

Summary
Genome-wide association studies have identified loci underlying human diseases, but the causal nucleotide changes and mechanisms remain largely unknown. Here we developed a fine-mapping algorithm to identify candidate causal variants for 21 autoimmune diseases from genotyping data. We integrated these predictions with transcription and cis-regulatory element annotations, derived by mapping RNA and chromatin in primary immune cells, including resting and stimulated CD4+ T-cell subsets, regulatory T-cells, CD8+ T-cells, B-cells, and monocytes. We find that ~90% of causal variants are noncoding, with ~60% mapping to immune-cell enhancers, many of which gain histone acetylation and transcribe enhancer-associated RNA upon immune stimulation. Causal variants tend to occur near binding sites for master regulators of immune differentiation and stimulus-dependent gene activation, but only 10–20% directly alter recognizable transcription factor binding motifs. Rather, most noncoding risk variants, including those that alter gene expression, affect non-canonical sequence determinants not well-explained by current gene regulatory models.
Introduction
Genome-wide association studies (GWAS) have revolutionized the study of complex human traits by identifying thousands of genetic loci that contribute susceptibility for a diverse set of diseases1,2. However, progress towards understanding disease mechanisms has been limited by difficulty in assigning molecular function to the vast majority of GWAS hits that do not affect protein-coding sequence. Efforts to decipher biological consequences of noncoding variation face two major challenges. First, due to haplotype structure, GWAS tend to nominate large clusters of single nucleotide polymorphisms (SNPs) in linkage disequilibrium (LD), making it difficult to distinguish causal SNPs from neutral variants in linkage. Second, even assuming the causal variant can be identified, interpretation is limited by incomplete knowledge of noncoding regulatory elements, their mechanisms of action, and the cellular states and processes in which they function.
Inflammatory autoimmune diseases, which reflect complex interactions between genetic variation and environment, are important systems for genetic investigation of human disease3. They share a substantial degree of immunopathology, with increased activity of auto-reactive CD4+ T-cells secreting inflammatory cytokines and loss of regulatory T-cell (Treg) function4. A critical role for B-cells in certain diseases has also been revealed with the therapeutic efficacy of anti-CD20 antibodies5. Immune homeostasis depends on a balance of CD4+ pro-inflammatory (Th1, Th2, Th17) cells and FOXP3+ suppressive Tregs, each of which expresses distinct cytokines and surface molecules6. Each cell type is controlled by a unique set of master transcription factors (TFs) that directly shape cell-type specific gene expression programs, which include genes implicated in autoimmune diseases7–9. Immune subsets also have characteristic cis-regulatory landscapes, including distinct sets of enhancers that may be distinguished by their chromatin states9–13 and associated enhancer RNAs (eRNA)14. Familial clustering of different autoimmune diseases suggests that heritable factors underlie common disease pathways, although disparate clinical presentations and paradoxical effects of drugs in different diseases support key distinctions15.
GWAS have discovered hundreds of risk loci for autoimmunity15. While most risk variants have subtle effects on disease susceptibility, they provide unbiased support for possible etiological pathways, including antigen presentation, cytokine signaling, and NFκB transcriptional regulation15. The associated loci are enriched for immune cell-specific enhancers10,16,17 and expansive enhancer clusters18,19, termed ‘super-enhancers’, implicating gene regulatory processes in disease etiology. However, as is typical of GWAS, the implicated loci comprise multiple variants in LD and rarely alter protein coding sequence, which complicates their interpretation.
Here, we integrated genetic and epigenetic fine-mapping to identify causal variants in autoimmune disease-associated loci and explore their functions. Based on dense genotyping data20, we developed a novel algorithm to predict for each individual variant associated with 21 autoimmune diseases, the likelihood that it represents a causal variant. In parallel, we generated cis-regulatory element maps for a spectrum of immune cell types. Remarkably, ~60% of likely causal variants map to enhancer-like elements, with preferential correspondence to stimulus-dependent CD4+ T-cell enhancers that respond to immune activation by increasing histone acetylation and transcribing noncoding RNAs. Although these enhancers frequently reside within extended clusters, their distinct regulatory patterns and phenotypic associations suggest they represent independent functional units. Causal SNPs are enriched near binding sites for immune-related TFs, but rarely alter their cognate motifs. Our study provides a unique resource for the study of autoimmunity, links causal disease variants with high probability to context-specific immune enhancers, and suggests that most non-coding causal variants act by altering non-canonical regulatory sequence rather than recognizable consensus TF motifs.
Fine-mapped Genetic Architecture of Disease
To explore the genetic architecture underlying common diseases, we first collected 39 well-powered GWAS studies (Methods). Clustering of diseases and traits based on their shared genetic loci revealed groups of phenotypes with related clinical features (Fig 1A). This highlighted a large cluster of immune-mediated diseases forming a complex network of shared genetic loci; on average, 69% of the associated loci for each disease were shared with other autoimmune diseases, although no two diseases shared more than 38% of their loci.
Figure 1. Genetic fine-mapping of human disease.

a, GWAS catalog loci were clustered to reveal shared genetic features of common human diseases and phenotypes. Color scale indicates correlation between phenotypes (high=red, low=blue). b, Association signal to MS for SNPs at the IFI30 locus. c, Scatter plot of SNPs at the IFI30 locus demonstrates the linear relationship between LD distance (r2) to rs1154159 (red) and association signal. d, Candidate causal SNPs were predicted for 21 autoimmune diseases using PICS. Histogram indicates genomic distance (bp) between PICS Immunochip lead SNPs and GWAS catalog index SNPs. e, Histogram indicates number of candidate causal SNPs per GWAS signal needed to account for 75% of the total PICS probability for that locus. f, Plot shows correspondence of PICS SNPs to indicated functional elements, compared to random SNPs from the same loci (error bars indicate standard deviation from 1000 iterations using locus-matched control SNPs).
We focused subsequent analysis on autoimmune diseases, reasoning that recent dense genotyping data combined with emerging approaches for profiling epigenomes of specialized immune cells would provide an opportunity to identify and characterize the specific causal SNPs. Prior studies that have integrated GWAS with epigenomic features focused on lead SNPs or multiple associated SNPs within a locus, of which only a small minority reflects causal variants10,16–19,21. Although these studies demonstrated enrichments within enhancer-like regulatory elements, they could not with any degree of certainty pinpoint the specific elements or processes affected by the causal variants. To overcome this limitation, we leveraged dense genotyping data to refine a statistical model for predicting causal SNPs from genetic data alone. Rare recombination events within haplotypes can provide information on the identity of the causal SNP, provided sufficient genotyping density and sample size. We therefore examined a cohort of 14,277 cases with multiple sclerosis (MS) and 23,605 healthy controls genotyped using the Immunochip, which comprehensively covers 1000 Genomes Project SNPs22 within 186 loci associated with autoimmunity20. We developed an algorithm, Probabilistic Identification of Causal SNPs (PICS), that estimates the probability that an individual SNP is a causal variant given the haplotype structure and observed pattern of association at the locus (Methods, Extended Data Figs 1–4).
The IFI30 locus (Fig 1B–C) presents an illustrative example of the LD problem and the PICS strategy. The most strongly associated SNP at the locus is rs11554159 (R76Q, G>A; minor allele is protective), a missense variant in IFI30, a lysosomal enzyme that processes antigens for MHC presentation23. Although dozens of SNPs at the locus are significantly associated with disease, the association for each additional SNP follows a linear relationship with its linkage to rs11554159/R76Q, suggesting they owe their association solely to linkage with this causal variant. We used permutation to estimate the posterior probability for each SNP in the locus to be the causal variant, given the observed patterns of association. Interestingly, prior GWAS studies24 had attributed the signal at this locus to a missense variant in a neighboring gene, MPV17L2 (rs874628, r2 = 0.9 to R76Q), with no known immune function. However, we find that the R76Q variant is ~10 times more likely than rs874628 to be the causal SNP and three times more likely than the next closest SNP (a noncoding variant), providing compelling evidence that the IFI30 missense variant is the causal variant in the locus.
We next generalized PICS to analyze 21 autoimmune diseases, using Immunochip data when they were available or imputation to the 1000 Genomes Project22 when they were not (Methods; Supplementary Table S1). We mapped 636 autoimmune GWAS signals to 4950 candidate causal SNPs (mean probability of representing the causal variant responsible for the GWAS signal: ~10%). PICS indicates that index SNPs reported in the GWAS catalog have on average only a 5% chance of representing a causal SNP. Rather, GWAS catalog index SNPs are typically some distance from the PICS lead SNP (median 14 kb), and many are not in tight LD (Fig 1D, Extended Data Figure 5). PICS identified a single most likely causal SNP (>75% probability) at 12% of loci linked to autoimmunity. However, most GWAS signals could not be fully resolved due to LD and thus contain several candidate causal SNPs (Fig 1E).
To confirm the functional significance of fine-mapped SNPs, we compared PICS SNPs against a strict background of random SNPs drawn from the same loci. Candidate causal SNPs derived by PICS were strongly enriched for protein-coding (missense, nonsense, frameshift) changes, which account for 14% of the predicted causal variants compared to just 4% of the random SNPs. Modest enrichments over the locus background were also observed for synonymous substitutions (5%), 3′ UTRs (3%), and splice junctions (0.2%) (Fig 1F). Although these results support the efficacy of PICS for identifying causal variants, ~90% of GWAS hits for autoimmune diseases remain unexplained by protein-coding variants. Candidate causal SNPs and the PICS algorithm are available through an accompanying online portal (www.broadinstitute.org/pubs/finemapping).
Causal Autoimmunity Variants Map to Immune Cell-Specific Enhancers
To investigate the functions of predicted causal noncoding variants, we generated a resource of epigenomic maps for specialized immune subsets (Extended Data Figure 6). We examined primary human CD4+ T-cell populations from pooled healthy donor blood, including FOXP3+CD25highCD127low/−regulatory (Tregs), CD25−CD45RA+CD45RO− naïve (Tnaïve) and CD25−CD45RA−CD45RO+ memory (Tmem) T-cells, and ex vivo phorbol myristate acetate (PMA)/ionomycin stimulated CD4+ T-cells separated into IL-17-positive (CD25−IL17A+; Th17) and IL-17-negative (CD25−IL17A−; Thstim) subsets. We also examined naïve and memory CD8+ T-cells, B-cell centroblasts from pediatric tonsils (CD20+CD10+CXCR4+CD44−), and peripheral blood B-cells (CD20+) and monocytes (CD14+). We mapped six histone modifications by chromatin immunoprecipitation-sequencing (ChIP-seq) for all ten populations, and performed RNA-sequencing (RNA-seq) for each CD4+ T-cell population. We also incorporated data for B-lymphoblastoid cells17, Th0, Th1 and Th2 stimulated T-cells10, and non-immune cells from the NIH Epigenomics Project25 and ENCODE26, for a total of 56 cell types.
For each cell type, we computed a genome-wide map of cis-regulatory elements based on H3 lysine 27 acetylation (H3K27ac), a marker of active promoters and enhancers12. We then clustered cell types based on these cis-regulatory element patterns (Extended Data Figure 7). Fine distinctions could be drawn between CD4+ T-cell subsets based on quantitative differences in H3K27ac at thousands of putative enhancers (Fig 2A). These cell type-specific H3K27ac patterns correlate with the expression of proximal genes. In contrast, H3 lysine 4 mono-methylation (H3K4me1) was more uniform across subsets, consistent with its association to open or ‘poised’ sites shared between related cell types12.
Figure 2. Epigenetic fine-mapping of enhancers.

a, Heatmaps show H3K27ac and H3K4me1 signals for 1000 candidate enhancers (rows) in 12 immune cell types (columns). Enhancers are clustered by the cell type-specificity of their H3K27ac signals. Adjacent heatmap shows average RNA-seq expression for the genes nearest to the enhancers in each cluster. Gray-scale (right) depicts the enrichment of PICS autoimmunity SNPs in each enhancer cluster (hypergeometric p-values calculated based on the number of PICS SNPs overlapping enhancers from each cluster, relative to random SNPs from the same loci). The AP-1 motif is over-represented in enhancers preferentially marked in stimulated T-cells, compared to naïve T-cells. b, Candidate causal SNPs displayed along with H3K27ac and RNA-seq signals at the PTGER4 locus. A subset of enhancers with disease variants (shaded) shows evidence of stimulus-dependent eRNA transcription. c, Stacked bar graph indicates percentage overlap with immune enhancers and coding sequence for PICS SNPs at different probability thresholds, compared to control SNPs drawn from the entire genome (All SNPs) or the same loci (Locus CTRL). d, Venn diagram compares PICS SNPs to GWAS catalog SNPs with indicated r2 thresholds. e, Bar graph indicates percentage overlap with annotated T-cell enhancers for PICS SNPs, GWAS SNPs at indicted thresholds, locus control SNPs, and three subsets of SNPs defined and shaded as in panel d.
Mapping of autoimmune disease PICS SNPs to these regulatory annotations revealed enrichment in B-cell and T-cell enhancers (Fig 2A). A disproportionate correspondence to enhancers activated upon T-cell stimulation prompted us to examine such elements more closely. Substantial subsets of immune-specific enhancers markedly increase their H3K27ac signals upon ex vivo stimulation, often in conjunction with noncoding eRNA transcription, and induction of proximal genes (Figs 2A–B). Compared to naïve T-cells, enhancers in stimulated T-cells are strongly enriched for consensus motifs recognized by AP-1 TFs, master regulators of cellular responses to stimuli. PICS SNPs are strongly enriched within stimulus-dependent enhancers (p<10−20 for combined PMA/Iono; p<10−11 for combined CD3/CD28), while enhancers preferentially marked in unstimulated T-cells show no enrichment for causal variants. Candidate causal SNPs were further enriched in T-cell enhancers that produce noncoding RNAs upon stimulation (1.6-fold; p<0.01).
The association of candidate causal SNPs to immune enhancers increases with PICS probability score (Fig 2C). We estimate that immune enhancers overall account for ~60% of candidate causal SNPs, whereas promoters account for another ~8% of these variants (Extended Data Figure 7). When we compared these statistics against GWAS catalog SNPs, which were the focus of prior studies linking GWAS to regulatory annotations10,16–19,21, we found that the subset of associated SNPs that do not correspond to a PICS SNP fail to show any enrichment for T-cell enhancers, relative to locus controls (Figs 2D–E). These data support the efficacy of PICS and link likely causal autoimmune disease variants to specific enhancers activated upon immune stimulation.
Cell-Type Specific Signatures of Complex Diseases
Along with the 21 autoimmune diseases, we predicted causal SNPs for 18 other traits and diseases (Methods). Comparing SNP locations with chromatin maps for 56 cell types revealed the cell type-specificities of cis-regulatory elements that coincide with PICS SNPs, thus predicting cell types contributing to each phenotype (Fig 3). The patterns are more informative than the expression patterns of genes targeted by coding GWAS hits (Extended Data Figure 8). Notable examples include SNPs associated with Alzheimer’s disease and migraine, which map to enhancers and promoters active in brain tissues, and SNPs associated with fasting blood glucose, which map to elements active in pancreatic islets. Nearly all of the autoimmune diseases preferentially mapped to enhancers and promoters active in CD4+ T-cell subpopulations. However, a few diseases, such as systemic lupus erythematosus, Kawasaki disease, and primary biliary cirrhosis, preferentially mapped to B-cell elements. Notably, ulcerative colitis also mapped to gastrointestinal tract elements, consistent with its bowel pathology. While the primary signature of type 1 diabetes SNPs is in T-cell enhancers, there is also enrichment in pancreatic islet enhancers (p<10−7). Thus, while immune cell effects may be shared among autoimmune diseases, genetic variants affecting target organs such as bowel and pancreatic islets may shape disease-specific pathology.
Figure 3. Cell-type specificity of human diseases.

Heatmap depicts enrichment (red=high; blue=low) of PICS SNPs for 39 diseases/traits in acetylated cis-regulatory elements of 33 different cell types.
Causal Variants Map to Discretely-Regulated Elements Within Super-Enhancers
Genomic loci that encode cellular identity genes frequently contain large regions with clustered or contiguous enhancers bound by transcriptional co-activators and marked by H3K27ac. Recent studies showed that such ‘super-enhancer’ regions are enriched for GWAS catalog SNPs, including those related to autoimmunity18,19. Consistently, we find that PICS SNPs are 7.5-fold enriched in CD4+ T-cell super-enhancers, relative to random SNPs from the genome. We therefore parsed the topography of super-enhancers in immune cells using our genetic and epigenetic data.
The IL2RA locus exemplifies the complex landscape of enhancer regulation. IL2RA encodes a receptor with key roles in T-cell stimulation and Treg function15. The super-enhancer in this locus comprises a cluster of elements recognizable as distinct H3K27ac peaks (Fig 4A). Although the region meets the super-enhancer definition in multiple CD4+ T-cell types18, sub-elements are preferentially acetylated in Treg, Th17 and/or Thstim T-cells, consistent with differential regulation. Some sub-elements appear bound by T-cell master regulators, including FOXP3 in Tregs, TBET in Th1 cells, and GATA3 in Th2 cells. A systematic analysis indicates PICS SNPs are most enriched at distinct stimulus-dependent H3K27ac peaks within super-enhancer regions (Extended Data Figure 7).
Figure 4. Disease variants map to discrete elements in super-enhancers.

a, Candidate causal SNPs for autoimmune diseases are displayed along with H3K27ac, RNA-seq and TF binding profiles for the IL2RA locus, which contains a super-enhancer (pink shade). b, For all SNPs in the IL2RA locus, scatter plot compares strength of association with MS versus autoimmune thyroiditis. Immunochip data resolve rs706779 (red) as the lead SNP for autoimmune thyroiditis and rs2104286 (blue) as the lead SNP for MS. c, LD matrix displaying r2 between lead SNPs for different diseases at the IL2RA locus confirms distinct and independent genetic associations within the super-enhancer.
PICS SNPs for eight autoimmune diseases map to distinct segments of the IL2RA super-enhancer. For example, Immunochip data identify a candidate causal SNP for MS that has no effect on autoimmune thyroiditis disease risk. Conversely, a candidate causal SNP for autoimmune thyroiditis has no effect on MS risk, despite the proximity of the two SNPs within the super-enhancer (Fig 4B). Furthermore, index SNPs for multiple other diseases are not in LD, suggesting that multiple sites of nucleotide variation in the locus have separable disease associations (Fig 4C). The distribution of PICS SNPs and the partially discordant regulation of sub-regions suggest that super-enhancers may comprise multiple discrete units with distinct regulatory signals, functions and phenotypic associations.
Disease Variants Map to TF Binding Sites, but Infrequently Disrupt Consensus Motifs
The enrichment of candidate causal variants within enhancers suggests that they affect disease risk by altering gene regulation, but does not distinguish the underlying mechanisms. Enhancer activity is dependent on complex interplay between TFs, chromatin, noncoding RNAs and tertiary interactions of DNA loci27. A straightforward hypothesis is that disease SNPs alter TF binding. Indeed, PICS SNPs tend to coincide with nucleosome-depleted regions, characterized by DNase hypersensitivity and localized (~150 bp) dips in H3K27ac signal26, which are indicative of TF occupancy (Fig 5A).
Figure 5. Causal variants map to regions of TF binding.

a, Plot depicts composite H3K27ac and DNase signals26 in immune cells over PICS autoimmunity SNPs. PICS SNPs overall coincide with nucleosome-depleted, hypersensitive sites, indicative of TF binding. b, Bar plot indicates TFs whose binding is enriched near PICS SNPs for all 21 autoimmune diseases26. Heatmap depicts enrichment of these TFs near variants associated with specific diseases (red:high; blue:low). c, H3K27ac, DNaseI26 and conservation signals, and selected TF binding intervals are shown in a SMAD3 intronic locus. rs17293632, a noncoding candidate causal SNP for Crohn’s disease, disrupts a conserved AP-1 binding motif in an enhancer marked by H3K27ac in CD14+ monocytes. Summing of ChIP-seq reads overlapping the SNP in the heterozygous HeLa cell line shows that only the intact motif binds AP-1 TFs, Jun and Fos. d, Bar graph shows the fraction of PICS SNPs (black) versus random SNPs from the same locus (white) that create or disrupt one of the significantly enriched motifs, any Selex motif, or any conserved K-mer. Error bars indicate standard deviation from 1000 iterations using locus-matched control SNPs.
We therefore overlapped PICS SNPs with 31 TF binding maps generated by ENCODE26 (Fig 5B). Candidate causal SNPs are strongly enriched within binding sites for immune-related TFs, including NFκB, PU1, IRF4, and BATF. Variants associated with different diseases correlate to different combinations of TFs that control immune cell identity and response to stimulation. For example, MS SNPs preferentially coincide with NFκB, EBF1 and MEF2A-bound regions, whereas rheumatoid arthritis and celiac disease SNPs preferentially coincide with IRF4 regions.
Next, we examined whether causal variants disrupt or create cognate sequence motifs recognized by these TFs. We focused on 823 of the highest-likelihood noncoding PICS SNPs, an estimated 30% of which represent true causal variants. We identified PICS SNPs that alter motifs for NFκB (n=2), AP-1 (n=8), or ETS/ELF1 (n=5). Overall, we identified 7 known TF motifs and 6 conserved sequence motifs28,29 with a significant tendency to overlap causal variants likely to alter binding affinity. Of the highest-likelihood SNPs, 7% affected one of these over-represented motifs, with a roughly equal distribution between motif creation and disruption (Extended Data Fig 9).
A notable motif-disrupting PICS SNP is the Crohn’s disease-associated variant rs17293632 (C>T, minor allele increases disease risk; PICS probability~54%), which resides in an intron of SMAD3 (Fig 5C). SMAD3 encodes a TF downstream of transforming growth factor-β (TGF-β) with pleiotropic roles in immune homeostasis30. The SNP disrupts a conserved AP-1 consensus site. ChIP-seq data for AP-1 factors (Jun, Fos) in a heterozygous cell line reveal robust binding to the reference sequence, but not to the variant sequence created by the SNP. As described above, a prominent AP-1 signature is associated with enhancers activated upon immune stimulation (Fig 2A). This suggests that rs17293632 may increase Crohn’s disease risk by directly disrupting AP-1 regulation of the TGF-β-SMAD3 pathway.
Despite this and other compelling examples, only ~7% of the highest-likelihood noncoding PICS SNPs alter an over-represented TF motif. Scanning a large database of TF motifs, we found that ~13% of high-likelihood causal SNPs create or disrupt some known consensus sequence derived by in vitro selection28, while ~27% create or disrupt a putative consensus sequence derived from phylogenetic analysis29. However, these proportions are similar to the rate for background SNPs (Fig 5D). Even extrapolating for uncertainty in causal SNP assignments, our data suggest that at most 10–20% of noncoding GWAS hits act by altering a recognizable TF motif.
Notwithstanding their infrequent coincidence to the precise TF motifs, noncoding PICS SNPs have a strong tendency to reside in close proximity to such sequences. Candidate causal variants are most significantly enriched in the vicinity of NFκB, RUNX1, AP1, ELF1, and PU1 motifs (Extended Data Fig 9), with 26% residing within 100 bp of such a motif. These findings parallel recent studies of genetic variation in mice, where DNA variants affecting NFκB binding are dispersed in the vicinity of the actual binding sites31. Our results suggest that many causal noncoding SNPs modulate TF-dependent enhancer activity (and confer disease risk) by altering adjacent DNA bases whose mechanistic roles are not readily explained by existing gene regulatory models.
Functional Effects of Disease Variants on Gene Expression
To assess the effects of autoimmunity-associated genetic variation on gene regulation, we incorporated a recent study that mapped variants associated with heritable differences in peripheral blood gene expression32. We used PICS to predict causal expression quantitative locus (eQTL) SNPs, which we compared against random SNPs from the same loci. These eQTL SNPs are strongly enriched in promoters (9%) and 3′ UTRs (25%), but show relatively modest preference for immune enhancers (14%), compared to GWAS SNPs (Fig 6A). Overall, ~12% of causal noncoding autoimmune disease variants also score as eQTL SNPs (Extended Data Figure 10). Disease SNPs that did not score as eQTLs in peripheral blood may score in more precise immune subsets in relevant regulatory contexts. Nonetheless, their modest overlap with eQTLs and their striking correspondence to enhancers suggest that most disease variants exert subtle and highly context-specific effects on gene regulation.
Figure 6. Functional Effects of Disease Variants on Gene Expression.
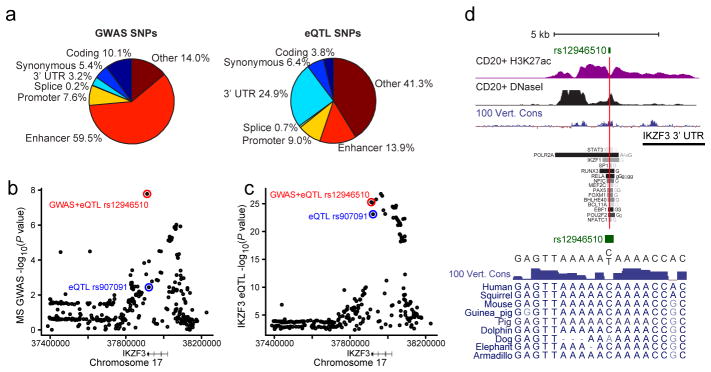
a, Pie charts show the fraction of PICS autoimmunity SNPs (left) or peripheral blood eQTLs (right) explained by the indicated genomic features. b, GWAS signal for MS risk at the IKZF3 locus. The minor allele of rs12946510 (red) is associated with both disease risk and eQTL effect (decreased IKZF3 expression), while the minor allele of rs907091 (blue) scored as eQTL only (increased IKZF3 expression). c, eQTL association signal for IKZF3 shown for the same regions as in b. d, H3K27ac, DNaseI and conservation signals, and selected TF binding intervals are shown in the vicinity of rs12946510, which occurs in a conserved site marked by H3K27ac in multiple cell types, including CD20+ B-cells, and bound by multiple TFs. The C/T variation at this SNP does not disrupt any clearly defined DNA motif, but coincides with a degenerate MEF2 motif.
Incorporation of eQTL SNPs allowed us to link causal noncoding disease variants to specific genes. For example, PICS fine-mapping identified two SNPs in the IKZF3 locus with independent effects on IKZF3 expression, rs12946510 and rs907091. IKZF3 is an IKAROS family TF with key roles in lymphocyte differentiation and function33. Interestingly, the minor allele of rs12946510 is associated with decreased IKZF3 expression and increased MS risk (Fig 6B–C), whereas the minor allele of rs907091 is associated with increased IKZF3 expression, but does not affect disease risk. This suggests that disease risk is dependent on the specific mode and context in which a variant influences gene expression.
Despite strong evidence from fine-mapping that rs12946510 is the causal SNP affecting MS risk and IKZF3 expression, the underlying sequence does not reveal a clear mechanism of action. The disease SNP resides within a conserved element with enhancer-like chromatin in immune cells. It coincides with a nucleosome-depleted, DNase hypersensitive site bound by multiple TFs, including immune-related factors RUNX3, RELA (NFκB family member), EBF1, POU2F2 and MEF2 (Fig 6D). The C/T variation at this site does not create or disrupt a readily recognizable consensus DNA motif, but overlaps a highly degenerate MEF2 motif and might thus modulate TF binding despite incomplete sequence specificity. This example illustrates the value of integrative functional genomic analysis for investigating the complex mechanisms by which noncoding variants modulate gene expression and disease risk.
Discussion
Interpretation of noncoding disease variants, which comprise the vast majority of GWAS hits, remains a momentous challenge due to haplotype structure and our limited understanding of the mechanisms and physiological contexts of noncoding elements. Here we addressed these issues through combination of high-density genotyping and epigenomic data. Focusing on autoimmune diseases, we triaged causal variants based solely on genetic evidence and integrated chromatin and TF binding maps to distinguish their likely functions and physiologic contexts. We found that most causal variants map to enhancers and frequently coincide with nucleosome-depleted sites bound by immune-related TFs. The resulting resource highlights specific TFs, target loci and pathways with disease-specific or general roles in autoimmunity.
Yet despite their close proximity to immune TF binding sites, only a fraction of causal noncoding variants alter recognizable TF sequence motifs. Moreover, disease variants have a distinct functional distribution and infrequently overlap peripheral blood eQTLs, which suggests that they exert highly contextual regulatory effects. Although these features of noncoding disease variants further challenge GWAS interpretation, they might not be unexpected. Biochemical and genetic manipulations have established the potential of motif-adjacent sequences to influence TF activity34. Roles for such non-canonical sequences are also supported by the extended nucleotide conservation at many enhancers, most of which lies outside of known motifs, and the complex structural interactions and looping events that underlie gene regulation27. Furthermore, common variants contributing to polygenic autoimmunity are expected to have modest, context-restricted effects, given that strongly deleterious mutations would be eliminated from the population1. Compared to mutations that disrupt TF motifs, alterations to non-canonical determinants may produce subtle but pivotal alterations to the immune response, without reaching a level of disruption that would result in strong negative selection.
Systematic integration of fine-mapped genetic and epigenetic data implies a nuanced complexity to disease variant function that will continue to push the limits of experimental and computational approaches. Much work remains to be done to characterize SNPs whose causality can be firmly established through genotyping and to facilitate efforts to resolve GWAS signals that remain refractory to fine-mapping due to haplotype structure. Understanding their regulatory mechanisms could have broad implications for autoimmune disease biology and treatment, given genetic links to immune regulators, such as NFκB, IL2RA and IKZF3 (AIOLOS), and implied transcriptional and epigenetic aberrations, all of which are candidates for therapeutic intervention.
Supplementary Methods
Cell Isolation and Culture
Purification and Culture of Human CD4+ T-cell Subsets
Cells were obtained from the peripheral blood of pooled healthy subjects in compliance with Institutional Review Board (Yale University and Partners Human Research Committee) protocols. Untouched CD4+ T cells were isolated by gradient centrifugation (Ficoll-Hypaque; GE Healthcare) using the RosetteSep Human CD4+ T cell Enrichment kit (Stemcell Technologies). CD4+ T cells were next subjected to anti-CD25 magnetic bead labeling (Miltenyi Biotech), to allow magnetic cell separation (MACS) of CD25+ and CD25− cells. Subsequently CD25+ cells were stained with fluorescence-labeled monoclonal antibodies to CD4, CD25 and CD127 (BD Pharmingen), and sorted using a FACS ARIA (BD Biosciences) for CD25highCD127low/− Treg cells, which express FOXP3 (Biolegend) as confirmed by intracellular post-sort analysis by FACS (Extended Data Fig. 6). Dead cells were excluded by Propidium iodide (BD). An aliquot of CD25− cells was labeled with fluorescence-labeled monoclonal antibodies to CD4, CD45RA and CD45RO (BD Pharmingen), and sorted on a FACS ARIA to isolate CD45RO+CD45RA− memory (Tmem) and CD45RO−CD45RA+ naïve (Tnaive) CD4+ T-cell populations. Dead cells were excluded by Propidium iodide. Highly pure human Th17 cells were isolated with modifications as previously described35. In brief, CD25− cells were stimulated in serum-free X-VIVO15 medium (BioWhittaker) with PMA (50ng ml−1) and ionomycin (250ng ml−1; both from Sigma-Aldrich) for 8 hours and sorted by a combined MACS and FACS cell sorting strategy based on surface expression of IL-17A. Stimulated cells were stained with anti-IL-17A-PE (Miltenyi) and labeled with anti-PE microbeads (Miltenyi) and subsequently pre-enriched over an LS column (Miltenyi). The IL-17A negative fraction was used as control population (Thstim). MACS-enriched Th17 cells were further sorted on a FACS ARIA (BD) for highly pure IL-17A+ cells (Th17).
Purification of Human Naïve and Memory CD8+ T cells
Leukocyte-enriched fractions of peripheral blood (byproduct of Trima platelet collection) from anonymous healthy donors were obtained from the Kraft Family Blood Donor Center (DFCI, Boston, MA) in compliance with the institutional Investigational Review Board (Partners Human Research Committee) protocol. For two independent purifications of each cell subset, blood fractions from 7 and 8 donors were pooled. Total T cells were isolated by immunodensity negative selection using the RosetteSep Human T-cell Enrichment Cocktail (STEMCELL Technologies, Vancouver, Canada) and gradient centrifugation on Ficoll-Paque PLUS (GE Healthcare, Pittsburgh, PA), according to the manufacturer’s instructions. Subsequently, T cells were stained at 4°C for 30 min using fluorescently-labeled monoclonal anti-human CD8 (FITC, 2.5 μg/ml, clone RPA-T8, Biolegend, San Diego, CA), CD4 (PE, 1.25 μg/ml, clone RPA-T4, Biolegend), CD45RA (PerCP-Cy5.5, 2.4 μg/ml, clone HI100, eBioscience, San Diego, CA) and CD45RO (APC, 0.6 μg/ml, clone UCHL1, eBioscience) antibodies diluted in staining buffer (PBS supplemented with 2% fetal bovine serum, FBS). 4′,6-diamidino-2-phenylindole (DAPI, 2.5 μg/ml, Life Technologies, Grand Island, NY) was also included to stain for dead cells. After washing with staining buffer, naïve (CD45RA+CD45RO−) and memory (CD45RA−CD45RO+) CD8+ or CD4+ were isolated using a BD FACSAria 4-way cell sorter (BD Biosciences, San Jose, CA). Cell subsets were identified using a BD FACSDiva Software (BD Biosciences) after gating on lymphocytes (by plotting forward versus side scatters) and excluding aggregated (by plotting forward scatter pulse height versus pulse area), dead (DAPI+), and CD8/CD4 double positive cells (Extended Data Fig. 6). Cell purity was 90-94% CD8+ or 97–99% CD4+, and >99% naïve or memory.
Purification of Human B Centroblasts
Cells were obtained in compliance with Institutional Review Board (Partners Human Research Committee) protocols. For purification of human centroblasts, bulk mononuclear cells were isolated from fresh pediatric tonsillectomy specimens by mechanical disaggregation and Ficoll-Paque centrifugation36. MACS enrichment of germinal center cells was performed using anti-CD10-PE-Cy7 (BD Biosciences), and anti-PE microbeads (Miltenyi Biotec). Centroblasts37 (CD19+CD10+CXCR4+CD44−CD3−) were purified from the enriched germinal center cells by FACS antibodies for CD19 (APC, clone SJ25C1, BD), CD3 (BV606, clone OKT3, Biolegend), CD10 (PE-Cy7, clone HI10A, BD), CD44 (FITC, clone L178, BD) and CXCR4 (PE, clone 12G5, eBioscience) (Extended Data Fig. 6).
Purification of Adult Human Peripheral Blood B Cells and Monocytes
Human peripheral B cells and monocytes were provided by the S. Heimfeld Laboratory at the Fred Hutchinson Cancer Research Center. The cells were obtained from human leukapheresis product using standard procedures. Briefly, Peripheral B cells (CD20+CD19+) and monocytes (CD14+) were isolated by immunomagnetic separation using the CliniMACS affinity-based technology (Miltenyi Biotec GmbH, Bergisch Gladbach, Germany) according to the manufacturer’s recommendation. Reagents, tubing sets, and buffers are purchased from Miltenyi Biotec.
ChIP-Seq
Following isolation (+/− ex vivo stimulation), cells were cross-linked in 1% formaldehyde at room temperature or 37°C for 10 minutes in preparation for ChIP. Chromatin immunoprecipitation and sequencing were performed as previously described38. Datasets were publicly released upon verification at epigenomeatlas.org.
RNA-Seq
RNA was extracted from CD4+ T-cell subsets with Trizol. Briefly, polyadenylated RNA was isolated using Oligo dT beads (Invitrogen) and fragmented to 200–600 base pairs and then ligated to RNA adaptors using T4 RNA Ligase (NEB), preserving strand of origin information as previously described39,40.
Enhancer Annotation and Clustering
ChIP-seq data were processed as previously described38. Briefly, ChIP-seq reads of 36 bp were aligned to the reference genome (hg19) using the Burroughs-Wheeler Alignment tool (BWA)41. Reads aligned to the same position and strand were only counted once. Aligned reads were extended by 250 bp to approximate fragment sizes and then a 25-bp resolution chromatin map was derived by counting the number of fragments overlapping each position. H3K27ac and H3K4me1 peaks were identified by scanning the genome for enriched 1kb windows and then merging all enriched windows within 1kb, using as a threshold 4 genome-normalized reads per base pair38. Adjacent windows separated by gaps less than 500bp in size were joined. H3K27ac peaks that do not overlap ±2.5 kb region of an annotated TSS were defined as candidate distal regulatory elements. In order to define the cell-specific H3K27ac peaks, we calculated the mean signal in 5Kb regions centered at distal H3K27ac peaks and sorted the peaks by the ratio of signal in one cell type to all remaining cell types. For each cell type, the top 1000 distal H3K27ac peaks with highest ratio were cataloged as the cell-specific distal H3K27ac peaks. The heatmaps for H3K27ac and H3K4me1 signal were plotted over 10kb regions surrounding all distal cell-specific H3K27ac peaks.
The distal H3K27ac peaks were assigned to their potential target genes if they locate in the gene body or within 100kb regions upstream the TSS. Expression levels of the target genes were derived from RNA-seq data. Paired-end RNA-seq reads were aligned to RefSeq transcripts using Bowtie242. RNA-seq data for B cells, B centroblast, Macrophages, Th1, Th2 and Th0 were retrieved from NCBI GEO and SRA database (Bnaive: GSE45982; BgerminalCenter: GSE4598243; Macrophages: GSE3695244; Th0, Th1 and Th2: SRA08267010). RNA-seq data for lymphoblastoid (GM12878) was retrieved from ENCODE project26. The number of reads per kilobase per million reads (RPKM) was calculated for each gene locus. Heatmap of RNA-seq data shows the average relative expression of all potential target genes for each cluster of cell type-specific regulatory elements.
Shared Genetic Loci for Common Human Diseases
Publicly available GWAS catalog data were obtained from the NHGRI website, http://www.genome.gov/gwastudies/, current as of July 201345,46. Studies were included based on the criteria that they had at least 6 hits at the genome-wide significant level of p ≤ 5 ×10−8. From a set of 21 autoimmune diseases and 18 representative non-autoimmune diseases/traits, we included index SNPs with significance p ≤ 10−6 for downstream analysis.
In some cases, the same disease had multiple index SNPs mapping to the same locus (defined as within 500kb of each other), due to independently conducted GWAS studies identifying different lead SNPs within the same region. For these loci, only the most significant GWAS index SNP was kept for downstream analysis, resulting in 1170 GWAS index SNPs for 39 diseases/traits. For each pair of diseases/traits, we compared their respective lists of index SNPs to find instances of common genetic loci (defined as the two diseases sharing index SNPs within 500kb of each other). The number of overlapping loci was calculated for each disease pair. To measure the genetic similarity between two diseases/traits, a disease-by-disease correlation matrix was calculated based on the number of overlapping loci for each disease/trait with each of the other diseases, and the results are shown in Fig. 1A.
Sources of Immunochip and Non-Immunochip GWAS Data
Summary statistics for published Immunochip studies of celiac disease47, autoimmune thyroiditis48, primary biliary cirrhosis49, and rheumatoid arthritis50 were downloaded from the Immunobase website, http://www.immunobase.org/. Full genotype data and PCA analysis for the multiple sclerosis Immunochip GWAS study20 was provided by the International Multiple Sclerosis Genetics Consortium. For ankylosing spondylitis51, atopic dermatitis52, primary sclerosing cholangitis53, juvenile idiopathic arthritis54, and psoriasis55, Immunochip studies had been previously been published, but only the lead SNPs from associated Immunochip regions were available. We also included GWAS of autoimmune diseases that had not been studied using Immunochip, including asthma, allergy, Kawasaki disease, Behcet’s disease, vitiligo, alopecia arreata, systemic lupus erythematosis, systemic sclerosis, type 1 diabetes, Crohn’s disease, and ulcerative colitis. For these diseases and the 18 representative non-immune diseases, index SNPs from the GWAS catalog were used46. In addition, full genotype data and PCA analysis for the inflammatory bowel disease Immunochip GWAS study were provided by the International Inflammatory Bowel Diseases Genetics Consortium for purposes of calculating the statistical models used in PICS. Because the results for the IBD Immunochip analysis are unpublished, we used the previously published index SNP results for inflammatory bowel disease from the GWAS catalog.
Probabilistic Identification of Causal SNPs (PICS)
We developed a fine-mapping algorithm, which we call Probabilistic Identification of Causal SNPs (PICS), that makes use of densely-mapped genotyping data to estimate each SNP’s probability of being a causal variant, given the observed pattern of association at the locus. We developed PICS on large multiple sclerosis (MS) (14277 cases, 23605 controls20) and inflammatory bowel disease (IBD) cohorts (34594 cases, 28999 controls; unpublished data) that were genotyped using the Immunochip, a targeted ultra-dense genotyping array with comprehensive coverage of 1000 Genomes Project SNPs22 within 189 autoimmune disease-associated loci.
Analysis of IBD risk associated with SNPS at the IL23R locus presents an illustrative example of the LD problem and the potential for PICS to overcome this challenge (Extended Data Fig. 1). The most strongly associated SNP is rs11209026, a loss of function missense variant that changes a conserved arginine to glutamine at amino acid position 381 (R381Q) and decreases downstream signaling through the STAT3 pathway56,57. Association with IBD decreases with physical distance along the chromosome, due to rare recombination events that break up the haplotype and distinguish the causal missense mutation from other tightly linked neutral variants. These rare informative recombination events would be missed by standard genotyping arrays with probes spread thinly across the entire genome.
For neutral SNPs whose association signal is only due to being in LD with a causal SNP, the strength of association, as measured by chi-square (or log-pvalue, since chi-square and log-pvalue are asymptotically linear) scales linearly with their r2 to the causal SNP. This is because strength of association is linear with r2 by the formula for the Armitage Trend Test58:
where χ2 is the chi-square association test statistic, n is the sample size, and r2 is the square of the correlation coefficient.
This linear trend is observed at the IL23R locus, consistent with a model where R381Q is the causal variant, and neutral SNPs demonstrate association signal in proportion to their LD to the causal variant (Extended Data Fig. 1). SNPs in linkage to R381Q do not perfectly fall on the expected line, due to statistical fluctuations. Independent association studies for the same disease tend to nominate different SNPs within a given locus as their best association, due to statistical fluctuation pushing a different SNP to the forefront in each subsequent study59–62. Note that a group of SNPs that are strongly associated to disease but are not in linkage with rs11209026 (R381Q) represent independent association signals at the locus.
Although we know from functional studies that R381Q is the likely causal variant, we sought additional statistical evidence to support R381Q as the causal variant, and to refute the null hypothesis that the prominent association of R381Q (compared to other SNPs in the haplotype) is due to chance. We simulated 1000 permutations by fixing the association signal at R381Q, but with all other SNPs being neutral, while preserving the LD relationships between SNPs in the locus. An odds ratio of 1.2 was used rather than the ~2-fold odds ratio naturally observed at R381Q, because this was more representative of the modest association signal strengths observed at other GWAS loci. For each round of permutation, we obtained the association signal at all SNPs in the locus. Because only the association signal at R381Q is fixed, the signal at the remaining neutral SNPs in the locus are free to vary due to statistical fluctuations; four typical examples of simulated association results at the R381Q locus are shown (Extended Data Fig. 1), including two examples where the causal variant is not the most strongly associated SNP in the locus. From these 1000 iterations, we calculated the standard deviation in the association signal for each of the SNPs in the IL23R locus (Extended Data Fig. 2). We show that the distribution of association signals for each SNP approximates a normal distribution, centered at the expected value based on that SNP’s r2 to the causal variant (Extended Data Fig. 2).
These permutations demonstrate that the causal variant need not be the most strongly associated SNP within the locus, due to statistical fluctuations. Rather, given the observed pattern of association at a locus, we are interested in knowing the probability of each SNP within the locus to be the causal variant. We can use Bayes’ theorem to infer the probability of each SNP being the causal variant, by using information derived from the permutations. Since the prior probability of each SNP to be the causal variant is equal, the SNP most likely to be the causal variant is therefore the SNP whose simulated signal most closely approximates the observed association at the locus. By performing permutations of a simulated association signal at each SNP within the locus, we can estimate the probability that the SNP could lead to the observed association at the locus.
For example, consider a two SNP example where SNP A and SNP B are in LD, and SNP A is the lead SNP in the locus (Extended Data Fig. 2). If we are interested in knowing P(Bcausal|Alead), i.e. the probability that SNP B is the causal variant given that SNP A is the top signal in the locus, then by Bayes’ theorem:
Where P(Alead|Bcausal) is the probability of SNP A being the top signal in the locus, given that SNP B is the causal variant. P(Alead|Bcausal) is straightforward to calculate by performing permutations with a simulated signal at SNP B, and measuring the number of permutations where SNP A emerges as the top signal in the locus despite SNP B being the actual causal variant. We have assumed that the prior probability of each SNP to be the causal variant or the lead SNP is equal, although this could be adjusted based on external information, such as functional annotation of the SNP to be a coding variant.
Using the formula above, we calculate both P(Bcausal|Alead) and P(Acausal|Alead), and then normalize both of these probabilities so that P(Bcausal|Alead) + P(Acausal|Alead) = 1. In cases where there are more than two SNPs to consider, we similarly normalize the probabilities so that they sum to 1. Probabilities were calculated for all SNPs with r2 > 0.5 to the lead SNP.
Because the calculation of thousands of permutations is computationally expensive and requires full genotype data, we sought to generalize the results of the permutation-based method in order to extend it to the analysis of autoimmune diseases for which Immunochip data were not available, or only the identity of the lead index SNPs was reported, such as from the GWAS catalog. We developed a general model, where PICS was able to calculate P(Bcausal|Alead), where B is a SNP within a locus, and A is the lead SNP in the locus, by using LD relationships from the Immunochip where these were available, and from the 1000 Genomes Project otherwise. Since the distribution of association signal at neutral SNPs in the locus approximates a normal distribution, given the lead SNP in the locus, we need to be able to estimate the mean expected association for a neutral SNP in LD with the lead SNP, and the standard deviation for that SNP.
The expected mean association signal for SNPs in the locus scales linearly with r2 to the causal SNP in the locus. We derived an approximation for the standard deviation for each SNP in the locus based on the results of empiric testing. We picked 30,000 random SNPs from densely-mapped Immunochip loci, with half coming from the MS Immunochip data, and half coming from the IBD Immunochip data. For each SNP, we simulated 100 permutations with that SNP being the causal variant. SNPs selected had minor allele frequency above 0.05, and the odds ratio used varied from 1.1 fold to 2.0 fold. The number of cases and controls and total sample size were also allowed to randomly vary from 1%–100% of the total number of samples in the original studies. These results indicated that the standard deviation for the association signal at a SNP in LD (with r2>0.5) to a causal variant in the locus was approximately:
where s is the standard deviation of the association signal at the SNP, m is the expected mean of the association signal at the SNP, indexpval is the −log10(p-value) of the causal SNP in the locus, r2 is the square of the correlation coefficient (a measure of LD) between the SNP and the causal SNP in the locus, and k is an empiric constant that can be adjusted to fit the curve; in practice, we found that choosing k from a wide range of values between 6 and 8 had little measurable effect on the candidate causal SNPs selected, and we used a value of k=6.4. The results of the 30,000 simulated iterations and the empiric curve fitted using the above equation is shown in Extended Data Fig. 3. To verify that our method was applicable to a wide range of case-control ratios and effect sizes, we performed six additional simulations, with the percentage of case samples fixed at 10%, 20%, and 50%, and the effect sizes of causal SNPs fixed at 1.2-fold, 1.5-fold, and 2.0-fold, which cover a broad range of parameters likely to be encountered in practical GWAS studies (Extended Data Fig. 3). We found that for all six scenarios, the relationship between r2 to the causal SNP and standard deviation similarly followed the empirically fitted curve.
For each SNP in the locus, we used the estimated mean and standard deviation of the association signal at each neutral SNP in LD (r2 > 0.5) to the lead SNP in the locus to calculate the probability of each SNP to be the causal variant relative to the lead SNP. We then normalized the probabilities so that the total of their probabilities summed to 1.
For diseases where summary SNP information was available, but the r2 relationships between SNPs was unknown, the r2 relationship was estimated based on the ratio between the association signal at the lead SNP versus the SNP in question. For diseases where only the lead SNP was known, r2 values were drawn from the LD relationships from the MS Immunochip study if the SNP was from an Immunochip, or from the 1000 Genomes Project otherwise. 1000 Genomes European LD relationships were used for diseases, except for Kawasaki disease, for which 1000 Genomes East Asian LD relationships were used. For diseases which had both GWAS catalog results and Immunochip results, we used Immunochip results whenever possible, and GWAS catalog results in regions outside Immunochip dense-mapping coverage.
Multiple Independent Association Signals
For the MS data, we were able to use full genotyping information to distinguish multiple independent signals. We used stepwise regression to condition away SNPs one at a time until no associations remain at the p<10−6 level, which is an effective method for separating independent signals, when LD between the independent causal variants is low. We then treated each independent signal separately for the purpose of using PICS to derive the likely causal variants.
Missing Immunochip Data
For the minority of SNPs that were missing from the Immunochip, we used 1000 Genomes SNPs LD relations to the index SNP to estimate their probability of being the causal SNP. For the diseases with only Immunochip summary statistic data, we could not be certain of the LD relationships, and therefore we estimated the LD to the index SNP from the difference between the association at the lead SNP and the SNP in question, since these follow a linear relationship. For the diseases that only had Immunochip index SNP data, we used Immunochip LD relationships where available from the MS data, and 1000 Genomes SNPs LD relations to the index SNP where these were not available.
Distance between GWAS Catalog SNPs and Lead SNPs
For Immunochip regions that were previously studied by non-Immunochip studies, we examined the performance of prior non-fine-mapped studies at correctly determining the lead SNP. GWAS catalog SNPs within 200kb of Immunochip regions were considered, and the LD and genomic distance between the catalog SNP and any Immunochip lead SNPs for that disease in the Immunochip region were measured and reported in the histograms in Fig. 1D and Extended Data Fig. 5. PICS was also used to calculate the probability of GWAS catalog SNPs to be causal variants; the probability was 5.5% on average.
Number of Candidate Causal SNPs per GWAS Signal
For each GWAS signal, we obtained a set of candidate causal SNPs, each with a probability of being the causal variant. For each signal, we asked what was the minimum number of candidate causal SNPs required to to cover at least 75% of the probability (Fig. 1E).
Distribution of GWAS Signals in Functional Genomic Elements: Signal to Background
For downstream analyses, we considered the set of 4905 candidate causal SNPs which had mean probability of greater than 10% of being the candidate causal SNP (the cutoff was probability > 0.0275). We performed 1000 iterations, picking 4905 minor-allele-frequency-matched random SNPs from the same loci (from genomic regions within 50kb of the candidate causal SNPs and excluding the actual causal SNPs). It was necessary to match for minor-allele-frequency because lower MAF SNPs are far more likely to be coding variants. Furthermore, it was necessary to match for locus, because GWAS SNPs are greatly enriched at gene bodies, and using a background of random 1000 genome SNPs for comparison results in massive nonspecific enrichment of all functional elements. Because we are comparing the candidate causal SNPs to a background set of control SNPs from the same regions, the observed enrichments at functional elements strongly argues that PICS effectively predicts causal variants within the loci. For each functional category (missense, nonsense, and frameshift were merged), we calculated the number of actual candidate causal SNPs above mean background (mean of 1000 random iterations), divided by the total number of GWAS signals represented (635), and used these results to populate the pie chart indicating the approximate percentage of GWAS signals that can be attributed to each assessed functional category (Fig. 6).
Analysis of ex vivo Stimulation-dependent Enhancers
We searched for motifs enriched in cell type-specific enhancers in the five stimulated T-cell subsets (PMA/Ionomycin stimulated Thstim and Th17 T-cells, anti-CD3/CD28 stimulated Th0, Th1, and Th2 T-cells) compared to enhancers in naïve T-cells, using the motif finding program HOMER (http://homer.salk.edu/homer/)63. AP-1 was the most strongly enriched motif in enhancers that gained H3K27ac in the stimulated T-cells (Fig. 2), whereas this enrichment was absent when comparing naïve T-cells with memory or regulatory T-cells. Additional motifs that were enriched in the stimulation-dependent enhancers included NFAT for the PMA/Ionomycin stimulation conditions and STAT for the anti-CD3/CD28 stimulation condition.
Enhancer Signal-to-noise Analysis
We focused on 14 immune cell types (8 CD4+ T-cell subsets, 2 CD8+ T-cell subsets, CD14+ monocytes, and 3 B-cell subsets) and 19 representative non-immune cell/tissue types from the Roadmap Epigenomics project. Enhancers were broken up into 1kb segments and immune specific enhancers were identified based on the following criteria: (1) # of normalized mean H3K27ac ChIP-seq extended reads/base > 4, and (2) mean H3K27ac in the top 15th percentile when comparing immune cells to non-immune cells/tissues. We measured the percentage of PICS SNPs (with different probability cutoffs) that either map to an immune enhancer or cause an amino acid coding change (Fig. 2). We next considered the 4300 candidate causal SNPs that were not associated with protein coding changes, and compared them against 1000 iterations of frequency and locus matched controls (picked from genomic regions within 50kb of the candidate causal SNPs and excluding the actual candidate causal SNPs; see discussion of background calculations above). Enhancers were enriched approximately 2:1 above background. We also measured the signal to backround ratio for GWAS signals that had been attributed to coding variants; these produced a much lower signal to background ratio for immune enhancers, as would be anticipated by the fact that most of these are acting on coding regions rather than enhancers (Extended Data Fig. 7). The mean signal above background was shown in a pie chart (Fig. 6).
Comparison to Other Methods for Determining Candidate Causal Variants
We compared the efficacy of PICS versus previously published methods used to determine candidate causal variants (Fig. 2D–E). We first considered studies that had used cutoffs of r2=1.0 and r2>0.8 to determine likely causal SNPs. Because prior studies had not made use of dense genotyping data, we used only the GWAS catalog results for this comparison, and applied PICS, and the two r2-cutoff criteria. In practice these were much more stringent than prior analyses, because we limited the GWAS catalog studies to those that produced 6 or more genome-wide significant hits, thereby pruning underpowered studies. We also required a significance of p<10−6 for index SNPs, and merged index SNPs at the same locus to use the strongest and most accurate lead SNP. We found that PICS autoimmunity SNPs were much more likely to map to immune enhancers than SNPs identified by the other statistics. In addition, when the PICS SNPs which overlapped the r2>0.8 and r2>1.0 sets were removed, the remaining SNPs did not show any enrichment above background. In contrast, the candidate causal SNPs identified by PICS, but missed by both of the other methodologies, were significantly enriched for immune enhancers. Background was calculated based on random SNPs drawn from the same loci (within 50kb, frequency-matched controls) as the candidate causal SNPs.
We also compared PICS with a recently reported Bayesian approach64, using a recently published study of MS20 that employed this methodology to call candidate causal SNPs. Because this published method required full genotypes to be available, this comparison was limited to only the MS dataset. Both PICS and the published method are Bayesian approaches, where each SNP within the locus is given equal prior consideration to be the causal variant, and the algorithm then weighs each SNP based on the likelihood of each model given the data. However, the PICS method provides two advantages. First, the probabilities assigned to each SNP by PICS are determined empirically using permutation, rather than using a theoretical estimate for the weight of each SNP. Second, PICS can be generalized to all GWAS data with publicly available summary statistic data and does not rely on genotype data.
For the same MS immunochip dataset, PICS called 434 candidate causal SNPs, while the prior method called 4070 candidate causal SNPs; 177 SNPs were shared between the two analyses. Of the 434 PICS candidate causal SNPs, 26.5% overlapped immune enhancers, whereas 9.5% of the SNPs from the other method overlapped immune enhancers; the background rate of random SNPs from the same loci overlapping immune enhancers was 8% (Extended Data Fig. 4). Because the Wellcome Trust (Maller et al.) method is clearly less stringent than PICS, we also tried using a high confidence set of SNPs derived by that method, by selecting the top SNPs such that their average probability of being a causal variant was 10% (the same cutoff used for the PICS SNPs). There were 165 SNPs in this high confidence set, compared to 434 for PICS, with an overlap of 65 SNPs. 20.3% of the candidate causal SNPs in the high confidence Maller et al. set overlapped immune enhancers. Although anecdotal, these results suggest that PICS performs at least as well as the prior method.
Tissue-specificity of Diseases
We used PICS fine-mapping to determine the set of candidate causal SNPs for each of 39 different diseases, and examined whether they were enriched within the enhancers most specific to each cell type (defined as being in the top 15th percentile of H3K27ac signal compared to other cell types, and with > 1 normalized mean H3K27ac ChIP-seq extended reads/base). To compare enhancer regions across different cell types, we first subdivided regions of the genome that were marked as enhancers into enhancer segments ~1kb in size. Next, H3K27ac read density at each enhancer segment in the genome was compared across all 33 cell types to determine the cell types in the top 15th percentile (H3K27ac signal was quantile normalized across the cell types prior to comparison). The heatmap (Fig. 3) depicts p-values for the enrichment of PICS SNPs for each disease in H3K27ac elements for each cell type, as calculated by the chi square test. For this comparative analysis, enrichment of PICS SNPs was measured against a background of all common 1000 Genomes SNPs. We used this approach because the goal was to highlight cell type-specificity of the diseases, which would have been normalized out by the rigorous locus controls used above, and given that the specificity of PICS SNPs for enhancers within the loci was already established. We also mapped the expression patterns of genes with PICS candidate causal coding SNPs associated with Crohn’s disease, MS and rheumatoid arthritis (Extended Data Fig. 8).
Super-enhancer Enrichment
The full set of loci called as super-enhancers18 in CD4+ T-cell subsets (Tnaive, Tmem, Th17, ThStim) were merged and identified as CD4+ T-cell super-enhancer regions. These regions often contain clusters of discrete enhancers marked with H3K27ac, separated by non-acetylated regions. We assessed if PICS SNPs mapping to super-enhancers were more likely to occur in H3K27ac-marked enhancer regions than in intervening regions. Within CD4+ T-cell super-enhancer regions, we compared overlap of PICS candidate causal SNPs with CD4+ T-cell H3K27ac regions, compared to frequency-matched background SNPs drawn from these same regions (Extended Data Fig. 7). H3K27ac intervals in CD4+ T-cell super-enhancers were called based on being in the top 15th percentile in mean H3K27ac in T-cells compared to the other 25 cell types. In addition, we assessed overlap between PICS SNPs and H3K27ac elements preferential to either stimulated or unstimulated CD4+ T-cells. Stimulated CD4+ T-cell elements were defined as those with a mean increase of >25% in H3K27ac in the (average of) Th17, ThStim, Th0, Th1, Th2 cells, compared to the Tnaive, Tmem, Treg; the remainder of the CD4+ T-cell set were defined as unstimulated elements.
Fig. 4 shows that some sub-elements within IL2RA super-enhancer locus appear bound by T-cell master regulators based on published ChIP-Seq data, including FOXP3 in Tregs, TBET in Th1 cells, and GATA3 in Th2 cells65,66.
Noncoding RNA Analysis
We next examined the set of disease-associated enhancers, i.e. immune enhancers containing PICS autoimmunity SNPs, and their association with noncoding RNAs. Noncoding RNA transcripts were called based on a RNA-seq read density of 0.5 genome-normalized reads per bp over a window size of at least 2kb, excluding RNA transcripts overlapping annotated exons or gene bodies. We found that enhancers containing PICS autoimmunity SNPs were enriched for noncoding transcript production, primarily consistent with unspliced enhancer-associated RNAs. Candidate causal SNPs were enriched 1.6-fold within T-cell enhancers that transcribed noncoding RNAs, compared to T-cell enhancers overall (p<0.01).
H3K27ac and DNase Profiles
We measured H3K27ac profiles and DNase hypersensitivity profiles in a 12kb window centered around candidate causal SNPs, taking the average signal for the 14 immune cell types for which H3K27ac was available, and immune cell types from ENCODE26 for which DNase was available (CD14+, GM12878, CD20+, Th17, Th1, Th2). Average normalized reads for H3K27ac and DNase centered at PICS SNPs are displayed in Fig. 5A.
Transcription Factor ChIP-Seq Binding Site Analysis
We compared the enrichment of PICS autoimmunity SNPs at TF binding sites identified by ENCODE ChIP-Seq67, relative to random SNPs drawn from the same loci (50kb window around the candidate causal SNPs, frequency matched). We show the results for the 31 TFs whose binding sites are most significantly enriched for PICS SNPs (Fig. 5B).
Motif Creation / Disruption Analysis
We downloaded consensus motifs from Selex28 and Xie et al.29 (represented as degenerate nucleotide codes). We used the 853 highest probability non-coding PICS SNPs (mean probability = 0.30, cutoff >0.1187), representing 403 different GWAS signals. For each candidate causal SNP, we examined whether it created or disrupted a known motif from Selex or Xie et al. For comparison, we ran 1000 iterations using frequency-matched random SNPs drawn from the same loci (within 50kb of the PICS SNPs). We found several known motifs (Extended Data Fig. 9) to be significantly enriched, including AP1, ETS, NFKB, SOX, PITX, as well as several unknown conserved motifs (Extended Data Fig. 9). Subtracting the number of motifs found to be disrupted against that expected by background, and dividing by the total number of GWAS signals, we estimate that approximately 11% of noncoding GWAS hits can be attributed to direct disruption or creation of TF binding motifs.
Neighboring Motif Analysis
We compared the sequence within 100nt of high-likelihood PICS SNPs (cutoff >0.1187) against random flanking sequence (10kb away on either side from the causal SNPs) and looked for enriched motifs using HOMER (http://homer.salk.edu/homer/)63. We found significant enrichments for NFKB, RUNX, AP1, ELF1, and PU1 (Extended Data Fig. 9). Interestingly, there was a palindromic unknown motif TGGCWNNNWGCCA (p<10−4) that was significant both in this method and in the motif disruption/creation analysis, suggesting a yet uncharacterized TF motif with relevance in autoimmunity.
Expression Quantitative Trait Loci (eQTL) Analysis
We used PICS to predict causal SNPs from a peripheral blood eQTL dataset with 1000 Genomes summary statistic data available for all cis-eQTLs. We required a gene to have a cis-eQTL with a p-value < 10−6 for this analysis, giving us 4136 genes. For each gene we applied PICS. We considered a GWAS hit to score as an eQTL if any GWAS PICS SNP in the locus coincided with an eQTL PICS SNP with average probability >0.01%. We found that 11.6% (74/636) of GWAS hits were also eQTL hits. In addition, 18.5% (15/81) of coding GWAS hits also showed eQTL effects, suggesting that they may actually operate at the transcriptional level, in addition to any coding effects they may have.
To quantify overlap of candidate causal eQTL SNPs with functional elements, we compared PICS eQTL SNPs against frequency-matched background SNPs drawn from the same loci (within 50kb) in 1000 iterations. These comparisons are shown in signal-to-background bar graphs for both coding/transcript-related functional elements and for enhancers and promoters (Extended Data Fig. 10). The signal above mean background was calculated for each functional category, and these results were compared against the results for GWAS hits in the pie charts shown in Fig. 6A.
We further examined whether the magnitudes of disease-associated eQTLs differed, compared to the space of all eQTLs (Extended Data Fig. 10). Disease-associated variants had modestly larger effects on gene expression (p<10−6 by rank-sum test), but did not necessarily correspond to the strongest eQTLs.
Extended Data
Extended Data Figure 1. GWAS Result for IBD Immunochip data at IL23R locus (actual data a–c, simulated permutationd d–e).

(a) Each of the 500 SNPs in the IL23R densely genotyped locus is plotted according to its association signal and position along the chromosome. The R381Q missense variant is circled in red. (b) Each of the 500 SNPs in the IL23R densely genotyped locus is plotted according to its association signal and r2 linkage to R381Q. (c) Same as (b), but showing the association signal on the y-axis in chi-square units. Over the range of values typically encountered in GWAS analyses, chi-square units and log p-value are asymptotically linear. (d) Simulated permutation analysis of signal at IL23R locus. 1.2-fold odds ratio signal was simulated at the R381Q SNP by fixing the association signal at R381Q, but permuting cases and controls such that all other SNPs are neutral and vary only with statistical noise. Four representative results from the simulations are shown, with the panels on the left showing the association signal in genomic space, and the panels on the right (e) showing the association signal for each SNP in relation to r2.
Extended Data Figure 2. Calculating the Relative Likelihood of Being the Causal SNP from Standard Deviation in Association Signal.

(a) For each SNP in the IL23R locus, the mean association signal and the standard deviation, calculated across 1000 permutations (using a 1.2-fold odds ratio at the R381Q SNP), are shown in genomic space and (b) in terms of each SNP’s r2 linkage disequilibrium to the causal R381Q variant. (c) The distribution of association signals at rs77319898 (r2 = 0.71 to the causal variant) for 1000 permutations is shown. The distribution of association signal values at each SNP approximated a normal distribution. (d) PICS analysis of a two SNP case to determine the relative likelihood of each to explain the pattern of association at the locus. The SNPs represented here are R381Q (SNP A) and rs77319898 (SNP B), which has an r2 = 0.71 to R381Q. The signal at SNP B is well-explained by LD to SNP A, in a model where SNP A is treated as the putative causal variant. The error bars indicate the standard deviation in the association signal expected for SNP B, under the assumption that SNP A is causal. (e) The signal at SNP A is poorly explained by LD to SNP B, in a model where SNP B is treated as the putative causal variant. The error bars indicate the standard deviation in the association signal expected for SNP A, under the assumption that SNP B is causal.
Extended Data Figure 3. Simulated Permutations and Empiric Curve Fitting for 30,000 GWAS Signals at Immunochip Loci.

(a) We simulated 30,000 causal SNPs in densely mapped Immunochip regions. Plot shows the relationship between standard deviation in the association signal of neutral SNPs and their r2 to the causal SNP (neutral SNPs within r2 > 0.5 of the simulated causal variant are shown). The red line indicates the expected values derived from the empiric equation for the standard deviation of the association signal at neutral SNPs in LD with the causal SNP. (b) Plot shows the relationship between standard deviation in the association signals of neutral SNPs and the association signal of the causal SNP. Each panel represents the set of neutral SNPs with the indicated r2 to the causal variant. (c) Simulated permutations over a range of case-control ratios. We plotted the relationship between standard deviation at neutral SNPs and their r2 to the causal SNP. Plots are shown for three series of simulations, with the percentage of cases fixed at 10%, 20%, and 50% of the total sample size, and a causal SNP p-value of 10−20. Red line indicates the expected values derived from the empiric equation for the standard deviation of the association signal at neutral SNPs in LD with the causal SNP in the locus. (d) Simulated permutations over a range of effect sizes. Plots are shown for three series of simulations, with the effect size fixed at 1.2-fold, 1.5-fold, and 2.0-fold, and the corresponding lead SNP p-values fixed at 10−20, 10−70, and 10−150 respectively.
Extended Data Figure 4. Comparison of PICS with prior Bayesian fine-mapping method.

Bar graph shows the percentage of MS SNPs overlapping immune enhancers using different algorithms for calling candidate causal SNPs. The dotted line indicates the background rate at which random 1000 Genomes Project SNPs drawn from the same loci intersect immune enhancers (~8%). The categories shown are (from top to bottom): 257 SNPs called only by PICS, 3812 SNPs called only by the Bayesian method, 177 SNPs called by both PICS and the Bayesian method, all 434 SNPs called by PICS, 165 called by the Bayesian using a cutoff that only includes the highest confidence SNPs, and all 4070 SNPs called by Bayesian method.
Extended Data Figure 5. LD Distance Between PICS lead SNPs and GWAS catalog index SNPs.
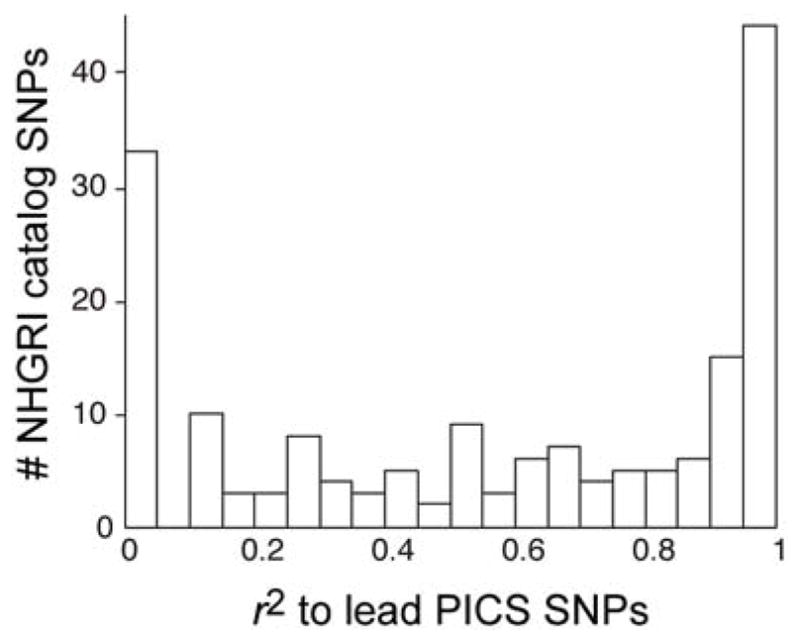
Histogram indicates LD distance (in r2) between PICS fine-mapped Immunochip lead SNPs and previously reported GWAS catalog index SNPs from the same loci.
Extended Data Figure 6. Purification of Human Immune Cell Subsets.

(a) Immune populations subjected to epigenomic profiling in this study (red labels) or prior publications. (b) CD4+ cells were enriched based on CD25 expression (MACS) and subsequently sorted based on CD25highCD127low/− to isolate Treg cells; confirmed with FOXP3 intracellular staining. (c) CD4+CD25− cells were sorted to isolate Tmem (CD45RO+CD45RA−) and Tnaive (CD45RO−CD45RA+) cells. (d) CD4+CD25− cells were PMA/ionomycin stimulated and separated based on IL17 surface expression (MACS and FACS) to isolate Th17 cells (IL17+) and ThStim cells (IL17−). (e) Naïve (CD45RA+CD45RO−) and memory (CD45RA−CD45RO+) CD8+ T cells were isolated using a BD FACSAria 4-way cell sorter. Results are shown from one of two large-scale sorts. (f) Mononuclear cells were isolated from pediatric tonsils. Following CD10 enrichment (MACS), B centroblasts (CD19+CD10+CXCR4+CD44−CD3−) were purified by FACS.
Extended Data Figure 7. PICS SNPs Localize to Immune Enhancers and Stimulus-Dependent H3K27ac Peaks in Super-enhancers.

(a) Correlation matrix of 56 cell types, clustered by similarity of H3K27ac profiles (high=red, low=blue). (b) Enrichment of noncoding autoimmune disease candidate causal SNPs within immune enhancers and promoters compared to background. The background expectation is based on frequency-matched control SNPs drawn from within 50kb of the candidate causal SNPs. Candidate causal SNPs that produced coding changes or were in LD with a coding variant (paired bars on the right) showed a smaller degree of enrichment in immune enhancers and promoters compared to background. (c) Overlap of PICS SNPs with H3K27ac peaks within T-cell super-enhancers. Bar plot shows overlap of PICS SNPs with H3K27ac peaks in super-enhancers in CD4+ T-cells, compared to random SNPs drawn from within the same super-enhancers (All CD4+; left bar graph). Adjacent bars show overlap to H3K27ac peaks within CD4+ T-cell super-enhancers that do (Stim) or do not (Unstim) increase their acetylation upon stimulation.
Extended Data Figure 8. Expression Pattern of Genes with PICS Autoimmunity Coding SNPs.

Heatmap shows the relative expression levels of genes with coding SNPs associated with Crohn’s disease, multiple sclerosis, and rheumatoid arthritis.
Extended Data Figure. 9. Motifs Directly Altered by or Adjacent to Candidate Causal SNPs.

(a) Known motifs (identified by conservation or SELEX) created or disrupted by candidate causal SNPs at a higher frequency than expected by chance when compared to control SNPs drawn from the same loci. (b) Additional motifs, identified by conservation, created or disrupted by candidate causal SNPs more frequently than by chance. (c) Known motifs significantly enriched within 100bp of candidate causal SNPs, compared to background control SNPs drawn from the same loci.
Extended Data Figure 10. Enrichment of Candidate Causal eQTL SNPs in Functional Elements.

(a) PICS was used to identify candidate causal SNPs for 4136 eQTL signals in peripheral blood. Bar plot show their overlap with indicated functional genic annotations. Background expectation was calculated based on frequency-matched control SNPs drawn from within 50kb of the candidate causal SNPs. (b) Overlap of candidate causal eQTL SNPs with immune enhancers and promoters, versus background. (c) Magnitudes of disease-associated eQTLs compared to the space of all eQTLs. Histogram compares the magnitudes of PICS eQTL SNPs that overlap PICS autoimmunity SNPs against the full set of PICS eQTL SNPs.
Supplementary Material
Acknowledgments
We thank members of the NIH Epigenomics Consortium, Michael Greenberg, Hannah Chang and Genevieve Haliburton for constructive comments. We also thank IIBDGC and P.Sullivan for sharing data pre-publication, and G.Cvetanovich, S.Bhela, C.Hartnick, F.Preffer, D.Dombkowski and the Brigham and Women’s Hospital PhenoGenetic Project for assistance with data collection. This research was supported by the NIH Common Fund (ES017155), the National Human Genome Research Institute (HG004570), the National Institute of Allergy and Infectious Disease (AI045757, AI046130, AI070352, AI039671), the National Institute of Neurological Disorders and Stroke (NS2427, NS067305), the National Institute of General Medical Sciences (GM093080), the National Multiple Sclerosis Society (CA1061-A-18), the UCSF Sandler Fellowship, a gift from Jake Aronov, the Penates Foundation, the Nancy Taylor Foundation and the Howard Hughes Medical Institute.
Footnotes
Author Contributions: A.M., D.A.H. and B.E.B. designed the study. K.K.F. performed genetic analysis, PICS development and integration. M.J.D. supervised genetic analysis. J.Z, M.K., S.B., N.S, H.W., R.J.H.R., A.A.S., M.H., M.J.C.-A., D.M., C.J.H., V.K.K. and C.B.E. contributed to data collection and analysis. N.A.P. and P.L.D.J. contributed MS genotyping data. K.K.F., A.M., D.A.H. and B.E.B. wrote the manuscript.
Conflict of Interest: B.E.B. is a scientific advisor for Syros Pharmaceuticals and a founder and scientific advisor for HiFiBio SAS.
References
- 1.Altshuler D, Daly MJ, Lander ES. Genetic mapping in human disease. Science. 2008;322:881–888. doi: 10.1126/science.1156409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hindorff LA, et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:9362–9367. doi: 10.1073/pnas.0903103106. 0903103106 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vyse TJ, Todd JA. Genetic analysis of autoimmune disease. Cell. 1996;85:311–318. doi: 10.1016/s0092-8674(00)81110-1. [DOI] [PubMed] [Google Scholar]
- 4.Buckner JH. Mechanisms of impaired regulation by CD4(+)CD25(+)FOXP3(+) regulatory T cells in human autoimmune diseases. Nature reviews. Immunology. 2010;10:849–859. doi: 10.1038/nri2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Browning JL. B cells move to centre stage: novel opportunities for autoimmune disease treatment. Nature reviews. Drug discovery. 2006;5:564–576. doi: 10.1038/nrd2085. [DOI] [PubMed] [Google Scholar]
- 6.Zhou L, Chong MM, Littman DR. Plasticity of CD4+ T cell lineage differentiation. Immunity. 2009;30:646–655. doi: 10.1016/j.immuni.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 7.Ciofani M, et al. A validated regulatory network for Th17 cell specification. Cell. 2012;151:289–303. doi: 10.1016/j.cell.2012.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marson A, et al. Foxp3 occupancy and regulation of key target genes during T-cell stimulation. Nature. 2007;445:931–935. doi: 10.1038/nature05478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Samstein RM, et al. Foxp3 exploits a pre-existent enhancer landscape for regulatory T cell lineage specification. Cell. 2012;151:153–166. doi: 10.1016/j.cell.2012.06.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hawkins RD, et al. Global chromatin state analysis reveals lineage-specific enhancers during the initiation of human T helper 1 and T helper 2 cell polarization. Immunity. 2013;38:1271–1284. doi: 10.1016/j.immuni.2013.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vahedi G, et al. STATs shape the active enhancer landscape of T cell populations. Cell. 2012;151:981–993. doi: 10.1016/j.cell.2012.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rivera CM, Ren B. Mapping human epigenomes. Cell. 2013;155:39–55. doi: 10.1016/j.cell.2013.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ostuni R, et al. Latent enhancers activated by stimulation in differentiated cells. Cell. 2013;152:157–171. doi: 10.1016/j.cell.2012.12.018. [DOI] [PubMed] [Google Scholar]
- 14.Lam MT, et al. Rev-Erbs repress macrophage gene expression by inhibiting enhancer-directed transcription. Nature. 2013;498:511–515. doi: 10.1038/nature12209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parkes M, Cortes A, van Heel DA, Brown MA. Genetic insights into common pathways and complex relationships among immune-mediated diseases. Nature reviews. Genetics. 2013;14:661–673. doi: 10.1038/nrg3502. [DOI] [PubMed] [Google Scholar]
- 16.Maurano MT, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 337:1190–1195. doi: 10.1126/science.1222794. science.1222794 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ernst J, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hnisz D, et al. Super-enhancers in the control of cell identity and disease. Cell. 2013;155:934–947. doi: 10.1016/j.cell.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parker SC, et al. Chromatin stretch enhancer states drive cell-specific gene regulation and harbor human disease risk variants. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:17921–17926. doi: 10.1073/pnas.1317023110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.International Multiple Sclerosis Genetics Consortium. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nature genetics. 2013;45:1353–1360. doi: 10.1038/ng.2770. ng.2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trynka G, et al. Chromatin marks identify critical cell types for fine mapping complex trait variants. Nature genetics. 2013;45:124–130. doi: 10.1038/ng.2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.1000 Genomes Project Consortium. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.West LC, Cresswell P. Expanding roles for GILT in immunity. Current opinion in immunology. 2013;25:103–108. doi: 10.1016/j.coi.2012.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.International Multiple Sclerosis Genetics Consortium and Wellcome Trust Case Control Consortium 2. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature. 2011;476:214–219. doi: 10.1038/nature10251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bernstein BE, et al. The NIH Roadmap Epigenomics Mapping Consortium. Nature biotechnology. 28:1045–1048. doi: 10.1038/nbt1010-1045. nbt1010-1045 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.ENCODE Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bulger M, Groudine M. Functional and mechanistic diversity of distal transcription enhancers. Cell. 2011;144:327–339. doi: 10.1016/j.cell.2011.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jolma A, et al. DNA-binding specificities of human transcription factors. Cell. 2013;152:327–339. doi: 10.1016/j.cell.2012.12.009. [DOI] [PubMed] [Google Scholar]
- 29.Xie X, et al. Systematic discovery of regulatory motifs in human promoters and 3′ UTRs by comparison of several mammals. Nature. 2005;434:338–345. doi: 10.1038/nature03441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li MO, Flavell RA. TGF-beta: a master of all T cell trades. Cell. 2008;134:392–404. doi: 10.1016/j.cell.2008.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heinz S, et al. Effect of natural genetic variation on enhancer selection and function. Nature. 2013;503:487–492. doi: 10.1038/nature12615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wright FA, et al. Heritability and genomics of gene expression in peripheral blood. Nature genetics. 2014;46:430–437. doi: 10.1038/ng.2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Quintana FJ, et al. Aiolos promotes TH17 differentiation by directly silencing Il2 expression. Nature immunology. 2012;13:770–777. doi: 10.1038/ni.2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gordan R, et al. Genomic regions flanking E-box binding sites influence DNA binding specificity of bHLH transcription factors through DNA shape. Cell reports. 2013;3:1093–1104. doi: 10.1016/j.celrep.2013.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brucklacher-Waldert V, et al. Phenotypical characterization of human Th17 cells unambiguously identified by surface IL-17A expression. Journal of immunology. 2009;183:5494–5501. doi: 10.4049/jimmunol.0901000. [DOI] [PubMed] [Google Scholar]
- 36.Johnston A, Sigurdardottir SL, Ryon JJ. Isolation of mononuclear cells from tonsillar tissue. In: Coligan John E, et al., editors. Current protocols in immunology. Unit 7. Chapter 7. 2009. p. 8. [DOI] [PubMed] [Google Scholar]
- 37.Caron G, Le Gallou S, Lamy T, Tarte K, Fest T. CXCR4 expression functionally discriminates centroblasts versus centrocytes within human germinal center B cells. Journal of immunology. 2009;182:7595–7602. doi: 10.4049/jimmunol.0804272. [DOI] [PubMed] [Google Scholar]
- 38.Zhu J, et al. Genome-wide chromatin state transitions associated with developmental and environmental cues. Cell. 2013;152:642–654. doi: 10.1016/j.cell.2012.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gifford CA, et al. Transcriptional and epigenetic dynamics during specification of human embryonic stem cells. Cell. 2013;153:1149–1163. doi: 10.1016/j.cell.2013.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Engreitz JM, et al. The Xist lncRNA exploits three-dimensional genome architecture to spread across the X chromosome. Science. 2013;341:1237973. doi: 10.1126/science.1237973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nature methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Beguelin W, et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer cell. 2013;23:677–692. doi: 10.1016/j.ccr.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Beyer M, et al. High-resolution transcriptome of human macrophages. PloS one. 2012;7:e45466. doi: 10.1371/journal.pone.0045466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Welter D, et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic acids research. 2014;42:D1001–1006. doi: 10.1093/nar/gkt1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hindorff LA, MJEBI, Morales J, Junkins HA, Hall PN, Klemm AK, Manolio TA European Bioinformatics Institute. [Accessed July, 2013];A Catalog of Published Genome-Wide Association Studies. Available at: http://www.genome.gov/gwastudies.
- 47.Trynka G, et al. Dense genotyping identifies and localizes multiple common and rare variant association signals in celiac disease. Nature genetics. 2011;43:1193–1201. doi: 10.1038/ng.998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cooper JD, et al. Seven newly identified loci for autoimmune thyroid disease. Human molecular genetics. 2012;21:5202–5208. doi: 10.1093/hmg/dds357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu JZ, et al. Dense fine-mapping study identifies new susceptibility loci for primary biliary cirrhosis. Nature genetics. 2012;44:1137–1141. doi: 10.1038/ng.2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Eyre S, et al. High-density genetic mapping identifies new susceptibility loci for rheumatoid arthritis. Nature genetics. 2012;44:1336–1340. doi: 10.1038/ng.2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.International Genetics of Ankylosing Spondylitis Consortium. Identification of multiple risk variants for ankylosing spondylitis through high-density genotyping of immune-related loci. Nature genetics. 2013;45:730–738. doi: 10.1038/ng.2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ellinghaus D, et al. High-density genotyping study identifies four new susceptibility loci for atopic dermatitis. Nature genetics. 2013;45:808–812. doi: 10.1038/ng.2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu JZ, et al. Dense genotyping of immune-related disease regions identifies nine new risk loci for primary sclerosing cholangitis. Nature genetics. 2013;45:670–675. doi: 10.1038/ng.2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hinks A, et al. Dense genotyping of immune-related disease regions identifies 14 new susceptibility loci for juvenile idiopathic arthritis. Nature genetics. 2013;45:664–669. doi: 10.1038/ng.2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tsoi LC, et al. Identification of 15 new psoriasis susceptibility loci highlights the role of innate immunity. Nature genetics. 2012;44:1341–1348. doi: 10.1038/ng.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pidasheva S, et al. Functional studies on the IBD susceptibility gene IL23R implicate reduced receptor function in the protective genetic variant R381Q. PloS one. 2011;6:e25038. doi: 10.1371/journal.pone.0025038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Duerr RH, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314:1461–1463. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Armitage P. Tests for Linear Trends in Proportions and Frequencies. Biometrics. 1955;11:375–386. [Google Scholar]
- 59.Franke A, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nature genetics. 2010;42:1118–1125. doi: 10.1038/ng.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.UK IBD Genetics Consortium. Genome-wide association study of ulcerative colitis identifies three new susceptibility loci, including the HNF4A region. Nature genetics. 2009;41:1330–1334. doi: 10.1038/ng.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Barrett JC, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nature genetics. 2008;40:955–962. doi: 10.1038/ng.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Anderson CA, et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nature genetics. 2011;43:246–252. doi: 10.1038/ng.764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Heinz S, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Molecular cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wellcome Trust Case Control Consortium. Bayesian refinement of association signals for 14 loci in 3 common diseases. Nature genetics. 2012;44:1294–1301. doi: 10.1038/ng.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Birzele F, et al. Next-generation insights into regulatory T cells: expression profiling and FoxP3 occupancy in Human. Nucleic acids research. 2011;39:7946–7960. doi: 10.1093/nar/gkr444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kanhere A, et al. T-bet and GATA3 orchestrate Th1 and Th2 differentiation through lineage-specific targeting of distal regulatory elements. Nature communications. 2012;3:1268. doi: 10.1038/ncomms2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gerstein MB, et al. Architecture of the human regulatory network derived from ENCODE data. Nature. 2012;489:91–100. doi: 10.1038/nature11245. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.