

Abstract
Saccadic oscillations are continuous back-to-back saccades that cause excessive image motion across fovea and threaten clear vision. Acquired processes, related to immune or metabolic mechanisms, are common culprits. Saccadic oscillations are also seen in degenerative cerebellar disease or as a part of a familial syndrome of saccadic oscillations and limb tremor. Some normal individuals have innate ability to voluntarily trigger saccadic oscillations (i.e. voluntary nystagmus). Contemporary theory for the pathogenesis for saccadic oscillations has emphasized hyperexcitable or disinhibited state of the brainstem saccadic burst neuron membrane. This review discusses etiologies and treatment of saccadic oscillations in light of novel cell membrane based theory.
Keywords: ion channels, saccade, opsoclonus, computational simulation, oscillopsia, genetic, paraneoplastic
Gaze stabilization on the object of interest is a fundamental requirement for clear vision. The brain has a number of mechanisms to achieve gaze stabilization. Vestibulo-ocular reflex (VOR) keeps the eyes on target by shifting them with an equal amplitude and velocity as head movements, but in the opposite direction [1]. Cerebellar and brainstem mediated gaze holding mechanisms keep the eyes steady when the head is not in motion. Saccadic eye movements rapidly shift the gaze from one target to the other and facilitate scanning of the environment [1]. Abnormal spontaneous eye movements, such as nystagmus and saccadic oscillations, are threats to steady fixation and clear vision. There are two types of nystagmus. Jerk nystagmus is comprised of the drift of the eyes away from the desired target followed by a corrective rapid movement (quick phase) back to the target. Pendular nystagmus features sinusoidal or quasi sinusoidal eye movement trajectory [1]. Saccadic oscillations, back-to-back saccades (without inter-saccadic interval), also appear to have sinusoidal trajectory [1]. The key difference between the clinical appearance of saccadic oscillations and pendular nystagmus is the frequency of oscillations. The oscillations in pendular nystagmus are generally less than 10 Hz (typically 2-6 Hz) , whereas the frequency of saccadic oscillations is more than 15 Hz (typically 20-40 Hz)[1]. The differences are attributed discrete pathophysiology of saccadic oscillations and pendular nystagmus [1]. Saccadic oscillations can be unidimensional (typically horizontal), or multi-dimensional. When saccadic oscillations are only present in the horizontal plane, they are called ocular flutter. If saccadic oscillations are present in all three planes, they are called opsoclonus [1]. Here we have restricted the use of the term ‘saccadic oscillations’ for both ocular flutter and opsoclonus.
The aim of this review is to discuss the etiology and contemporary proposals for the pathophysiology of saccadic oscillations.
Etiology of saccadic oscillations
Saccadic oscillations are analogous to common tremor disorders of the head and limbs[2]. Some tremor disorders have clear relationship to an anatomical abnormality (e.g. Parkinson's disease or degenerative cerebellar disease), yet others occur despite the central nervous system appears structurally sound, e.g. essential tremor, physiological tremor and its exaggerated forms in hyperthyroidism. Similarly, saccadic oscillations are present in patients with gross structural deficits that impair the function of specific cell populations or their connections. For example saccadic oscillations are seen in degenerative disorders of cerebellar Purkinje neurons [3], demyelinating disease [4], or intracranial mass [5]. Saccadic oscillations can also occur in subjects without structural neurological deficit. Examples of these include physiological saccadic oscillations during eye closure and with convergence[6-8], familial saccadic oscillations [2], saccadic oscillations secondary to drug intoxications[9,10], transient saccadic oscillations in the newborn[11], and saccadic oscillations associated with migraine[12].
Mechanisms of saccadic oscillations
There are at least two independent hypotheses that describe saccadic oscillations[3,13-15]. The first hypothesis accounts for saccadic oscillations in patients with gross structural lesions. The second hypothesis describes saccadic oscillations in the absence of structural lesions emphasizing the role of neuronal hyperexcitability due to transient (or permanent) deficits of the ion channel or neurotransmitter receptors at the membranes of saccade generators[13]. The functional organization of the saccade generator is the backbone of the both hypotheses. The subsequent sections will first outline the principles of saccade generation and then discuss the mechanisms of saccadic oscillations.
Functional organization of the neural circuit that generates saccadic oscillations
Commands to shift gaze are initiated within the frontal and parietal eye fields of the cerebral cortex, from here saccade-related signals follow two parallel pathways (Figure 1). Both pathways are destined for omnidirectional pause neurons (OPN) in the saccadic burst generator area at the nucleus raphe interpositus of the midline pons [16, 17]. One pathway projects to OPN directly via the superior colliculus (SC), but the other projects to the nucleus reticularis tegmenti pontis (NRTP) of the pontine reticular formation, then to the oculomotor vermis (OMV, lobules 5-7) of the cerebellar cortex. Oculomotor vermis sends GABAergic inhibitory signals to the underlying caudal fastigial nucleus (FOR, Figure 1). FOR then projects to OPN [18]. OPNs inhibit excitatory burst neurons (EBN), located in the caudal pontine reticular formation and inhibitory burst neurons (IBN) located in the nucleus paragigantocelluraris dorsalis. Normally this inhibition is critical to induce fast velocities of saccades during desired gaze shifts and prevent unwanted intrusions when steady gaze holding is required [2,13,17,18].
Figure 1.
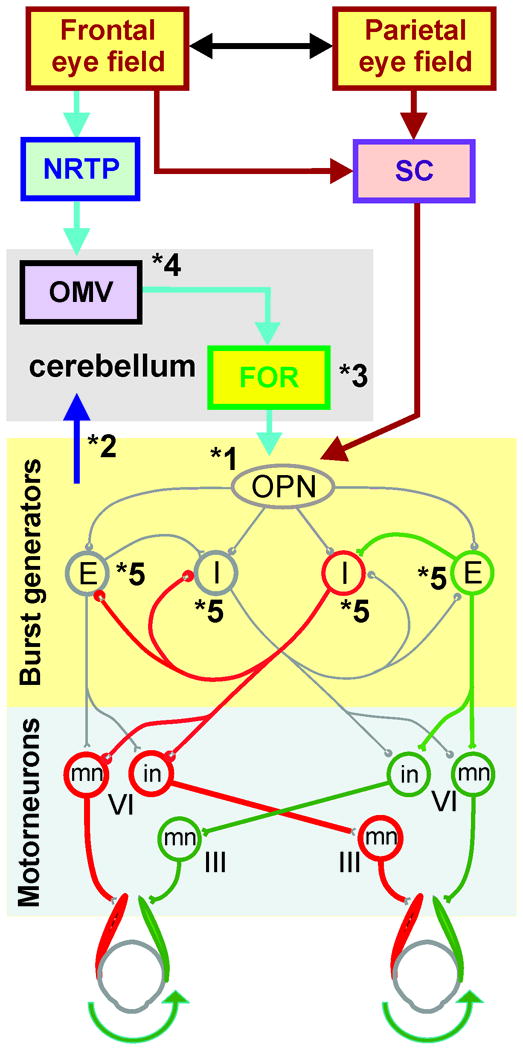
This schematic diagram summarizes central saccade generating pathways. NRTP: Nucleus reticularis tegmenti pontis; SC: superior coliculus; OMV: oculomotor vermis; FOR: fastigial oculomotor region, FOR; OPN: omnipause neurons; E: excitatory burst neurons; I: inhibitory burst neurons; III: oculomotor nucleus; VI: abducens nucleus; in: interneuron; mn: motoneuron, Dashed line: midline, green pathway: excitatory projection; red pathway: inhibitory projections. (Modified from Shaikh, et al., 2008 with permission [13])
The EBNs directly excite the ipsilateral motoneurons (mn) and interneurons (in) of the abducens nucleus (VIth nucleus) (green pathway Figure 1). The interneurons (in) project to the contralateral oculomotor nucleus (IIIrd nucleus) via medial longitudinal fasciculus (MLF). The IBNs inhibit the contralateral abducens motor neurons and ipsilateral oculomotor nucleus motor neurons (red pathways Figure 1). Thus for a rightward saccade (as shown in Figure 1), the inhibition of the right IBN is released. Latter facilitates the inhibition of the left abducens motoneurons and right oculomotor motonerons. Simultaneously, right EBNs excite the right abducens motoneurons and left oculomotor motoneurons. The reciprocal innervations of yoked pair of ocular muscles rapidly shifts the gaze to the desired position.
Unfortunately, reciprocal innervation makes the circuit of inhibitory and excitatory burst neurons vulnerable to internally generated oscillations. These oscillations are normally suppressed by the inhibition through the OPNs [2,14]. Disinhibition of the reciprocally innervated burst neurons is cardinal to generation of saccadic oscillations[2,13].
Mechanisms of saccadic oscillations secondary to gross structural deficits
Structural injury to the brainstem saccade generator, cerebellar cortex, deep cerebellar nuclei, or the white matter tracks connecting these areas can result in saccadic oscillations. It was hypothesized that the structural injury due to any etiology ultimately leads to hyperexcitability or disinhibition of the burst neurons causing saccadic oscillations[12,13,19]. Immune-mediated lesion of OPN can cause paraneoplastic opsoclonus[20]. It is intuitive that damage of OPN would cause opsoclonus by disinhibiting the reciprocally innervated circuit of the burst neurons. The caveat to this hypothesis is that in primate model, injection of muscimol (agonist of chloride channel that reversibly inactivates the neuron) did not cause saccadic oscillations but slowed saccades[21]. In contrast, in human subjects transient physiological inhibition of OPN during active eye lid closure caused saccadic oscillations superimposed upon the slow saccades[6]. Therefore it is speculated that lack of saccadic oscillations in primate model could be due to species related differences in the neuronal infrastructure between monkeys and humans.
Degenerative cerebellar disorders often cause saccadic oscillations [3]. We know that FOR participates in the feedback loop for the brainstem saccade generation (Figure 1). Degenerative cerebellar disease causes secondary disinhibition of the FOR leading to the instability of the saccadic burst generators and saccadic oscillations[22,23].
Patients with demyelinating disease or developmental absence of myelination can present with saccadic oscillations[4,11]. It is hypothesized that saccadic oscillations in patients with lack of myelin is due to the abnormal delay in the conduction through the crossing tracks of the reciprocally innervating pontine burst neurons.
Mechanisms of saccadic oscillations in a structurally-intact nervous system
Saccadic burst generators comprise reciprocally innervating circuit to facilitate prompt initiation, rapid velocity, and abrupt cessation of saccades [1]. This efficient organization of the system is associated with a caveat. During certain pathological conditions, when disinhibited, this circuit internally generates oscillations. When transmitted further downstream, these reverberations cause saccadic oscillations. A number of etiologies can transiently or permanently disinhibit the burst generators causing saccadic oscillations. In the subsequent section the mechanisms of saccadic oscillations in various etiologies will be discussed starting with the general concept underlying the internally generated oscillations.
A hyperexcitable state of the saccadic burst generators as well as abnormally decreased inhibition (via OPN) of the burst neurons can trigger internally generated oscillations within the circuit. The neuronal excitability after cessation of transient inhibition is determined by a rebound increase in neuronal membrane discharge after sustained membrane hyperpolarization (post-inhibitory rebound; PIR). It was proposed that the strength of PIR is fundamental to generate rapid velocities of saccades in healthy subjects and saccadic oscillations during diseased states[2,6]. Hyperpolarization-activated mixed cation currents (Ih), and low threshold calcium currents (IT) are key determinants of neuronal excitability and PIR [24-26] (schematized in Figure 2). The ion channels carrying Ih and IT were also identified in the EBNs, IBNs, and OPNs of human tissue[27]. Hence any etiology that directly or indirectly increases the Ih or IT can lead to hyperexcitable state and subsequently saccadic oscillations.
Figure 2.
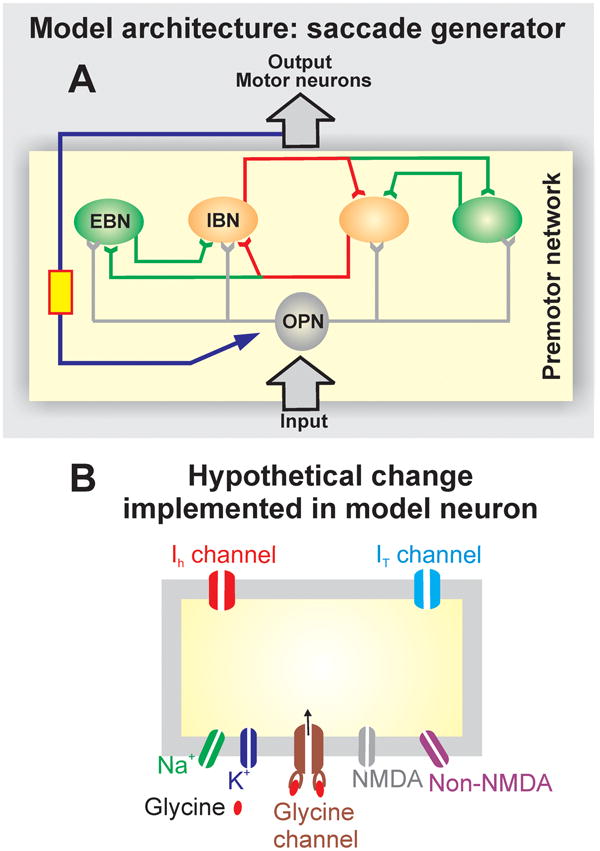
This is the outline of neuromimetic model of saccadic burst generators (see [2] for deails). The model is based on traditional local-feedback model of the burst neuron [19]. Red is inhibitory and green is excitatory local feedback (A). Such feedback is fundamental to efficiently accomplish accurate yet high velocity saccades. (B) Schematic illustration of the neuromimetic model of the burst neuron and its membrane. The neuromimetic model obeys the classic Hodgkin-Huxley equations for membrane potential, in addition, it also has parameters for hyperpolarization activated cation current (Ih), low-threshold calcium current (IT), NMDA and AMPA sensitive glutamate receptors and glycine receptor (Modified from Shaikh, et al., 2008 with permission [13]).
This hypothesis was tested in a neuromimetic model of saccade generator[2]. The model featured anatomically realistic local-feedback loops between the Hodgkin-Huxley type burst neurons (red is inhibitory and green is excitatory local feedback, schematized in Figure 2A) [2,14,19,28-30]. Under physiological circumstances the model simulated normal saccades. However increased neuronal excitability of the modeled burst neurons, by increasing the membrane parameters that describes increases in Ih and/or IT caused saccadic oscillations [2]. Transient reduction in the inhibition via simulated OPNs caused slow saccades and superimposed saccadic oscillations[6].
The model had two key predictions: 1) Increases in IT increases the amplitude but decreases the frequency of saccadic oscillations; and 2) Increases in Ih increases the frequency but decreases the amplitude of oscillations[2]. These hypotheses were later tested pharmacologically on physiological saccadic oscillation in two healthy subjects. In both subjects, selective blockade of IT with ethosuximide decreased the amplitude of oscillations but increased their frequency[31]. The model also predicted that the oscillation amplitude is more sensitive to the changes in IT as compared to the frequency. The effects of ethosuximide on the physiological saccadic oscillations also validated this prediction[31].
This neuromimetic model for saccades emphasized the role of increased excitability or disinhibition of the pontine burst neurons in pathophysiology of saccadic oscillations. In the subsequent section the contemporary neuromimetic model is put into clinical perspective to describe the mechanism of saccadic oscillations due to various etiologies and physiological saccadic oscillations that are seen in normal subjects.
Saccadic oscillations in cocaine abuse
Transient saccadic oscillations are seen in patients with cocaine intoxication[9]. Cocaine reduces the reuptake of norepinephrine, hence elevating its synaptic concentration[32]. Synaptic norepinephrine increases the maximal conductance through Ih channels at the post-synaptic membrane, hence increasing the excitability of the post-synaptic neuron[24,25,33]. In light of the recent neuromimetic model, increased strength of PIR and hyperexcitability of post-synaptic neuron due to cocaine intoxication explains saccadic oscillations[13].
Hyperammonemic and uremic states also cause saccadic oscillations[1]
Increased neural excitability again explains saccadic oscillations in such metabolically challenged states. Uremia or hyperammonemia decreases the extracellular pH. The acidic pH increases the maximal conductance through the Ih channel, inducing the hyperexcitable state[24,25,34]. Increased neuronal excitability triggers reverberations in the reciprocally innervated circuit of burst neurons.
Saccadic oscillations have been reported in organophosphate poisoning [35]
Organophosphate poisoning is known for inducing cholinergic excess. This cholinergic surge may increase the activation of FOR[35]. The hyperexcitability of FOR is then transmitted to saccadic burst generators via excitatory connections [18]. Hyperexcitable burst neurons will induce saccadic oscillations.
Strychnine poisoning is associated with saccadic oscillations[10]
Strychnine, a known blocker of glycine channel, may disinhibit the burst neurons. Reciprocally innervated circuit of disinhibited burst neurons will generate saccadic oscillations.
Saccadic oscillations are also associated with migraine[12]
Contemporary theories have described migraine as hyperexcitable state[36-38]. The circuit of hyperexcitable burst neurons can cause saccadic oscillations.
A number of paraneoplastic and autoimmune states are associated with saccadic oscillations
Paraneoplastic degeneration of the cerebellar cortex is one of the proposed hypotheses[39]. In an alternate hypothesis, it was proposed that immune-mediated “channelopathy” at the cell membranes of the saccadic burst generators cause saccadic oscillations [2,13]. The “channelopathy” increases the excitability of the burst neurons causing saccadic oscillations. This hypothesis explains saccadic oscillations in patients with paraneoplastic or post-immune disease state but normal brain MRI, the situation routinely encountered in the clinical practice.
Physiological saccadic oscillations are present in most healthy subjects
Some healthy subjects can voluntarily trigger robust physiological saccadic oscillations (voluntary nystagmus) by converging the eyes followed by making a small amplitude saccade at a distant target. Physiological saccadic oscillations are also seen during blinks or sustained eye closure[6,8]. Normally while making saccades, the oscillations are present along the axes orthogonal to that of saccades. For example, torsional and vertical saccadic oscillations are seen during horizontal saccades[2,6]. Transient removal of OPN inhibition on the reciprocally innervated circuit of burst neurons, during all of these three conditions, is the basis for physiological saccadic oscillations.
Genetics and saccadic oscillations
Some of the disorders mentioned above are common, why do all affected individuals not develop saccadic oscillations? The answer might remain within the genetic profile of the given individual. It is speculated that the genetic profile affects the expression of membrane ion-channels and cellular biology. We know that the state of membrane function affects the circuit behavior; latter determines the phenomenology of the disease. Hence, genetically determined variations in the expression profile of normally functioning ion-channels could alter the semiology of the disease. The same concept also explains why only some normal subjects can generate voluntary nystagmus. Genetic influence of this innate ability also explains relatively wide range of frequencies of saccadic oscillations that are seen across normal individuals, but within families, the frequency of saccadic oscillations is similar[40]. In a broader sense, there are number of known genetic mutations that manifest with wide spectrum of deficits, not all patients present with equal severity or constellation of symptoms, but latter might be similar in two genetically related individuals [41-43].
Clinical presentation and diagnosis of saccadic oscillations
Clinically the saccadic oscillations can present with a variety of symptoms. Some patients experience “shaking” of visual surround more so while shifting the gaze from one object to the other. Transient episodes of blurred vision lasting for seconds are also a common symptom of saccadic oscillations. It is very unusual, rather unlikely, for saccadic oscillations to present with vertigo (spinning of the environment). It is noteworthy that symptoms of saccadic oscillations are often masked by the symptoms of the primary etiology. Large saccadic oscillations can be diagnosed at bed-side with careful eye movement examination. In these patients, the oscillations are often transiently exaggerated after gaze shifts (see movie clip). Ophthalmoscopy or slit-lamp examination is often required to appreciate fine saccadic oscillations. High-resolution video-oculography and scleral search coils are the techniques of choice to quantitatively assess the saccadic oscillations.
Treatment of saccadic oscillations
The pharmacological treatment of saccadic oscillations has been described in isolated case reports, randomized drug trials are however lacking. The rationale of pharmacotherapy of saccadic oscillations is based upon the selective blockade of ion channels. Carbamazepine, a blocker of IT channel, reduces saccadic oscillations [44]. Selective blocker of IT channel, ethosuximide, also reduces saccadic oscillation[31]. Propranolol has successfully treated familial saccadic oscillations in one patient [31].
Expert commentary
Saccadic oscillations, back-to-back saccades without inter-saccadic interval, pose threat to clear vision. Neural correlates of saccadic oscillations could be either related to gross structural deficits in the saccadic burst neurons and their connections or related to the abnormal membrane properties of the saccadic burst generators. The oscillations are the result of unsuppressed instability in reciprocally innervating circuit of burst neurons. The instability could be due to lack of inhibition or excessive excitation of the burst neurons. Ion conductance such as Ih or IT determines the membrane excitability and thus the kinematic properties of saccadic oscillations. Successful therapeutic intervention depends on the blockade of membrane ion channels that determine resting membrane properties and excitability of the burst neurons.
Five-year view
Modern neuroscience research is heavily geared towards delineating the molecular biology, membrane physiology, and designing selective modulators of ion channels. Such progress will enhance target-directed therapy, customized to the etiology of saccadic oscillations. At the forefront of basic science understanding the physiology of saccades and saccadic oscillations has been supported by behavioral experiments and the outcomes of the pharmacotherapy, thus far. It is desirable to generate the animal model of saccadic oscillations using selective ion channel modulators to further confirm the role of a given ion channel in the pathophysiology of saccadic oscillations. The role of PIR has also been heavily emphasized in the membrane-based model of saccadic oscillations[2]. This emphasis is based upon the anatomical observations of the candidate ion channels in the human brain and computational modeling. In vitro experiments confirming the presence of PIR at saccade generating burst neurons are clearly justified. Although case reports have established the application of selective ion channel blockers in the treatment of saccadic oscillations systematic drug trials investigating the usefulness of these agents are justified in larger cohort of patients.
Supplementary Material
Executive summary.
Saccadic oscillations are back-to-back saccades without intersaccadic interval.
Clinical presentation of saccadic oscillations includes transient episodes of blurred vision or the “jitters” of the visual surround.
Disinhibited or hyperexcitable circuit of reciprocally innervating neurons in brainstem saccade generator could cause saccadic oscillations.
Saccadic oscillations are seen in toxic metabolic states, present as paraneoplastic disorder, or they could be familial.
Beta-blockers can be used to treat saccadic oscillations.
Acknowledgments
I thank Dr. David S. Zee for providing the movie clip and Dr. Benjamin Miller for reviewing the manuscript.
Reference List
- 1.Leigh RJ, Zee DS. The Neurology of Eye Movements. Oxford; New York: 2006. [Google Scholar]
- 2**.Shaikh AG, Miura K, Optican LM, Ramat S, Leigh RJ, Zee DS. A new familial disease of saccadic oscillations and limb tremor provides clues to mechanisms of common tremor disorders. Brain. 2007;130(Pt 11):3020–3031. doi: 10.1093/brain/awm240. Reference describes novel pathophysiology of familial saccadic oscillations. [DOI] [PubMed] [Google Scholar]
- 3.Shaikh AG, Marti S, Tarnutzer AA, et al. Gaze fixation deficits and their implication in ataxia-telangiectasia. J Neurol Neurosurg Psychiatry. 2009;80(8):858–864. doi: 10.1136/jnnp.2008.170522. [DOI] [PubMed] [Google Scholar]
- 4.Francis DA, Heron JR. Ocular flutter in suspected multiple sclerosis: a presenting paroxysmal manifestation. Postgrad Med J. 1985;61(714):333–334. doi: 10.1136/pgmj.61.714.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Keane JR, Devereaux MW. Opsoclonus associated with an intracranial tumor. A clinicopathologic case report. Arch Ophthalmol. 1974;92(5):443–445. doi: 10.1001/archopht.1974.01010010455017. [DOI] [PubMed] [Google Scholar]
- 6*.Shaikh AG, Wong AL, Optican LM, Miura K, Solomon D, Zee DS. Sustained eye closure slows saccades. Vision Res. 2010;50(17):1665–1675. doi: 10.1016/j.visres.2010.05.019. Describes role of omnipause neuron in saccade velocity and generation of oscillations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ramat S, Somers JT, Das VE, Leigh RJ. Conjugate ocular oscillations during shifts of the direction and depth of visual fixation. Invest Ophthalmol Vis Sci. 1999;40(8):1681–1686. [PubMed] [Google Scholar]
- 8*.Hain TC, Zee DS, Mordes M. Blink-induced saccadic oscillations. Ann Neurol. 1986;19(3):299–301. doi: 10.1002/ana.410190315. Shows that transient shut-down of omnipause neurons (during blink) causes saccadic oscillations. [DOI] [PubMed] [Google Scholar]
- 9.Scharf D. Opsoclonus-myoclonus following the intranasal usage of cocaine. J Neurol Neurosurg Psychiatry. 1989;52(12):1447–1448. doi: 10.1136/jnnp.52.12.1447-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blain PG, Nightingale S, Stoddart JC. Strychnine poisoning: abnormal eye movements. J Toxicol Clin Toxicol. 1982;19(2):215–217. doi: 10.3109/15563658208990383. [DOI] [PubMed] [Google Scholar]
- 11.Hoyt CS. Neonatal opsoclonus. J Pediatr Ophthalmol. 1977;14(5):274–277. [PubMed] [Google Scholar]
- 12.Ashe J, Hain TC, Zee DS, Schatz NJ. Microsaccadic flutter. Brain. 1991;114(Pt 1B):461–472. doi: 10.1093/brain/114.1.461. [DOI] [PubMed] [Google Scholar]
- 13**.Shaikh AG, Ramat S, Optican LM, Miura K, Leigh RJ, Zee DS. Saccadic burst cell membrane dysfunction is responsible for saccadic oscillations. J Neuroophthalmol. 2008;28(4):329–336. doi: 10.1097/WNO.0b013e31818eb3a5. Descriptive review of pathophysiology of saccadic oscillations in various neurological disorders. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ramat S, Leigh RJ, Zee DS, Optican LM. Ocular oscillations generated by coupling of brainstem excitatory and inhibitory saccadic burst neurons. Exp Brain Res. 2005;160(1):89–106. doi: 10.1007/s00221-004-1989-8. [DOI] [PubMed] [Google Scholar]
- 15.Leigh RJ, Robinson DA, Zee DS. A hypothetical explanation for periodic alternating nystagmus: instability in the optokinetic-vestibular system. Ann N Y Acad Sci. 1981;374:619–635. doi: 10.1111/j.1749-6632.1981.tb30906.x. [DOI] [PubMed] [Google Scholar]
- 16.Buttner-Ennever JA. Mapping the oculomotor system. Prog Brain Res. 2008;171:3–11. doi: 10.1016/S0079-6123(08)00601-8. [DOI] [PubMed] [Google Scholar]
- 17.May PJ, Hartwich-Young R, Nelson J, Sparks DL, Porter JD. Cerebellotectal pathways in the macaque: implications for collicular generation of saccades. Neuroscience. 1990;36(2):305–324. doi: 10.1016/0306-4522(90)90428-7. [DOI] [PubMed] [Google Scholar]
- 18.Noda H, Sugita S, Ikeda Y. Afferent and efferent connections of the oculomotor region of the fastigial nucleus in the macaque monkey. J Comp Neurol. 1990;302(2):330–348. doi: 10.1002/cne.903020211. [DOI] [PubMed] [Google Scholar]
- 19.Zee DS, Robinson DA. A hypothetical explanation of saccadic oscillations. Ann Neurol. 1979;5(5):405–414. doi: 10.1002/ana.410050502. [DOI] [PubMed] [Google Scholar]
- 20.Hormigo A, Dalmau J, Rosenblum MK, River ME, Posner JB. Immunological and pathological study of anti-Ri-associated encephalopathy. Ann Neurol. 1994;36(6):896–902. doi: 10.1002/ana.410360615. [DOI] [PubMed] [Google Scholar]
- 21.Soetedjo R, Kaneko CR, Fuchs AF. Evidence that the superior colliculus participates in the feedback control of saccadic eye movements. J Neurophysiol. 2002;87(2):679–695. doi: 10.1152/jn.00886.2000. [DOI] [PubMed] [Google Scholar]
- 22.Lefevre P, Quaia C, Optican LM. Distributed model of control of saccades by superior colliculus and cerebellum. Neural Netw. 1998;11(7-8):1175–1190. doi: 10.1016/s0893-6080(98)00071-9. [DOI] [PubMed] [Google Scholar]
- 23.Dean P. Modelling the role of the cerebellar fastigial nuclei in producing accurate saccades: the importance of burst timing. Neuroscience. 1995;68(4):1059–1077. doi: 10.1016/0306-4522(95)00239-f. [DOI] [PubMed] [Google Scholar]
- 24.Shaikh AG, Finlayson PG. Excitability of auditory brainstem neurons, in vivo, is increased by cyclic-AMP. Hear Res. 2005;201(1-2):70–80. doi: 10.1016/j.heares.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 25.Shaikh AG, Finlayson PG. Hyperpolarization-activated (I(h)) conductances affect brainstem auditory neuron excitability. Hear Res. 2003;183(1-2):126–136. doi: 10.1016/s0378-5955(03)00224-7. [DOI] [PubMed] [Google Scholar]
- 26.Perez-Reyes E. Molecular physiology of low-voltage-activated t-type calcium channels. Physiol Rev. 2003;83(1):117–161. doi: 10.1152/physrev.00018.2002. [DOI] [PubMed] [Google Scholar]
- 27.Zerrette M, Shaikh AG, Zee DS, Pardo C. Society for Neuroscience. Atlanta, GA: 2006. Ion channel profile of human brainstem saccadic burst neurons. Ed.ˆ(Eds) Program: 35.33/R12. [Google Scholar]
- 28.Jurgens R, Becker W, Kornhuber HH. Natural and drug-induced variations of velocity and duration of human saccadic eye movements: evidence for a control of the neural pulse generator by local feedback. Biol Cybern. 1981;39(2):87–96. doi: 10.1007/BF00336734. [DOI] [PubMed] [Google Scholar]
- 29.Scudder CA, Kaneko CS, Fuchs AF. The brainstem burst generator for saccadic eye movements: a modern synthesis. Exp Brain Res. 2002;142(4):439–462. doi: 10.1007/s00221-001-0912-9. [DOI] [PubMed] [Google Scholar]
- 30.Miura K, Optican LM. Membrane channel properties of premotor excitatory burst neurons may underlie saccade slowing after lesions of omnipause neurons. J Comput Neurosci. 2006;20(1):25–41. doi: 10.1007/s10827-006-4258-y. [DOI] [PubMed] [Google Scholar]
- 31.Shaikh AG, Zee DS, Optican LM, Miura K, Ramat S, Leigh RJ. The effects of ion channel blockers validate the conductance-based model of saccadic oscillations. Ann N Y Acad Sci. 2011;1233:58–63. doi: 10.1111/j.1749-6632.2011.06130.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Whitby LG, Hertting G, Axelrod J. Effect of cocaine on the disposition of noradrenaline labelled with tritium. Nature. 1960;187:604–605. doi: 10.1038/187604a0. [DOI] [PubMed] [Google Scholar]
- 33.McCormick DA, Pape HC. Noradrenergic and serotonergic modulation of a hyperpolarization-activated cation current in thalamic relay neurones. J Physiol. 1990;431:319–342. doi: 10.1113/jphysiol.1990.sp018332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McCormick DA, Pape HC. Properties of a hyperpolarization-activated cation current and its role in rhythmic oscillation in thalamic relay neurones. J Physiol. 1990;431:291–318. doi: 10.1113/jphysiol.1990.sp018331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liang TW, Balcer LJ, Solomon D, Messe SR, Galetta SL. Supranuclear gaze palsy and opsoclonus after Diazinon poisoning. J Neurol Neurosurg Psychiatry. 2003;74(5):677–679. doi: 10.1136/jnnp.74.5.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aurora SK, Welch KM. Brain excitability in migraine: evidence from transcranial magnetic stimulation studies. Curr Opin Neurol. 1998;11(3):205–209. doi: 10.1097/00019052-199806000-00003. [DOI] [PubMed] [Google Scholar]
- 37.Aurora SK, Ahmad BK, Welch KM, Bhardhwaj P, Ramadan NM. Transcranial magnetic stimulation confirms hyperexcitability of occipital cortex in migraine. Neurology. 1998;50(4):1111–1114. doi: 10.1212/wnl.50.4.1111. [DOI] [PubMed] [Google Scholar]
- 38.van der Kamp W, Maassen VanDenBrink A, Ferrari MD, van Dijk JG. Interictal cortical hyperexcitability in migraine patients demonstrated with transcranial magnetic stimulation. J Neurol Sci. 1996;139(1):106–110. doi: 10.1016/s0022-510x(96)00044-5. [DOI] [PubMed] [Google Scholar]
- 39.Wong AM, Musallam S, Tomlinson RD, Shannon P, Sharpe JA. Opsoclonus in three dimensions: oculographic, neuropathologic and modelling correlates. J Neurol Sci. 2001;189(1-2):71–81. doi: 10.1016/s0022-510x(01)00564-0. [DOI] [PubMed] [Google Scholar]
- 40.Neppert B, Rambold H. Familial voluntary nystagmus. Strabismus. 2006;14(2):115–119. doi: 10.1080/09273970600701226. [DOI] [PubMed] [Google Scholar]
- 41.Crawford TO, Mandir AS, Lefton-Greif MA, et al. Quantitative neurologic assessment of ataxia-telangiectasia. Neurology. 2000;54(7):1505–1509. doi: 10.1212/wnl.54.7.1505. [DOI] [PubMed] [Google Scholar]
- 42.M N, T H. Genetic polymorphisms and arrhythmia susceptibility. Circulation Journal. 2007;71(Suppl A):A54–60. doi: 10.1253/circj.71.a54. [DOI] [PubMed] [Google Scholar]
- 43.Donaldson MR, Yoon G, Fu YH, Ptacek LJ. Andersen-Tawil syndrome: a model of clinical variability, pleotrophy, and genetic heterogenity. Annals of Medicine. 2004;36(Suppl 1):92–97. doi: 10.1080/17431380410032490. [DOI] [PubMed] [Google Scholar]
- 44.Sharpe JA, Feletcher WA. In: Disorders of Visual Fixation. Smith JL, editor. Year Book Medical Publishers; Chicago: 1986. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.