

Abstract
Purpose
To evaluate the tolerability and efficacy of poly(ADP-ribose) polymerase (PARP) inhibition by veliparib during cytotoxic topotecan administration with filgrastim or pegfilgrastim neutrophil support in women with persistent or recurrent uterine cervix cancer.
Experimental Design
This phase I–II trial examined twice-daily oral veliparib (10 mg) given during once-daily intravenous topotecan (0.6 mg/m2) on days 1–5 of each treatment cycle. Cycles were repeated every 21 days until disease progression or until toxicity prohibited further therapy. Toxicity and objective response rate were primary endpoints.
Results
Twenty-seven women were enrolled. Frequently reported grade 3 or higher treatment-related toxicities were anemia (59%), thrombocytopenia (44%), leukopenia (22%), and neutropenia (19%). There were 2 partial responses (7% [90% confidence interval: 1–22%]). Four patients had a disease progression date more than 6 months after the start of veliparib-topotecan therapy. Patients with low immunohistochemical expression (0–1+) of PARP-1 in their primary uterine cervix cancer were more likely to have a longer progression-free interval (hazard ratio: 0.25, P = 0.02) and survival (hazard ratio: 0.12, P = 0.005) after veliparib-topotecan therapy.
Conclusions
Clinical activity of a veliparib-topotecan combination was minimal in women with persistent or recurrent uterine cervix cancer. Women whose uterine cervix cancers express PARP-1 at low levels may benefit preferentially from PARP inhibitors combined with cytotoxic therapies, suggesting further study of PARP expression as an integral triage biomarker.
Keywords: veliparib, topotecan, cervical cancer, poly (ADP-ribose) polymerase
INTRODUCTION
Poly(ADP-ribose) polymerase (PARP) acts as a constitutively-expressed nuclear tankyrase enzyme primarily involved in mammalian base excision DNA repair [1–3]. PARP rapidly catalyzes the new synthesis of ADP-ribose branching polymers from its substrate nicotinamide adenine dinucleotide, creating negatively-charged, branching scaffolds upon which other nuclear proteins such as DNA polβ, X-ray repair cross-complementing factor 1 (XRCC1), and DNA ligase III co-localize to DNA [4,5]. As such, PARP functions as a key regulator of both base excision DNA damage repair and DNA duplication [5–10].
In PARP knockout mouse models, 80–90% of PARP-dependent base excision repair in DNA becomes significantly impaired after deletion of PARP-1 [8,9]. Residual DNA repair occurs through PARP-2 activity [9]. PARP activity rises 500-fold when the enzyme binds to DNA strand breaks—in the absence of such binding, poly(ADP-ribose) polymer synthesis is negligible. These findings suggest that only PARP-1 and PARP-2 need to be pharmacologically blocked to disrupt base excision repair in mammalian DNA [3]. PARP inhibition sensitizes cancer cells both to cytotoxic chemotherapy, such as alkylators (temozolomide, cyclophosphamide) or topoisomerase inhibitors (irinotecan, topotecan, camptothecin), and to ionizing radiation—all of which induce DNA damage requiring base excision repair [11–15]. Expression of PARP has been shown to be higher in cancer cells as compared to normal cells [16], and its overexpression associates with cytotoxic drug resistance. Two-fold elevated PARP-1 expression has been detected in uterine cervix cancer cells as compared to normal cells [16].
Veliparib (ABT-888) is an orally available equipotent small molecule pharmacological inhibitor of PARP-1 and PARP-2, whose single oral dose of 10 mg reduces PARP activity by at least 75–85% in cancer cells [12,13]. Preclinical pharmacologically-relevant veliparib-topotecan treatments in in vitro uterine cervix squamous cancer cells demonstrated enhanced cancer cell death after exposure to the combination [5]. A molecular mechanism for this finding involved collapsed topotecan-poisoned replication forks, formation of topotecan-related single-strand DNA nicks, and conversion of those nicks into lethal double-strand breaks when DNA repair was impeded by veliparib [5].
In a phase 0 trial of veliparib (10 mg twice daily) and topotecan (0.6–1.2 mg/m2/day) recruiting 13 patients with refractory solid tumors and lymphomas [13], veliparib lowered poly(ADP-ribose) levels and increased γH2AX signal (i.e., a biomarker of unrepaired double-strand DNA damage) in tumor cells and in circulating peripheral blood mononuclear cells. The phase 0 trial identified a maximum tolerated dose of veliparib 10 mg twice a day plus topotecan 0.6 mg/m2/day on days 1–5 of a 21-day cycle [13]. Our phase I–II trial used this recommended veliparib-topotecan dose and schedule to study the safety and efficacy of the combination in women with pretreated persistent or recurrent cervical cancer.
Materials and Methods
This phase I–II multicenter trial (NCT01266447) enrolled women with pretreated persistent or recurrent adenocarcinoma, adenosquamous, squamous cell, or non-squamous cell cancers of the uterine cervix between February 2011 and January 2013.
Patient selection
All included patients provided written informed consent and fulfilled the following criteria: age ≥18 years, at least 1 measurable unirradiated site of disease as defined by Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1 (or a tumor within a previously irradiated field demonstrating either radiographic disease progression or persistent disease by biopsy at least 90 days following completion of radiation therapy), a Gynecologic Oncology Group (GOG) performance status of 0–2, at least 1 systemic chemotherapy regimen with or without biologic therapy directed at persistent, recurrent, or metastatic disease (i.e., concurrent or adjuvant chemotherapies at the time of primary radiation were not counted) and adequate organ function including absolute neutrophil count >1,500/mcl, platelets >100,000/mcl, creatinine <1.5 x upper limit of normal (ULN), bilirubin ≤1.5 x ULN, aspartate aminotransferase ≤3 x ULN, alanine aminotransferase ≤3 x ULN, alkaline phosphatase ≤2.5 x ULN, and neuropathy ≤ grade 1. Patients must have had a negative pregnancy test or be postmenopausal. Patients must have had an ability to swallow pills whole. Exclusion criteria included prior therapy that included PARP inhibitors (including veliparib) or topotecan, active malignancy (except adequately treated non-melanoma skin cancer) within the previous 3 years, prior abdominal radiotherapy or chemotherapy other than for treatment of cervical cancer, and any history or evidence of central nervous system disease (i.e., primary brain tumor, uncontrolled seizures, brain metastases, or cerebrovascular accident [stroke], transient ischemic attack [TIA], or subarachnoid hemorrhage) within 6 months of the 1st date of trial treatment.
Study design and safety assessment
This phase I–II study was an open-label, single-arm trial with a safety lead-in to estimate the antitumor activity of the combination of veliparib administered orally with topotecan hydrochloride administered intravenously in women with pretreated persistent or recurrent cancers of the uterine cervix. Veliparib was supplied under a Collaborative Research and Development Agreement between the National Cancer Institute (NCI) Cancer Therapy Evaluation Program and Abbott Laboratories, Inc. Topotecan hydrochloride was obtained commercially. All patients gave written informed consent before study entry in compliance with local institutional review board, state, and federal regulations.
The safety lead-in was evaluated through a Bayesian approach [17], which assessed the posterior probability of the 1st-cycle dose-limiting toxicities being higher than a specified target in the first 6 patients who were treated and started cycle 2, or, had dose-limiting toxicities prior to completing the 1st cycle. The interested target for the probability of dose-limiting toxicity was 0.33. Once safety was assured, the trial opened group wide for accrual.
Following a recommended phase II dose and schedule [13], we administered oral veliparib at a dose of 10 mg twice a day given concurrently with intravenous topotecan (0.6 mg/m2) once daily on days 1–5 of a treatment cycle. Cycles were repeated every 21 days until disease progression or adverse effects prohibit further therapy. Filgrastim (dose according to institutional standard) was administered daily subcutaneously starting 24–72 hours after the last dose of topotecan and veliparib, and continued through hematopoietic recovery. Alternatively, pegfilgrastim (6 mg) was given subcutaneously, 1 dose per treatment cycle, 24–72 hours after the last dose of topotecan. Administration of growth factors on the same day as topotecan was not recommended.
Toxicities were graded according to NCI Common Toxicity Criteria for Adverse Events version 4.0. Since the majority of the patients on the current trial would have had prior pelvic radiation, this study enrolled 6 patients in a safety lead-in phase, monitored by every other week telephone calls attended by Phase I Developmental Therapeutics Committee members of the GOG and participating investigators. Enrollment was halted for a safety review after the 6th patient (who had undergone prior pelvic radiation) completed 1 cycle of treatment (or had a dose-limiting toxicity prior to completing the 1st cycle). During the safety lead-in, adverse events and their impact on tolerability of treatment were assessed in the 1st cycle. Because of the prior pelvic radiation, we had anticipated myelosuppression at the current study’s phase II dose level. Therefore, we included upfront a lower dose modification level of oral veliparib at a dose of 10 mg once a day given concurrently with intravenous topotecan (0.6 mg/m2) once daily on days 1–5 of treatment cycle.
Complete blood counts with differential and serum chemistries were acquired before the 1st and subsequent veliparib-topotecan treatment cycles. Subsequent cycles of therapy were not to begin until the absolute neutrophil count was ≥1500 cells/mcl and the platelet count was ≥100,000/mcl. A veliparib dose modification to the lower dose regimen was to be done for grade 4 neutropenia, febrile neutropenia persisting greater than 7 days, transient grade 2 or higher neuropathy or renal toxicities, or transient grade 3 elevations in bilirubin, aspartate aminotransferase, alanine aminotransferase, or alkaline phosphatase levels. Veliparib-topotecan therapy may have been delayed for a maximum of 2 weeks until eligibility range values were achieved. Patients who did not recover adequate counts or laboratory values after a 2-week delay were removed from study. No dose re-escalations were permitted.
An optimal flexible 2-stage design was employed to assess the efficacy of the study regimen [18]. This design limited the number of patients exposed to an inactive dose and schedule. The targeted accrual for the 1st stage was 25 patients, but was allowed to deviate for administrative purposes. If 27 patients were accrued and at least 5 of them had tumor response, this study would open to the second stage of accrual to further evaluate the regimen. The cumulative targeted accrual for the 2nd stage was to be 55 patients, but was allowed to deviate for administrative purposes. If at least 12 of the 55 patients had a tumor response, the regimen would be considered worthy of further investigation. Assuming study accrual with ranges of 22–29 (stage 1) and 53–60 (stage 2), the average probability of falsely declaring the regimen as active at the end of stage 2 was limited to 10 percent if the true response rate was 15 percent; the average probability of correctly classifying the regimen as active was 90 percent if the true response rate was 30 percent. The average probability of early termination when the agent was inactive was 59 percent.
Safety and efficacy evaluations
A complete patient history and physical examination was conducted prior to the 1st and each subsequent treatment cycle. Complete blood counts with differential and serum chemistries were acquired 14 days before the 1st and within 4 days prior to subsequent treatment cycles. Twice weekly blood counts were done when grade 4 neutropenia was observed to document duration of neutropenia and to determine recovery. Radiographic imaging was done prior to treatment and every 2 cycles for the first 6 months to assess for tumor response following Response Evaluation Criteria in Solid Tumors (RECIST version 1.1). Repeat radiographic imaging was done every 3 months thereafter. Follow-up was done every 3 months for the 1st 2 years after start of trial therapy, and then was to continue every 6 months for an additional 3 years.
Tumor immunohistochemistry
Nineteen patients granted permission for immunoreactivity assays of their archived, untreated primary (n=16), persistent/recurrent (n=5), or metastatic (n= 4) tumors of uterine cervix cancer. Adapting techniques described previously [19], immunohistochemistry was done using PARP-1 mouse monoclonal antibody (1:1600; Santa Cruz Biotechnology [Dallas, TX]), PARP-2 mouse monoclonal antibody (1:100; Millipore [Billerica, MA]) and ribonucleotide reductase M2b (p53R2) rabbit polyclonal antibody (1:8000; Abcam [Cambridge, MA]). The ribonucleotide reductase M2b subunit was studied here as a cellular marker of de novo DNA repair capacity [20,21] and as a potential indicator of treatment response in cervical cancer [22,23]. One pathologist (DD) selected negative immunoreactivity controls based on known PARP-1, PARP-2 and M2b protein expression in human tissues. Blinded to treatment and response, one pathologist (DD) scored samples for PARP-1, PARP-2 and M2b protein for staining intensity and percent positive cells: negative 0 (<5%), positive 1+ (5% to <25%), positive 2+ (25% to <75%), and positive 3+ (≥75%) [19].
Statistics
Median progression-free survival (PFS) and overall survival (OS) were estimated by Kaplan-Meier method [24]. PFS was defined as the duration of time from study entry to time of progression or death, whichever occurs first. PFS was censored in patients who were alive and had not progressed. OS was defined as the duration of time from study entry to death or the date of last contact in patients who were alive. An exploratory translational science analysis of GOG protocol 127W was done to evaluate the potential associations of PARP-1 or PARP-2 expression and of ribonucleotide reductase M2b expression with tumor response, PFS and OS. To take advantage of biological information provided by both the intensity of immunohistochemical staining and the relative number of cells positive for PARP-1, PARP-2, and M2b, a composite histological score was computed by multiplying staining intensity (i.e., 0, 1, 2, 3) with the mean of percent positive cells (e.g., if scored 50–75% cells positive, then multiply by 62.5). Exact tests for Spearman’s rank correlation coefficient were used to examine associations between the histological score and treatment response [25]. Due to small sample sizes, permutation-based log-rank tests were conducted to explore associations among dichotomized (high = equal to or greater than median, low = less than the median) PARP-1, PARP-2, and M2b histological scores and survival under the assumption of exchangeability [26,27]. The corresponding hazard ratios were estimated by Cox proportional hazards model [28]. All tests were 2-sided and the significance level was 0.05. There was no adjustment made for multiple tests since the purpose of these tests were exploratory. Statistical analyses were performed using SAS 9.3 (SAS Institute Inc., Cary, NC).
RESULTS
Patient demographics
A total of 27 patients were enrolled on this phase I–II trial (Table 1). The mean age was 50 years (standard deviation = 10 years). Among 27 enrollees, five (18%) identified themselves as African-American race, 15 (55%) had a performance status greater than zero, four (15%) had pelvic disease only, and eight (30%) had pelvic and extrapelvic disease at enrollment. Two of the 1st 6 patients did not complete the 1st cycle of veliparib-topotecan treatment, were not evaluable for dose-limiting treatment-related adverse events, and were replaced in the safety lead-in phase of the trial. These 2 patients did contribute to safety and efficacy analyses. All 27 patients had previously received initial therapeutic treatment, had received at least 1 chemotherapy regimen directed at persistent or recurrent disease, and then had subsequent evidence of disease progression. In the safety lead-in cohort, all 6 evaluable patients had received cisplatin-based radiochemotherapy involving pelvic radiation, and then, at least 1 chemotherapy regimen directed at persistent or recurrent disease.
Table 1.
Patient characteristics (n = 27).
| Characteristic | No. of patients | %* |
|---|---|---|
| Age Group | ||
| 30–39 | 5 | 18 |
| 40–49 | 8 | 30 |
| 50–59 | 10 | 37 |
| 60–69 | 4 | 15 |
| Race | ||
| White | 20 | 74 |
| Black or African American | 5 | 18 |
| American Indian/Alaskan | 1 | 4 |
| Undeclared | 1 | 4 |
| Ethnicity | ||
| Hispanic | 2 | 7 |
| Non-Hispanic | 23 | 85 |
| Undeclared | 2 | 7 |
| Performance Status | ||
| 0 | 12 | 44 |
| 1 | 12 | 44 |
| 2 | 3 | 11 |
| Histology | ||
| Adenocarcinoma | 9 | 33 |
| Adenosquamous | 6 | 22 |
| carcinoma | ||
| Squamous cell carcinoma | 11 | 41 |
| Small cell carcinoma | 1 | 4 |
May not total 100 due to rounding.
Toxicity
Adverse events regardless of attribution to study treatment are listed in Table 2. The 1st enrolled patient did not complete the 1st cycle of therapy due to pain from pelvic floor disease progression and grade 4 elevated creatinine. The 2nd enrolled patient did not complete the 1st cycle of therapy because of rapid accumulation of abdominal ascites causing symptomatic grade 3 dyspnea. Both patients were replaced in the safety lead-in cohort; both remained evaluable for safety and efficacy assessments.
Table 2.
Adverse events by grade with any relationship to protocol treatment according to Common Terminology Criteria for Adverse Events Version 4
| Adverse Effect | 0 | 1 | 2 | 3 | 4 | 5 | Total |
|---|---|---|---|---|---|---|---|
| Leukopenia | 12 | 2 | 7 | 5 | 1 | 0 | 27 |
| Thrombocytopenia | 4 | 9 | 2 | 6 | 6 | 0 | 27 |
| Neutropenia | 15 | 4 | 3 | 5 | 0 | 0 | 27 |
| Anemia | 0 | 4 | 7 | 13 | 3 | 0 | 27 |
| Other Investigations | 14 | 6 | 3 | 2 | 2 | 0 | 27 |
| Cardiac | 25 | 0 | 1 | 1 | 0 | 0 | 27 |
| Ear and labyrinth | 25 | 2 | 0 | 0 | 0 | 0 | 27 |
| Endocrine | 26 | 1 | 0 | 0 | 0 | 0 | 27 |
| Gastrointestinal1 | 4 | 10 | 6 | 6 | 0 | 1 | 27 |
| General and administration site2 | 2 | 7 | 13 | 3 | 0 | 2 | 27 |
| Infections/infestations | 18 | 0 | 5 | 2 | 2 | 0 | 27 |
| Injury/poisoning | 21 | 6 | 0 | 0 | 0 | 0 | 27 |
| Metabolism/nutrition | 7 | 6 | 11 | 3 | 0 | 0 | 27 |
| Musculoskeletal/connective tissue | 14 | 4 | 4 | 5 | 0 | 0 | 27 |
| Neoplasms benign/malignant3 | 26 | 0 | 0 | 0 | 0 | 1 | 27 |
| Peripheral sensory neuropathy | 19 | 7 | 1 | 0 | 0 | 0 | 27 |
| Nervous system | 14 | 9 | 3 | 1 | 0 | 0 | 27 |
| Psychiatric | 19 | 4 | 4 | 0 | 0 | 0 | 27 |
| Renal/urinary | 21 | 5 | 1 | 0 | 0 | 0 | 27 |
| Reproductive/breast | 21 | 4 | 0 | 2 | 0 | 0 | 27 |
| Respiratory/thoracic/mediastinal | 13 | 8 | 2 | 3 | 1 | 0 | 27 |
| Skin/subcutaneous | 14 | 7 | 6 | 0 | 0 | 0 | 27 |
| Surgical/medical procedures | 26 | 0 | 0 | 1 | 0 | 0 | 27 |
| Vascular disorders | 17 | 5 | 2 | 3 | 0 | 0 | 27 |
This death was thought unlikely attributable to study treatment. The death is attributable to small intestinal perforation.
One death was thought possibly attributable to study treatment, and another death was thought unlikely attributable to study treatment. Both deaths are attributable to death not otherwise specified.
This death was thought unrelated to study treatment. The death is attributable to neoplasms benign, malignant and unspecified (incl cysts and polyps)-disease progression.
We observed 1 patient with 1st cycle treatment-related grade 4 thrombocytopenia in the 1st 6 evaluable safety lead-in patients enrolled on the veliparib 10 mg twice daily given concurrently with topotecan 0.6 mg/m2/day treatment dose level. This patient tolerated a next cycle dose reduction to veliparib 10 mg once daily given concurrently with topotecan 0.6 mg/m2/day treatment. One patient in the safety lead-in had no treatment-related grade 4 thrombocytopenia in the 1st and 2nd cycles of treatment, but had treatment-related grade 4 thrombocytopenia in the 3rd cycle. The 3rd patient in the safety lead-in had no treatment-related grade 4 thrombocytopenia in the 1st cycle and no thrombocytopenia in the rest of the 6 cycles. The 4th patient in the safety lead-in cohort had grade 3 thrombocytopenia in the 5th treatment cycle. It was decided that 2 additional patients would be recruited to the veliparib 10 mg twice daily and topotecan 0.6 mg/m2/day treatment dose level. No treatment-related grade 4 thrombocytopenias were observed in the 1st cycle in these 2 additional patients, and no thrombocytopenia was observed in the rest of their treatment cycles for these 2 patients. As such, the initial dose level was considered tolerable for full complement patient accrual.
A total of 103 veliparib-topotecan treatment cycles were delivered (range 1 to 15 cycles per individual patient). Among the 27 patients, 6 (22%) patients needed dose-reductions after the 1st treatment cycle. Frequently reported grade 3 or higher treatment-related adverse events were anemia (59%), thrombocytopenia (44%), leukopenia (22%), and neutropenia (19%). Table 2 reports the frequencies of maximum grade of adverse events regardless of attribution to study treatment. There were 4 deaths reported during the period of active treatment or within 30 days of last study treatment. One sudden patient death occurred during the 15th cycle of veliparib-topotecan therapy thought to be possibly attributable to study treatment; the cause of death was not otherwise specified. One patient received a single dose of 1st cycle veliparib-topotecan therapy, had symptomatic grade 3 dyspnea, stopped veliparib-topotecan therapy, and died 33 days after the 1st cycle veliparib-topotecan therapy; the patient death was thought unlikely attributable to study treatment. The 3rd death was thought unlikely attributable to study treatment and the death is attributable to small intestinal perforation. The 4th death was thought unrelated to study treatment and the death is attributable to neoplasms benign, malignant and unspecified (including cysts and polyps) – disease progression.
Efficacy
There were no complete responses observed. Two (7%, 90% confidence interval 1–22%) partial responses were observed in target cervical cancer lesions after veliparib-topotecan treatment. One patient (squamous cell cancer) achieved a confirmed partial response after 4 treatment cycles and then received 3 more cycles of therapy prior to disease progression and disease-attributed death. This patient had response duration of 4 months. Another patient (adenosquamous cell cancer) had 11 cycles of veliparib-topotecan therapy at cessation of therapy. This patient had censored response duration of 6.7 months. Ten (37%) patients had stable disease. Three of them had duration of stable disease of more than 6 months. Twelve (44%) patients had progression of disease. Two patients had an indeterminate response status. The median PFS was 2 months (90% confidence interval: 1–3 months). The median OS was 8 months (90% confidence interval: 6–10 months).
PARP-1 expression
Topotecan inserts itself between bases of coiled and supercoiled DNA, halts DNA replication forks, impedes religation of single strand cleaved DNA, and thereby increases the probability of a cell-lethal DNA double strand break at a stalled replication fork [29]. PARP senses strand breaks in DNA [10] and accelerates DNA repair by erecting branching scaffolds of poly(ADP-ribose) polymers [5]. To investigate whether cervical cancer cells overexpress PARP enzyme that renders enhanced strand break repair possible, an immunohistochemical assay for PARP-1 was performed (Figure 1). Among 16 archived untreated primary cervical cancers, 12 (75%) had high (2–3+) PARP-1 nuclear staining intensity. By primary cervical cancer histology, 4 (67%) of 6 adenocarcinomas, 1 (50%) of 2 adenosquamous cancers, and 7 (88%) of 8 squamous cell cancers had high (2–3+) PARP-1 nuclear staining intensity. Of the 9 persistent/recurrent (n = 5) or metastatic (n = 4) cervical cancers representing disease treated by protocol veliparib-topotecan therapy, 8 (89%) had high (2–3+) PARP-1 nuclear staining intensity. The median PARP-1 histological score (i.e., product of staining intensity and percent positive cells) was 168 (range: 0–299) in the 16 primary cervical cancers. Among those 16 primary cervical cancers, a low PARP-1 histological score correlated with a likelihood of veliparib-topotecan treatment response (Spearman correlation coefficient: −0.63, P = 0.011). A patient with a low PARP-1 histological score dichotomized by its median was more likely to have longer PFS (HR: 0.25, P = 0.023) and OS after protocol veliparib-topotecan therapy (HR: 0.12, P = 0.005) compared to a patient with a higher histological score. Too few persistent/recurrent or metastatic samples were obtained for PARP-1 correlative analyses.
Figure 1.
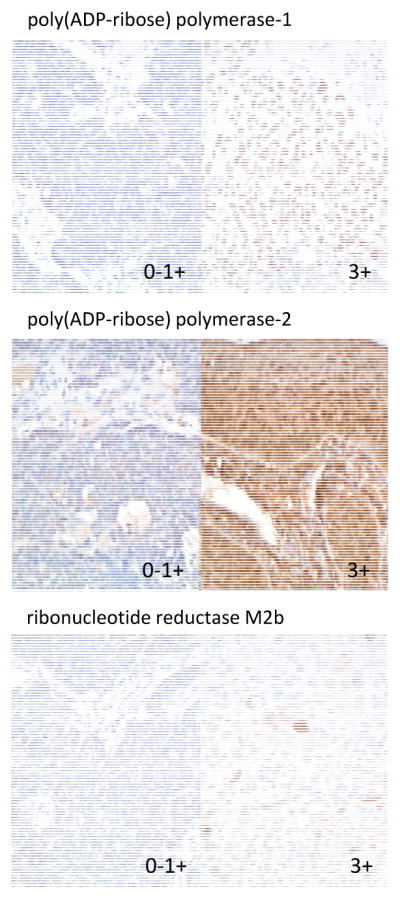
Representative cervical cancer immunohistochemistry for poly(ADP-ribose) polymerase-1, poly(ADP-ribose) polymerase-2, and ribonucleotide reductase M2b (p53R2). Magnification is 20x.
PARP-2 expression
PARP-2 shares a catalytic domain with PARP-1, is a smaller DNA-binding protein, erects poly(ADP-ribose) polymers in a manner similar to PARP-1, and activates base excision machinery in response to DNA damage [9]. To query whether cervical cancer cells express PARP-1 and PARP-2 similarly, an immunohistochemical assay for PARP-2 was performed on pair-matched samples from the PARP-1 assay (Figure 1). PARP-2 nuclear expression was high (2–3+) in only 6 (38%) of the 16 matched archived and untreated primary cervical cancers. Only six (38%) of the 16 cancers had both PARP-1 and PARP-2 highly expressed (2–3+). By primary cervical cancer histology, 2 (33%) of 6 adenocarcinomas and 4 (50%) of 8 squamous cell cancers had high (2–3+) PARP-2 nuclear staining intensity. Only 2 (22%) of the 9 persistent/recurrent or metastatic cervical cancers had high (2–3+) PARP-2 nuclear staining intensity. The median PARP-2 histological score was 90 (range: 0–299) for the 16 primary cervical cancers. Among those 16 primary cervical cancers, a low PARP-2 histological score correlated with a likelihood of veliparib-topotecan treatment response (Spearman correlation coefficient: −0.48, P = 0.061). A patient with a low PARP-2 histological dichotomized by its median score was more likely to have longer PFS (HR: 0.21, P = 0.013) and longer OS after protocol veliparib-topotecan therapy (HR: 0.38, P = 0.118) comparing to a patient with a higher histological score. Not enough persistent/recurrent or metastatic samples were submitted for PARP-2 correlative analyses.
Ribonucleotide reductase M2b expression
Cervical cancers have an overactive ribonucleotide reductase enzyme (in the form of M1-M2 or M1-M2b) generating deoxyribonucleotides used in DNA duplication and in DNA repair as a consequence of virally silenced or mutated p53 proteins [19–21]. The ribonucleotide reductase M2b subunit persists throughout the cell cycle [30], is regulated in the cytosol by a protein-protein bond with p53 [31], and once freed from p53 by p53 phosphorylation, may localize to the nucleus for immediate supply of deoxyribonucleotides when nuclear DNA is damaged [20,21]. To evaluate whether ribonucleotide reductase M2b expression correlated with treatment outcome, immunohistochemical analyses were conducted (Figure 1). M2b expression was high (2–3+) in 9 (56%) of the 16 archived and untreated cervical cancers, consistent with a 59 percent high (2–3+) M2B expression rate from other uterine cervix cancers similarly screened [19]. By primary cervical cancer histology, 4 (67%) of 6 adenocarcinomas, 2 (10%) of 2 adenosquamous cancers, and 3 (38%) of 8 squamous cell cancers had high (2–3+) M2b staining intensity. Only 4 (44%) of the 9 persistent/recurrent or metastatic cervical cancers had high (2–3+) M2b staining intensity. M2b histological score (i.e., computed using the cytoplasm staining intensity singularly) did not correlate with response (Spearman correlation coefficient: 0.01, P =0.994), with PFS (HR: 1.13, P = 0.822), or OS (HR: 1.18, P = 0.772). An insufficient number of samples were procured for M2b correlative analyses in persistent/recurrent or metastatic tissue.
DISCUSSION
This trial evaluated the adverse events and efficacy of the PARP inhibitor veliparib given as an oral 10 mg tablet given twice daily in combination with the topoisomerase I inhibitor topotecan administered intravenously at 0.6 mg/m2/day on days 1–5 of a 21-day cycle. Treatment-related anemia (59%) and thrombocytopenia (44%) were encountered most often in this heavily pretreated cervical cancer patient cohort. Two patients with objective partial tumor responses out of 27 eligible and evaluable patients did not meet the predetermined critical value to open to second stage trial accrual.
This phase I–II clinical trial was modeled on concepts vetted in preclinical [5] and phase 0 clinical trial [13] forums. Topotecan becomes lethal to cells when DNA replication forks stall at topotecan-DNA foci, DNA strand nicks accumulate, and unrepaired nicks become cell death-provoking DNA double-strand breaks. While the precise mechanism of how poly(ADP-ribose) polymers stabilize DNA nicks for base excision repair is not known, it is becoming more apparent that DNA not held together with poly(ADP-ribose) polymers allows an increasing probability that 2 “free” ends of a DNA double-strand break to separate irreparably [5]. Certainly when topotecan induces stalled replication forks, cells are challenged to fasten double-strand break free ends back together in a relatively short time period. Cervical cancer cells challenged in vitro with this task retain many unrepaired lethal double-strand breaks after veliparib-topotecan exposure [5]. On trial, veliparib-topotecan treatment produced significant myelosuppression and may be conceptually linked to replication fork collapse and protracted repair in myeloid precursor cells [32, 33]. Moreover, a 41 percent severe neutropenia and 18 percent severe thrombocytopenia rate was observed at the same veliparib-topotecan dose level in the phase 0 clinical trial [13]. Our data fall in line with this previous experience. On trial, anemia was frequently encountered, and while possibly attributed to veliparib-topotecan exposure, chronic bleeding occurring as a result of persistent or recurrent cervical cancer might obscure the overall anemia rate observed in this patient cohort.
Veliparib-topotecan treatment resulted in 2 (7%) partial responses, which did not meet the predetermined 15 percent response benchmark for 2nd-stage trial accrual. However, our data are informative. PARP-1 and PARP-2 are crucial for the initiation, elongation, and branching of poly(ADP-ribose) polymers. In this work, immunohistochemistry demonstrated predominantly high (2–3+) PARP-1 expression (75%) and low (0–1+) PARP-2 expression (62%) in untreated primary cancers of the uterine cervix. Looking for patterns of PARP expression, we noticed that adenocarcinomas and adenosquamous carcinomas of the uterine cervix most often expressed PARP-1 at high levels. Adenocarcinomas and adenosquamous carcinoma histologies of the uterine cervix are associated more often with advanced initial stage, incomplete first treatment response, and poorer OS [34, 35]. This trial accrued 15 (56%) of the 27 studied patients with heavily pretreated adenocarcinoma or adenosquamous carcinomas. We found that a low PARP-1 histological score correlated with a likelihood of veliparib-topotecan treatment response, longer PFS interval, and OS. Perhaps these findings are due to a sufficient veliparib 10 mg dose-related block of low-level PARP activity that subsequently imparts an enhanced cancer-cell lethal topotecan effect. Alternatively, given the low response rate and the frequency of high (2–3+) PARP-1 expression in untreated primary, persistent/recurrent, and metastatic cancers of the uterine cervix, perhaps not enough veliparib was administered on trial to overcome the likely enhanced capacity for repair of collapsed replication forks and DNA strand damage. The strength of the latter interpretation would be improved by an increased availability of adenocarcinoma and adenosquamous cancer tissue for PARP-1 immunohistochemical analyses. A clinical trial evaluating a PARP inhibitor and cytotoxic chemotherapy, enriched for women with recurrent/persistent or metastatic adenocarcinoma or adenosquamous carcinoma, would be of interest. A third interpretive possibility involves a gradual selective process of PARP-1, PARP-2, or M2b protein becoming more or less expressed in a cancer cell population as a differential function of prior anticancer treatment. This process may be a key mechanism of subsequent treatment resistance as biological traits adapt in surviving cells.
The p53-regulated ribonucleotide reductase M2b subunit appears elevated (2–3+) in more than one-half of the patients’ primary cervical cancers (56%), most often in the subset of squamous cell cancers. Perhaps the most intriguing feature of M2b subunit expression observed here is its agreement with an earlier contention that nearly 60% of cervical cancers exhibit disruption of a M2b-p53 protein-protein interaction by human papillomavirus viral elimination of p53 or by mutational modification of p53 [19]. In this study, pretherapy M2b expression status did not predict response to veliparib-topotecan treatment; however, in other clinical trials elevated pretherapy M2b expression may predict response to ribonucleotide reductase inhibitors given during cisplatin radiochemotherapy [22, 23]. Ribonucleotide reductase in its M1-M2b form has been considered vital to the nuclear and mitochondrial DNA damage response [19]. As is germane to this trial, topotecan only slightly increases the number of DNA double-strand breaks in squamous cell cervical cancers in vitro [5]. Because the number of deoxyribonucleotides demanded to fix topotecan-stalled replication forks may be relatively small, a biomarker like M2b may not necessarily predict treatment response to veliparib, a treatment that biologically targets poly(ADP-ribose) polymer construction and deconstruction kinetics and not ribonucleotide reductase deoxyribonucleotide payout. It is of interest that radiation exposure damages thousands of DNA bases and creates numerous single strand and double strand breaks in DNA. Enhanced cell lethality occurs after a veliparib-radiation treatment as a consequence of veliparib protracting PARP-related DNA base excision repair [5]. Such observations raise the intriguing possibility of a clinical trial evaluating PARP inhibition during upfront cisplatin radiochemotherapy for patients with advanced-stage adenocarcinoma or adenosquamous carcinomas of the uterine cervix.
In this trial, a veliparib-topotecan combination was investigated in patients with pretreated persistent or recurrent cancers of the uterine cervix, a poor prognostic group with an unmet therapeutic need for an active biologically-targeted treatment regimen. Myelosuppression occurred frequently, with thrombocytopenia prompting dose-reductions in veliparib daily dose. The investigated treatment had insufficient activity to warrant further investigation. However, this trial adds to the growing knowledge that a PARP inhibitor paired with a topoisomerase inhibitor effects cancer, but a pharmacodynamic treatment schedule that widens therapeutic antitumor response without myelosuppression is needed.
Acknowledgments
Financial support: This study was supported by National Cancer Institute grants to the Gynecologic Oncology Group (GOG) Administrative Office (CA 27469), the Gynecologic Oncology Group Statistical Office (CA 37517) and NRG Oncology (1 U10 CA180822).
We thank Adam Kresak for assistance in PARP-1, PARP-2, and ribonucleotide reductase M2b immunohistochemistry and analysis.
References
- 1.Chambon P, Weill J, Mandel P. Nicotinamide mononucleotide activation of a new DNA-dependent polyadenylic acid synthesising nuclear enzyme. Biochem Biophys Res Commun. 1963;11:39–43. doi: 10.1016/0006-291x(63)90024-x. [DOI] [PubMed] [Google Scholar]
- 2.Dantzer F, de La Rubia G, Menissier-De Murcia J, Hostomsky Z, de Murcia G, Schreiber V. Base excision repair is impaired in mammalian cells lacking Poly(ADP-ribose) polymerase-1. Biochemistry. 2000;39:7559–7569. doi: 10.1021/bi0003442. [DOI] [PubMed] [Google Scholar]
- 3.Schreiber V, Ame J, Dollie P, Schultz I, Rinaldi B, Fraulob V, et al. Poly(ADP-ribose) polymerase-2 (PARP-2) is required for efficient base excision repair in association with PARP-1 and XRCC1. J Biol Chem. 2002;277:23028–23036. doi: 10.1074/jbc.M202390200. [DOI] [PubMed] [Google Scholar]
- 4.Krishnakumar R, Kraus WL. The PARP side of the nucleus: molecular actions, physiological outcomes, and clinical targets. Mol Cell. 2010;39:8–24. doi: 10.1016/j.molcel.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shunkwiler L, Ferris G, Kunos C. Inhibition of Poly(ADP-Ribose) Polymerase Enhances Radiochemosensitivity in Cancers Proficient in DNA Double-Strand Break Repair. International journal of molecular sciences. 2013;14:3773–3785. doi: 10.3390/ijms14023773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chalmers AJ. Poly(ADP-ribose) polymerase-1 and ionizing radiation: Sensor, signaller, and therapeutic target. Clin Oncol. 2004;16:29–39. doi: 10.1016/s0936-6555(03)00223-1. [DOI] [PubMed] [Google Scholar]
- 7.Loser DA, Shibata A, Shibata AK, Woodbine LJ, Jeggo PA, Chalmers AJ. Sensitization to radiation and alkylating agents by inhibitors of poly(ADP-ribose) polymerase is enhanced in cells deficient in DNA double-strand break repair. Mol Cancer Ther. 2010;9:1775–1787. doi: 10.1158/1535-7163.MCT-09-1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fernet M, Ponette V, Deniaud-Alexandre E, Menissier-de Murcia J, de Murcia G, Glocanti N, et al. Poly(ADP-ribose) polymerase, a major determinant of early cell response to ionizing radiation. Int J Radiation Oncol Biol Phys. 2000;76:1621–1629. doi: 10.1080/09553000050201118. [DOI] [PubMed] [Google Scholar]
- 9.Ame J, Rolli V, Schreiber V, Niedergang C, Apiou F, Decker P, et al. PARP-2, a novel mammalian DNA damage-dependent poly(ADP-ribose) polymerase. J Biol Chem. 1999;274:17860–17868. doi: 10.1074/jbc.274.25.17860. [DOI] [PubMed] [Google Scholar]
- 10.Ame J, Spenlehauer C, de Murcia G. The PARP superfamily. Bioessays. 2004;26:882–893. doi: 10.1002/bies.20085. [DOI] [PubMed] [Google Scholar]
- 11.Delaney C, Wang L, Kyle S, White A, Calvert A, Curtin N, et al. Potentiation of temozolomide and topotecan growth inhibition and cytotoxicity by novel poly(adenosine diphosphoribose) polymerase inhibitors in a panel of human tumor cell lines. Clin Cancer Res. 2000;6:2860–2867. [PubMed] [Google Scholar]
- 12.Donawho C, Luo Y, Luo Y, Penning T, Bauch J, Bouska J, et al. ABT-888, an orally active poly (ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clin Cancer Res. 2007;13:2728–2737. doi: 10.1158/1078-0432.CCR-06-3039. [DOI] [PubMed] [Google Scholar]
- 13.Kummar S, Kinders R, Gutierrez ME, Rubinstein L, Parchment RE, Phillips LR, et al. Phase 0 clinical trial of the poly(ADP-ribose) polymerase inhibitor ABT-888 in patients with advanced malignancies. J Clin Oncol. 2009;27:2705–2711. doi: 10.1200/JCO.2008.19.7681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Patel AG, Flatten KS, Schneider PA, Dai NT, McDonald JS, Poirier GG, et al. Enhanced killing of cancer cells by poly(ADP-ribose) polymerase inhibitors and topoisomerase I inhibitors reflects poisoning of both enzymes. J Biol Chem. 2012;287:4198–4210. doi: 10.1074/jbc.M111.296475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.D’Onofrio G, Tramontano F, Dorio AS, Muzi A, Maselli V, Fulgione D, et al. Poly(ADP-ribose) polymerase signaling of topoisomerase 1-dependent DNA damage in carcinoma cells. Biochem Pharmacol. 2011;81:194–202. doi: 10.1016/j.bcp.2010.09.019. [DOI] [PubMed] [Google Scholar]
- 16.Fukushima M, Kuzuya K, Ota K, Ikai K. Poly(ADP-ribose) synthesis in human cervical cancer cell-diagnostic cytological usefulness. Cancer Lett. 1981;14:227–236. doi: 10.1016/0304-3835(81)90148-8. [DOI] [PubMed] [Google Scholar]
- 17.Gonen M. A Bayesian evaluation of enrolling additional patients at the maximum tolerated dose in Phase I trials. Contemporary clinical trials. 2005;26:131–140. doi: 10.1016/j.cct.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 18.Chen TT, Ng TH. Optimal flexible designs in phase II clinical trials. Statistics in medicine. 1998;17:2301–2312. doi: 10.1002/(sici)1097-0258(19981030)17:20<2301::aid-sim927>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 19.Kunos C, Winter K, Dicker A, Small WJ, Abdul-Karim FW, Dawson D, et al. Ribonucleotide reductase expression in cervical cancer: a radiation therapy oncology group translational science analysis. Int J Gynecol Cancer. 2013;23:615–621. doi: 10.1097/IGC.0b013e31828b4eb5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kunos C, Chiu S, Pink J, Kinsella T. Modulating radiation resistance by inhibiting ribonucleotide reductase in cancers with virally or mutationally silenced p53 protein. Radiation Res. 2009;172:666–676. doi: 10.1667/RR1858.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kunos C, Radivoyevitch T, Pink J, Chiu S, Stefan T, Jaccobberger J, et al. Ribonucleotide reductase inhibition enhances chemoradiosensitivity of human cervical cancers. Radiation Res. 2010;174:574–581. doi: 10.1667/RR2273.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kunos C, Radivoyevitch T, Waggoner S, DeBernardo R, Zanotti K, Resnick K, et al. Radiochemotherapy plus 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (3-AP, NSC #663249) in advanced-stage cervical and vaginal cancers. Gynecol Oncol. 2013;130:75–80. doi: 10.1016/j.ygyno.2013.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kunos C, Waggoner S, Von Gruenigen V, Eldermire E, Pink J, Dowlati A, et al. Phase I trial of intravenous 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (3-AP, NSC #663249) in combination with pelvic radiation therapy and weekly cisplatin chemotherapy for locally advanced cervical cancer. Clin Cancer Res. 2010;16:1298–1306. doi: 10.1158/1078-0432.CCR-09-2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaplan E, Meier P. Nonparametric estimation from incomplete observations. J Am Stat Assoc. 1958;53:457–481. [Google Scholar]
- 25.Snedecor G, Cochran W. Statistical Methods. Ames, IA: Iowa State University Press; 1989. [Google Scholar]
- 26.Good P. Permutation tests: a practical guide to resampling methods for testing hypotheses. New York: Springer; 2000. [Google Scholar]
- 27.Klein J, Moeschberger M. Survival analysis techniques for censored and truncated data. New York: Springer; 2003. [Google Scholar]
- 28.Cox D. Regression models and life tables (with discussion) J R Stat Soc. 1972;34:187–220. [Google Scholar]
- 29.Staker B, Hjerrild K, Feese M, Behnke C, Burgin AJ, Stewart L. The mechanism of topoisomerase I poisoning by a camptothecin analog. Proc Natl Acad Sci USA. 2002;99:15387–15392. doi: 10.1073/pnas.242259599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hakansson P, Hofer A, Thelander L. Regulation of mammalian ribonucleotide reduction and dNTP pools after DNA damage and in resting cells. J Biol Chem. 2006;281:7834–41. doi: 10.1074/jbc.M512894200. [DOI] [PubMed] [Google Scholar]
- 31.Xue L, Zhou B, Liu X, Qiu W, Jin Z, Yen Y. Wild-type p53 regulates human ribonucleotide reductase by protein-protein interaction with p53R2 as well as hRRM2 subunits. Cancer Res. 2003;63:980–986. [PubMed] [Google Scholar]
- 32.Zamboni WC, D’Argenio DZ, Stewart CF, MacVittie T, Delauter BJ, Farese AM, et al. Pharmacodynamic model of topotecan-induced time course of neutropenia. Clin Cancer Res. 2001;7:2301–2308. [PubMed] [Google Scholar]
- 33.Leger F, Loos WJ, Bugat R, Mathijssen RH, Goffinet M, Verweij J, et al. Mechanism-based models for topotecan-induced neutropenia. Clinical pharmacology and therapeutics. 2004;76:567–578. doi: 10.1016/j.clpt.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 34.Smith HO, Tiffany MF, Qualls CR, Key CR. The rising incidence of adenocarcinoma relative to squamous cell carcinoma of the uterine cervix in the United States--a 24-year population-based study. Gynecol Oncol. 2000;78:97–105. doi: 10.1006/gyno.2000.5826. [DOI] [PubMed] [Google Scholar]
- 35.Farley JH, Hickey KW, Carlson JW, Rose GS, Kost ER, Harrison TA. Adenosquamous histology predicts a poor outcome for patients with advanced-stage, but not early-stage, cervical carcinoma. Cancer. 2003;97:2196–2202. doi: 10.1002/cncr.11371. [DOI] [PubMed] [Google Scholar]