the development of scoliosis is more determined by early and disproportionate axial weakness1,3,13 rather than ambulatory state. Scoliosis surgery was overall well tolerated, with preoperative motor abilities and respiratory performance maintained. There were no perioperative deaths, probably reflecting the absence of severe cardiac involvement. Indeed, cardiac involvement was rare, mainly right ventricle involvement associated with restrictive pulmonary impairment in SEPN1-mutated cases.5,34 A relatively high incidence of talipes was observed mainly among patients with NM, most often with NEB mutation, reflecting distal involvement and genetic overlap between the NM and the distal arthrogryposis spectrum.
We identified consistent genotype-phenotype correlations of relevance to clinical practice. At the severe end of the spectrum, dramatic neonatal onset with bulbar and respiratory complications was observed in all patients with MTM1 mutation,18 but also in several patients with ACTA1 mutation,35,36 a few RYR1 cases, mainly with recessive inheritance,17,37 and some patients with KLHL40 mutation.38 Despite severe neonatal onset in some cases, all patients with RYR1 mutation survived, in keeping with only a few lethal cases previously reported in the literature.17,39,40,e1 We found that in RYR1-related myopathies, feeding difficulties can be transitory: the only 4 neonates in our cohort who were tube-fed, but did not subsequently require G/J placement, all had RYR1 mutations. In 2 additional patients with RYR1 mutation, gastrostomy insertion performed early in life was subsequently removed because of spontaneous swallowing improvement and/or improved weight gain. Whereas respiratory support at birth may be needed in a proportion of RYR1 patients, NNIV requirement over the following years is not a feature. Taken together, those data suggest an overall more benign course of congenital RYR1-related myopathies regarding bulbar complications and survival, in keeping with smaller studies.13,e2,e3
Within NMs, patients with NEB mutations are thought to be more mildly affected than those with ACTA1 mutations,15 although rare case reports have suggested a lethal course in patients with NEB mutations,e4 as well as much milder and adult phenotypes of ACTA1.8 Of note, in our cohort, ACTA1 and NEB subgroups required NNIV and G/J in similar proportion, highlighting that, with the exception of early lethal ACTA1 cases, bulbar and respiratory complications are comparable over time. Those findings are in contrast to a recent smaller survey13 in which patients with ACTA1 mutation most frequently required G/J insertion, probably because of a shorter period of observation.
In conclusion, our study expands the clinical spectrum of CMs, assesses the overall mortality, and suggests a certain decline of motor function particularly in the subset in which independent ambulation could only be achieved with difficulty. We provide valuable information for counseling according to genotype, and to improve management plans. Data were not prospectively collected, and only 14.4% of patients were adults at the last follow-up, which could be a limitation. This highlights the need for further prospective studies, including adults, focusing on validation of functional measures for CMs that may be useful for clinical trials.
Supplementary Material
ACKNOWLEDGMENT
The authors are grateful to Deborah Ridout, Centre for Paediatric Epidemiology and Biostatistics/MRC Centre of Epidemiology for Child Health, UCL Institute of Child Health, London, for statistical advice.
GLOSSARY
- ACTA1
skeletal muscle α-actin
- AD
autosomal dominant
- AR
autosomal recessive
- CM
congenital myopathy
- CNM
centronuclear myopathy
- DNIV
daily noninvasive ventilation
- DNM2
dynamin 2
- FTD
fiber type disproportion
- G/J
gastrostomy/jejunostomy
- KLHL40
kelch-like family member 40
- MTM1
myotubularin
- NEB
nebulin
- NGT
nasogastric tube
- NM
nemaline myopathy
- NNIV
nocturnal noninvasive ventilation
- NSMC
nonspecific myopathic changes
- RYR1
ryanodine receptor type 1
- SEPN1
selenoprotein N
- TPM2
β-tropomyosin
- TPM3
tropomyosin 3
Footnotes
Editorial, page 15
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
I. Colombo: design of the study, analysis and interpretation of the data, drafting the manuscript. M. Scoto: design of the study, interpretation of the data, revising the manuscript. A.Y. Manzur: design of the study, interpretation of the data. S.A. Robb: interpretation of the data, revising the manuscript. L. Maggi: interpretation of the data, revising the manuscript. V. Gowda: revising the manuscript. T. Cullup: analysis of the data, revising the manuscript. M. Yau: analysis of the data. R. Phadke: design of the study, analysis of the data. C. Sewry: analysis of the data, revising the manuscript. H. Jungbluth: design of the study, interpretation of the data, revising the manuscript. F. Muntoni: design of the study, interpretation of the data, revising the manuscript.
STUDY FUNDING
The Dubowitz Centre for Congenital Muscular Dystrophies and Myopathies is financed by the UK National Health System (NHS), Highly Specialised Services National Specialised Commissioning Team.
DISCLOSURE
I. Colombo was supported by ENS Fellowship 2012. M. Scoto, A. Manzur, S. Robb, L. Maggi, and V. Gowda report no disclosures relevant to the manuscript. T. Cullup was supported by grants from the Guy's and St. Thomas' Charitable Foundation and the Myotubular Trust. M. Yau, R. Phadke, and C. Sewry report no disclosures relevant to the manuscript. H. Jungbluth was supported by grants from the Guy's and St. Thomas' Charitable Foundation and the Myotubular Trust. F. Muntoni is supported by the Great Ormond Street Children's Charity and its Biomedical Research Centre. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Sewry C, Jimenez-Mallebrera C, Muntoni F. Congenital myopathies. Curr Opin Neurol 2008;21:569–575. [DOI] [PubMed] [Google Scholar]
- 2.Laing N. Congenital myopathies. Curr Opin Neurol 2007;20:583–589. [DOI] [PubMed] [Google Scholar]
- 3.North KN, Wang CH, Clarke N, et al. Approach to the diagnosis of congenital myopathies. Neuromuscul Disord 2014;24:97–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang CH, Dowling JJ, North K, et al. Consensus statement on standard of care for congenital myopathies. J Child Neurol 2012;27:363–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scoto M, Cirak S, Mein R, et al. SEPN1-related myopathies: clinical course in a large cohort of patients. Neurology 2011;76:2073–2078. [DOI] [PubMed] [Google Scholar]
- 6.Løseth S, Voermans NC, Torbergsen T, et al. A novel late-onset axial myopathy associated with mutations in the skeletal muscle ryanodine receptor (RYR1) gene. J Neurol 2013;260:1504–1510. [DOI] [PubMed] [Google Scholar]
- 7.Lamont PJ, Dubowitz V, Landon DN, et al. Fifty year follow-up of a patient with central core disease shows slow but definite progression. Neuromuscul Disord 1998;8:385–391. [DOI] [PubMed] [Google Scholar]
- 8.Jungbluth H, Sewry CA, Brown SC, et al. Mild phenotype of nemaline myopathy with sleep hypoventilation due to a mutation in the skeletal muscle alpha-actin (ACTA1) gene. Neuromuscul Disord 2001;11:35–40. [DOI] [PubMed] [Google Scholar]
- 9.Wallgren-Pettersson C, Lehtokari VL, Kalimo H, et al. Distal myopathy caused by homozygous missense mutations in the nebulin gene. Brain 2007;130:1465–1476. [DOI] [PubMed] [Google Scholar]
- 10.Darin N, Tulinius M. Neuromuscular disorders in childhood: a descriptive epidemiological study from western Sweden. Neuromuscul Disord 2000;10:1–9. [DOI] [PubMed] [Google Scholar]
- 11.Norwood FL, Harling C, Chinnery PF, et al. Prevalence of genetic muscle disease in Northern England: in-depth analysis of a muscle clinic population. Brain 2009;132:3175–3186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaplan JC, Hamroun D. The 2014 version of the gene table of monogenic neuromuscular disorders (nuclear genome). Neuromuscul Disord 2013;23:1081–1111. [DOI] [PubMed] [Google Scholar]
- 13.Maggi L, Scoto M, Cirak S, et al. Congenital myopathies—clinical features and frequency of individual subtypes diagnosed over a 5-year period in the United Kingdom. Neuromuscul Disord 2013;23:195–205. [DOI] [PubMed] [Google Scholar]
- 14.Herman GE, Finegold M, Zhao W, et al. Medical complications in long-term survivors with X-linked myotubular myopathy. J Pediatr 1999;134:206–214. [DOI] [PubMed] [Google Scholar]
- 15.Wallgren-Pettersson C, Pelin K, Nowak KJ, et al. Genotype-phenotype correlations in nemaline myopathy caused by mutations in the genes for nebulin and skeletal muscle alpha-actin. Neuromuscul Disord 2004;14:461–470. [DOI] [PubMed] [Google Scholar]
- 16.Catteruccia M, Fattori F, Codemo V, et al. Centronuclear myopathy related to dynamin 2 mutations: clinical, morphological, muscle imaging and genetic features of an Italian cohort. Neuromuscul Disord 2013;23:229–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Klein A, Lillis S, Munteanu I, et al. Clinical and genetic findings in a large cohort of patients with ryanodine receptor 1 gene-associated myopathies. Hum Mutat 2012;33:1310. [DOI] [PubMed] [Google Scholar]
- 18.McEntagart M, Parsons G, Buj-Bello A, et al. Genotype-phenotype correlations in X-linked myotubular myopathy. Neuromuscul Disord 2002;12:939–946. [DOI] [PubMed] [Google Scholar]
- 19.Ryan MM, Schnell C, Strickland CD, et al. Nemaline myopathy: a clinical study of 143 cases. Ann Neurol 2001;50:312–320. [DOI] [PubMed] [Google Scholar]
- 20.Anderson SL, Ekstein J, Donnelly MC. Nemaline myopathy in the Ashkenazi Jewish population is caused by a deletion in the nebulin gene. Hum Genet 2004;115:185–190. [DOI] [PubMed] [Google Scholar]
- 21.Scoto M, Cullup T, Cirak S. Nebulin (NEB) mutations in a childhood onset distal myopathy with rods and cores uncovered by next generation sequencing. Eur J Hum Genet 2013;21:1249–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dubowitz V, Sewry CA, Oldfors A. Muscle Biopsy: A Practical Approach, 4th ed Edinburgh: Saunders/Elsevier; 2013. [Google Scholar]
- 23.Blencowe H, Cousens S, Oestergaard MZ. National, regional, and worldwide estimates of preterm birth rates in the year 2010 with time trends since 1990 for selected countries: a systematic analysis and implications Lancet 2012;379:2162–2172. [DOI] [PubMed] [Google Scholar]
- 24.Dowling JJ, Lillis S, Amburgey K, et al. King-Denborough syndrome with and without mutations in the skeletal muscle ryanodine receptor (RYR1) gene. Neuromuscul Disord 2011;21:420–427. [DOI] [PubMed] [Google Scholar]
- 25.Amburgey K, McNamara N, Bennett LR, et al. Prevalence of congenital myopathies in a representative pediatric United States population. Ann Neurol 2011;70:662–665. [DOI] [PubMed] [Google Scholar]
- 26.Wallgren-Pettersson C, Jungbluth H. The congenital (structural) myopathies. In: Rimoin DL, Connor JM, Pyeritz RE, Korf B, editors. Emery and Rimoin's Principles of Practicing Medical Genetics. Edinburgh: Churchill Livingstone; 2007:2963–3000. [Google Scholar]
- 27.Sarnat HB. Cerebral dysgeneses and their influence on fetal muscle development. Brain Dev 1986;8:495–499. [DOI] [PubMed] [Google Scholar]
- 28.McClelland V, Cullup T, Bodi I, et al. Vici syndrome associated with sensorineural hearing loss and evidence of neuromuscular involvement on muscle biopsy. Am J Med Genet A 2010;152A:741–747. [DOI] [PubMed] [Google Scholar]
- 29.Cullup T, Kho AL, Dionisi-Vici C, et al. Recessive mutations in EPG5 cause Vici syndrome, a multisystem disorder with defective autophagy. Nat Genet 2013;45:83–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Awater C, Zerres K, Rudnik-Schöneborn S. Pregnancy course and outcome in women with hereditary neuromuscular disorders: comparison of obstetric risks in 178 patients. Eur J Obstet Gynecol Reprod Biol 2012;162:153–159. [DOI] [PubMed] [Google Scholar]
- 31.Wallgren-Pettersson C, Bushby K, Mellies U, Simonds A; ENMC. 117th ENMC workshop: ventilatory support in congenital neuromuscular disorders—congenital myopathies, congenital muscular dystrophies, congenital myotonic dystrophy and SMA (II) 4–6 April 2003, Naarden, The Netherlands. Neuromuscul Disord 2004;14:56–69. [DOI] [PubMed] [Google Scholar]
- 32.Akiyama C, Nonaka I. A follow-up study of congenital non-progressive myopathies. Brain Dev 1996;18:404–408. [DOI] [PubMed] [Google Scholar]
- 33.Gamble JG, Rinsky LA, Lee JH. Orthopaedic aspects of central core disease. J Bone Joint Surg Am 1988;70:1061–1066. [PubMed] [Google Scholar]
- 34.Ferreiro A, Estournet B, Chateau D, et al. Multi-minicore disease—searching for boundaries: phenotype analysis of 38 cases. Ann Neurol 2000;48:745–757. [PubMed] [Google Scholar]
- 35.Agrawal PB, Strickland CD, Midgett C, et al. Heterogeneity of nemaline myopathy cases with skeletal muscle alpha-actin gene mutations. Ann Neurol 2004;56:86–96. [DOI] [PubMed] [Google Scholar]
- 36.Sparrow JC, Nowak KJ, Durling HJ, et al. Muscle disease caused by mutations in the skeletal muscle alpha-actin gene (ACTA1). Neuromuscul Disord 2003;13:519–531. [DOI] [PubMed] [Google Scholar]
- 37.Bharucha-Goebel DX, Santi M, Medne L, et al. Severe congenital RYR1-associated myopathy: the expanding clinicopathologic and genetic spectrum. Neurology 2013;80:1584–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ravenscroft G, Miyatake S, Lehtokari VL, et al. Mutations in KLHL40 are a frequent cause of severe autosomal-recessive nemaline myopathy. Am J Hum Genet 2013;93:6–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Romero NB, Monnier N, Viollet L, et al. Dominant and recessive central core disease associated with RYR1 mutations and fetal akinesia. Brain 2003;126:2341–2349. [DOI] [PubMed] [Google Scholar]
- 40.Monnier N, Laquerrière A, Marret S, et al. First genomic rearrangement of the RYR1 gene associated with an atypical presentation of lethal neonatal hypotonia. Neuromuscul Disord 2009;19:680–684. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.