

The role of cellular metabolism in regulating cell proliferation and differentiation remains poorly understood1. For example, most mammalian cells cannot proliferate without exogenous glutamine supplementation even though glutamine is a non-essential amino acid1,2. Here we show that mouse embryonic stem cells (ESCs) grown under conditions that maintain naïve pluripotency3 are capable of proliferation in the absence of exogenous glutamine. Despite this, ESCs consume high levels of exogenous glutamine when the metabolite is available. In comparison to more differentiated cells, naïve ESCs utilize both glucose and glutamine catabolism to maintain a high level of intracellular α–ketoglutarate (αKG). Consequently, naïve ESCs exhibit an elevated αKG/succinate ratio that promotes histone/DNA demethylation and maintains pluripotency. Direct manipulation of the intracellular αKG/succinate ratio is sufficient to regulate multiple chromatin modifications, including H3K27me3 and Ten eleven translocation (Tet)-dependent DNA demethylation that contribute to the regulation of pluripotency-associated gene expression. In vitro, supplementation with cell-permeable αKG directly supports ESC self-renewal while cell-permeable succinate promotes differentiation. This work reveals that intracellular αKG/succinate levels can contribute to the maintenance of cellular identity and play a mechanistic role in the transcriptional and epigenetic state of stem cells.
Mouse ESCs can be maintained in two medium formulations: a serum-free medium reported to support a cellular phenotype that mimics “naïve” epiblast cells of the inner cell mass (ICM) (2i/LIF or 2i/L) or a serum-based medium that supports the proliferation of a more committed ESC phenotype (serum/LIF or S/L)4–11. To characterize ESC metabolism, we investigated whether cells cultured in these two media have different requirements for glucose and/or glutamine. ESCs cultured in either medium proliferated at equivalent rates when glucose and glutamine were abundant and cells cultured with or without 2i were unable to proliferate in the absence of glucose (Extended Data Fig. 1a,b). In contrast, cells cultured in 2i/L, but not S/L, proliferated robustly in the absence of exogenous glutamine (Fig. 1a and Extended Data Fig. 1c). Likewise, four newly-derived ESC lines (ESC-1-4) exhibited convincing glutamine-independent proliferation in 2i/L medium while retaining features of pluripotent cells, including ESC-like morphology, reactivity to alkaline phosphatase (AP) and the ability to form teratomas (Fig. 1b,c, Extended Data Fig. 1d). Cells cultured in 2i medium alone could also proliferate in the absence of exogenous glutamine (Extended Data Fig. 1e).
Figure 1. 2i is necessary and sufficient to confer glutamine independence.

a–f, Growth curves and representative images of ESCs grown in the absence of glutamine. Growth curves of ESC-V19 cells (a) and V6.5 ESC lines (ESC-1-4) (b) cultured in glutamine-free S/L or 2i/L medium. Phase images showing ESC-1 cells cultured in glutamine-free 2i/L (c) or S/L/2i (e) medium for 3 days. Top, brightfield (BF); bottom, alkaline phosphatase (AP) staining. Bar, 500 μm. d, Growth curve of ESC-V19 cells in glutamine-free S/L or S/L/2i medium. f, Growth curve of ESC-V19 cells cultured without glutamine in two serum-free media formulations containing N2 and B27 supplements, 2i/L and BMP4/L. g, Intracellular glutamate levels 8 hours after addition of medium with or without glutamine (Q). Data are presented as the mean ± s.d of triplicate wells from a representative experiment.
This effect was not due to differences in medium nutrient formulations as supplementing S/L medium with the GSK3β and ERK inhibitors present in 2i also enabled glutamine-independent proliferation while maintaining ESC morphology and markers of pluripotency (Fig. 1d,e). An alternative ESC medium containing BMP4 and LIF added to the same serum-free formulation as in 2i/L12 failed to support glutamine-independent growth (Fig. 1f). Likewise, epiblast stem cells (EpiSCs) could not proliferate in the absence of exogenous glutamine (Extended Data Fig. 1f,g). However, the ability to undertake glutamine-independent growth was not limited to embryonic pluripotency; fibroblast-derived induced pluripotent cells (iPSCs) were also able to proliferate in glutamine-free 2i/L medium (Extended Data Fig. 1h). These results indicate that the GSK3β and ERK inhibitors in 2i-containing medium are both necessary and sufficient to enable proliferation of pluripotent cells in the absence of exogenous glutamine.
The fact that cells proliferated in the absence of exogenous glutamine in 2i/L medium, albeit at a slower rate than cells cultured in glutamine-replete medium (Extended Data Fig. 1i), indicates that these cells must be capable of de novo glutamine synthesis. Indeed, chemical inhibition of glutamine synthase was sufficient to block proliferation of cells in glutamine-free 2i/L medium (Extended Data Fig. 1j). Likewise, addition of cell-permeable dimethyl-α-ketoglutarate (DM-αKG), a precursor for glutamine synthesis, was sufficient to enable glutamine-independent proliferation in both S/L and 2i/L conditions (Extended Data Fig. 1k), suggesting that the supply of precursors for glutamine synthesis determines the ability of ESCs to proliferate in the absence of glutamine. In support of this model, cells cultured in 2i/L preserved larger intracellular pools of glutamate following glutamine withdrawal than cells cultured in S/L (Fig. 1g). These results suggest that 2i/L cells can generate glutamate (and glutamine) from carbon sources other than glutamine itself.
Despite their different growth requirements, cells cultured in both S/L and 2i/L consumed high levels of glucose and glutamine, while excreting similar levels of lactate, consistent with the metabolic profile of most proliferating cells, including cancer cells and pluripotent cells (Fig. 2a)1,13. Oxidation of glucose and glutamine via the mitochondrial TCA cycle provides a critical source of the biosynthetic precursors required for cell proliferation. With the exception of α-ketoglutarate (αKG), steady-state levels of TCA cycle metabolites were reproducibly diminished in ESCs cultured in 2i/L (Fig. 2b).
Figure 2. 2i/L alters glucose and glutamine utilization.

a, Analysis of glucose uptake (left), glutamine uptake (center) and lactate secretion (right). b, Intracellular metabolite levels. Bars, mean of n = 4 (a) or n = 3 (b) replicate wells ± s.d. from representative experiments. c, Schematic of the TCA cycle including entry points for glucose- and glutamine-derived carbons. Isotope tracing was performed for metabolites shown in red. d,e, Fraction of each metabolite labeled by 13C derived from [U-13C]glutamine (13C-gln) (d) or derived from [U-13C]glucose (13C-glc) (e) over time (0–12 hours, h). Averages ± s.e.m. of three independent experiments are shown. f,g, Glutamine (f) and glucose (g) flux through αKG and malate pools. Averages ± s.e.m. of flux calculated for three independent experiments (shown in Figs. 2d,e) are shown. *, p < 0.05; **, p < 0.005; ***, p < 0.0005. p values determined by unpaired two-tailed Student’s t-tests.
In most cells, glutamine is catabolized to αKG to support TCA cycle anaplerosis (Fig. 2c). ESCs grown in S/L medium exhibited high levels of TCA cycle intermediates and virtually all intracellular glutamate, αKG and malate were rapidly labeled following addition of [U-13C]glutamine (Fig. 2d). In contrast, a substantial fraction of these metabolites failed to label with glutamine in ESCs grown in 2i/L. Instead, there was a rapid labeling of these three metabolite pools from [U-13C]glucose (Fig. 2e). Quantitation of metabolite fluxes revealed that although the flux of glutamine-derived carbons through αKG was similar in both conditions, glutamine flux through malate was significantly diminished in cells cultured in 2i/L, indicating that the entry of glutamine-derived αKG into the TCA cycle is repressed by culture in 2i/L (Fig. 2f). Instead, when cells are cultured in 2i/L, a substantial amount of both αKG and malate was produced from glucose (Fig. 2g).
Consistent with these results, cells cultured with 2i inhibitors demonstrated substantial glucose-dependent glutamate production (Extended Data Fig. 2a). Consequently, during conditions of glutamine depletion, cells cultured in 2i/L medium were able to use glucose-derived carbons to maintain elevated glutamate pools sufficient to support cell growth (Extended Data Fig. 2b). Moreover, in comparison to their S/L counterparts, 2i/L cells utilized more glucose-derived carbon and relatively less glutamine-derived carbon to support protein synthesis (Extended Data Fig. 2c), confirming that 2i promotes increased glucose-dependent amino acid synthesis.
Diminished glutamine entry into the TCA cycle, coupled with the observed efflux of glucose-derived carbons from the TCA cycle as glutamate, suggested that cells cultured in 2i/L might not be oxidizing all the αKG produced from glutamine in the mitochondria. Indeed, the αKG/succinate ratio was robustly elevated by 2i/L in every ESC line tested (Fig. 3a). Cellular αKG/succinate ratios have been implicated in the regulation of the large family of αKG-dependent dioxygenases14. As Jumonji-domain containing histone demethylases and the Tet family of DNA demethylases comprise a major subset of these enzymes, the elevated ratio of αKG/succinate observed in cells grown in 2i/L medium could have important implications for the regulation of chromatin structure.
Figure 3. Histone demethylation is regulated by intracellular α-ketoglutarate in embryonic stem cells.

a, GC-MS analysis of the αKG/succinate ratio in four ESC lines (ESC-1-4) grown in either S/L or 2i/L medium. b, Western blot of ESC-1 and ESC-2 cells grown in 2i/L medium with or without glutamine for three days. Molecular weight marker (in kDa) is shown. c, Simplified schematic of the reaction mechanism of αKG-dependent dioxygenases. d, ESC-1 cells grown in S/L in the presence of increasing amounts of GSK-J4 for 24 hours. e, H3K27me3 ChIP-PCR of ESC-1 cells cultured in S/L or 2i/L containing 30 μM of GSK-J4 for five hours. Values represent fold-change (GSK-J4/control) at individual bivalent domain genes (n=14). f, H3K27me3 ChIP-PCR of CRISPR/Cas9 edited cells JMJD3Δ/Δ-1 (left) and JMJD3Δ/Δ-2 (right) cultured in S/L or 2i/L. Values represent fold-change (JMJD3Δ/Δ cells relative to control cells) at individual bivalent domain genes (n=10). Bars represent mean values. p values determined by unpaired two-tailed Student’s t-test (e,f). g, The ratio of αKG/succinate in ESC-1 cells grown in S/L or 2i/L medium with 1 μM or 5 μM of GSK-J4 or GSK-J5 for three hours. **, p < 0.001; ***, p < 0.0001 as determined by 2-way ANOVA with Sidak’s multiple comparisons post-test (a,g). Data are presented as the mean ± s.d (a) or s.e.m. (g) of triplicate wells from a representative experiment.
Since αKG was largely derived from glutamine metabolism (Fig. 2d), we tested whether glutamine deprivation affected histone lysine methylations known to be regulated in part by αKG-dependent demethylases15. Cells cultured in glutamine-free medium exhibited increases in tri-methylation and decreases in mono-methylation on H3K9, H3K27, H3K36 and H4K20 while H3K4 methylations remained unchanged (Fig. 3b). DM-αKG reversed the increase in H3K27me3 and H4K20me3 observed in glutamine deficient medium (Extended Data Fig. 3a), confirming that these changes could be accounted for by the decline in glutamine-dependent αKG. Treatment with GSK-J416, a cell-permeable inhibitor that preferentially inhibits UTX and JMJD3, the two H3K27me3-specific JmjC-family histone demethylases (Fig. 3c), induced a dose-dependent increase in H3K27me3 with a concomitant reduction of H3K27me1 that was comparable in magnitude to the difference observed when cells were cultured in the presence or absence of glutamine (Fig. 3b,d). The above data suggest that the methylations of certain histone lysines, including H3K27, are actively suppressed by αKG-dependent histone demethylases in ESCs maintained in 2i/L medium.
In ESCs “bivalent domains” are developmentally regulated genomic regions characterized by the co-localization of H3K4me3 and H3K27me317–19. Recent genome-wide analysis of H3K27me3 in S/L and 2i/L cultured ESCs reported that H3K27me3 was specifically depleted at bivalent domain gene promoters in 2i/L cultured cells11. The present data suggest that the observed increase in αKG might promote αKG-dependent H3K27me3 demethylation in 2i/L ESCs. Indeed, cells cultured in 2i/L exhibited a greater increase in H3K27me3 at bivalent domain promoters when incubated with the H3K27me3 demethylase inhibitor GSK-J4 than cells cultured in S/L (Fig. 3e and Extended Data Fig. 3b,c). The average fold-change across 14 bivalent promoters tested showed a highly significant increase in 2i/L-cultured ESCs compared to S/L-cultured ESCs (Fig. 3e). Similarly, two independent cell lines with mutations in the Jumonji domain of the H3K27me3 demethylase JMJD3 (JMJD3Δ/Δ-1 and JMJD3Δ/Δ-2) (Extended Data Fig. 4a–c) demonstrated increases in H3K27me3 levels relative to control lines that were significantly elevated in cells cultured in 2i/L, reflecting enhanced demethylation at these loci in ESCs cultured in 2i/L (Fig. 3f). Furthermore, treatment with GSK-J4, but not the inactive isomer GSK-J5, increased the αKG/succinate ratio in cells cultured in 2i/L (Fig. 3g). These results indicate that 2i/L rewires glutamine metabolism to maintain αKG pools favoring active demethylation of a variety of histone marks.
In addition to reduced H3K27me3 at bivalent domain promoters, cells cultured in 2i/L exhibit DNA hypomethylation5,7–9. Incubating cells with ascorbic acid, a cofactor for αKG-dependent dioxygenases, activates Tet-dependent gene expression and promotes DNA demethylation20. Therefore, we tested whether αKG treatment could exert similar effects (Extended Data Fig. 5a). Total DNA methylation was reduced in cells cultured with cell-permeable αKG (Extended Data Fig. 5b) and treatment with αKG, but not succinate, induced expression of ICM and germline-associated genes previously identified as targets of Tet-mediated activation (Extended Data Fig. 5c)20,21. The effects of αKG persisted upon extended passaging (Extended Data Fig. 5d) and were largely abrogated in Tet1/Tet2 double knockout ESCs (Extended Data Fig. 5e). These results suggest that intracellular αKG production may stimulate the activity of multiple αKG-dependent dioxygenases in order to coordinately regulate the epigenetic marks characteristic of naïve pluripotency.
To test whether modulation of the αKG/succinate ratio can influence pluripotent cell fate decisions, we performed colony-forming assays with S/L-cultured ESCs in the presence of αKG or succinate. S/L+DM-αKG colonies had brighter AP staining and retained the compact colony morphology typical of undifferentiated ESCs (Fig. 4a). While the total number of colonies was similar in all three conditions, the S/L+DM-αKG wells contained more than double the number of fully undifferentiated colonies compared to S/L and S/L+DM-succinate (Fig. 4b). As a further test of the ability of αKG to promote maintenance of ESCs, we utilized a knock-in Nanog-GFP reporter line22 and found that αKG was sufficient to enhance Nanog expression in a dose-dependent manner (Figure 4c and Extended Data Fig. 6). These results support the conclusion that αKG promotes the self-renewal of ESCs in vitro.
Figure 4. α-ketoglutarate promotes the maintenance of pluripotency.

a,b, Colony formation assay using ESC-1 cells. Cells were plated at clonal density and media changed to experimental media containing either DM-αKG or DM-succinate on day 2 and then analyzed 4 days later by alkaline phosphatase (AP) staining and scored for number of differentiated, mixed and undifferentiated colonies. a, Representative brightfield images of AP-stained colonies. b, Quantification of colonies. DM-αKG has more undifferentiated colonies than vehicle or DM-succinate treated wells, ***, p < 0.0001, calculated by 2-way ANOVA with Tukey’s multiple comparisons post-test. c, Mean GFP intensity of Nanog-GFP cells treated for three days with or without DM-αKG. Data are presented as the mean ± s.e.m. (b) or 95% confidence intervals (c) of triplicate wells from a representative experiment.
The above data demonstrate that the cellular αKG/succinate ratio contributes to the ability of ESCs to suppress differentiation. The rewiring of cellular metabolism by inhibitors of GSK3β and MAPK/ERK signaling results in a reprogramming of glucose and glutamine metabolism that leads to accumulation of αKG and favors demethylation of repressive chromatin marks such as DNA methylation and H3K9me3, H3K27me3, and H4K20me3 (see Supplementary Discussion). Future studies will investigate the mechanisms through which these inhibitors influence the nuclear/cytosolic accumulation of αKG derived from glucose and glutamine. While we cannot rule out chromatin-independent effects of αKG supplementation on ESCs, our results support the notion that chromatin in pluripotent ESCs is responsive to alterations in intracellular metabolism. Indeed, recent clonal analysis of pluripotent cells revealed that DNA methylation is highly dynamic, balancing the antagonistic processes of removal and addition23. Together, these results suggest that continued elucidation of the interconnections between signal transduction and cellular metabolism will shed important light on stem cell biology, organismal development and cellular differentiation.
Methods
Cell lines
ESC1-4 lines are V6.5 ESCs derived from C57BL6 X 129S4/SvJae F1 embryos in 2i/L medium. Cells were derived from E3.5 blastocysts following standard ESC isolation procedures24. Flushed blastocysts were plated onto laminin-coated dishes (20 μg/ml, Stemgent 06-0002) in 2i/L medium. Mice were purchased from Jackson Labs, Bar Harbor, ME (C57BL/6 JAX 000664 and 129S4/SvJae JAX 009104). Tet1/2 double knockout ESCs25, V19 ESCs (ESC-V19) and OKS iPS cells26 were a kind gift from Rudolf Jaenisch (MIT/Whitehead Institute Cambridge, MA). All cells were routinely tested for mycoplasma contamination.
Cell culture
Maintenance media for ESCs were as follows: serum/LIF (S/L) maintenance medium contained Knockout DMEM (Gibco) supplemented with 15% ESC-qualified FBS (Gemini), penicillin/streptomycin (Life Technologies), 0.1 mM 2-mercaptoethanol, 2 mM L-glutamine (Life Technologies) and leukemia inhibitory factor (LIF) plated onto irradiated feeder mouse embryonic fibroblasts (MEFs); 2i/LIF (2i/L) maintenance conditions used a base medium made from a 1:1 mix of DMEM/F12 (Life Technologies 11302-033) and Neurobasal (Life Technologies 21103-049) containing N2 and B27 supplements (Life Technologies 17502-048 and 17504-044, 1:100 dilutions), penicillin/streptromycin, 0.1 mM 2-mercaptoethanol, 2 mM L-glutamine, LIF, CHIR99021 at 3 μM (Stemgent) and PD0325901 at 1 μM (Stemgent). Experimental media utilized for all experiments (except growth curves without glucose, 13C isotope tracing experiments and 14C labeling experiments) contained 1:1 mix of glutamine-free DMEM (Life Technologies 11960-051) and Neurobasal (Life Technologies 21103-049) with or without 2 mM glutamine. With the exception of 15% dialyzed FBS (Gemini 100-108) in S/L experimental medium, all other supplements were equivalent to maintenance media (S/L or 2i/L). For growth curves without glucose, 13C isotope tracing experiments and 14C labeling experiments, medium contained 1:1 mix of glutamine- and glucose-free DMEM (Invitrogen A14430-01) and glutamine- and glucose-free Neurobasal (Invitrogen 0050128DJ) containing either 20 mM [U-13C]glucose or 2 mM [U-13C]glutamine (Cambridge Isotope Labs) and either 20 mM unlabeled glucose or 2 mM unlabeled glutamine as necessary; all supplements were the same as experimental media described above (S/L or 2i/L). All experiments were performed using feeder-free conditions. ESC-1 EpiSCs were cultured feeder-free on fibronectin (Sigma) coated plates in EpiSC maintenance medium including DMEM/F12, N2 and B27 supplements, penicillin/streptromycin, 0.1 mM 2-mercaptoethanol, L-glutamine, 75 μg/ml BSA (Gibco) supplemented with human activin A (20 ng/ml; Peprotech) and bFgf (10 ng/ml; Invitrogen). EpiSCs were passaged 1:2 or 1:4 using Accutase every other day. For ESC to EpiSC differentiation, ESC-1 cells were plated onto fibronectcin-coated dishes. Twenty-four hours after plating the medium was changed to EpiSC maintenance medium supplemented with 6 μM JAK inhibitor (Calbiochem) for five passages. Analysis was performed on passage 7 EpiSCs. GSK-J4 and GSK-J5 were purchased from Tocris Bioscience.
Teratomas
ESC-1 cells were plated in maintenance medium at a concentration of 2.5 × 105 cells per T25 dish. The following day medium was changed to 2i/L experimental medium with or without glutamine. 72 hours later, 1 × 106 cells were harvested from each group and mixed 1:1 with experimental medium (without glutamine) plus Matrigel Basement Membrane Matrix (BD) or experimental medium alone and injected into the flanks of recipient SCID mice aged 8–12 weeks (NOD scid gamma JAX 005557 purchased from Jackson Labs, Bar Harbor, ME). All conditions produced tumors in 4–8 weeks. Mice were euthanized before tumor size exceeded 1.5 cm in diameter. Tumors were excised and fixed in 4% paraformaldehyde overnight at 4°C. Tumors were paraffin-embedded and sections were stained with hematoxylin and eosin according to standard procedures by Histoserv Inc. All animal procedures were designed following NIH guidelines and approved by the Institutional Animal Care and Use Committee (IACUC) at The Rockefeller University.
Glucose, glutamine and lactate measurements
Glucose, glutamine and lactate levels in culture medium were measured using a YSI 7100 multichannel biochemistry analyzer (YSI Life Sciences). Fresh medium was added to 12-well plates of sub-confluent cells and harvested 48 hours later. Changes in metabolite concentrations relative to fresh media were normalized to protein content of each well. These experiments were performed independently at least two times.
Metabolite profiling
For all metabolite experiments, cells were seeded in their standard culture medium in 6-well plates and the next day were changed into experimental medium. Medium was changed again at the indicated time before harvest (usually 1–24 hours). Metabolites were extracted with 1 mL ice-cold 80% methanol supplemented with 20 μM deuterated 2-hydroxyglutarate (D-2-hydroxyglutaric-2,3,3,4,4-d5 acid, d5-2HG) as an internal standard. After overnight incubation at −80°C, lysates were harvested and centrifuged at 21,000g for 20 minutes to remove protein. Extracts were dried in an evaporator (Genevac EZ-2 Elite) and resuspended by incubation at 30°C for 2 hours in 50 μL of 40 mg/mL methoxyamine hydrochloride in pyridine. Metabolites were further derivatized by addition of 80 μL of MSTFA + 1% TCMS (Thermo Scientific) and 70 μL ethyl acetate (Sigma) and incubated at 37°C for 30 minutes. Samples were analyzed using an Agilent 7890A GC coupled to Agilent 5975C mass selective detector. The GC was operated in splitless mode with constant helium gas flow at 1 mL/min. 1 μL of derivatized metabolites was injected onto an HP-5MS column and the GC oven temperature ramped from 60°C to 290°C over 25 minutes. Peaks representing compounds of interest were extracted and integrated using MassHunter software (Agilent Technologies) and then normalized to both the internal standard (d5-2HG) peak area and protein content of duplicate samples as determined by BCA protein assay (Thermo Scientific). Ions used for quantification of metabolite levels are as follows: d5-2HG m/z 354; αKG, m/z 304; aspartate, m/z 334; glutamate, m/z 363; malate, m/z 335 and succinate, m/z 247. All peaks were manually inspected and verified relative to known spectra for each metabolite. For isotope tracing studies, experiments were set up as described above using glucose- and glutamine-free DMEM:NB media base supplemented with 12C-glucose (Sigma) and 12C-glutamine (Gibco) or the 13C versions of each metabolite, [U-13C]glucose or [U-13C]glutamine (Cambridge Isotope Labs). Enrichment of 13C was assessed by quantifying the abundance of the following ions: αKG, m/z 304-315; aspartate, m/z 334-346; glutamate, m/z 363-377 and malate, m/z 335-347. Correction for natural isotope abundance was performed using IsoCor software27. Flux was calculated as the product of the first order rate constant of the kinetic labeling curve and relative metabolite pool size (normalized to mean S/L values for each experiment)28. The flux from glucose- and glutamine-derived carbons was calculated for each of three independent experiments and the average flux for each metabolite was shown. Flux experiments represent the average of three independent experiments; all other experiments were performed independently at least twice and a representative experiment is shown.
Protein labeling
ESCs were plated at 7.5 × 105 per 6-well plate into experimental medium (S/L or 2i/L) containing 0.01% unenriched D-[U-14C]-glucose (Perkin Elmer NEC042V250UC) or L-[U-14C]-glutamine (Perkin Elmer NEC451050UC). 48 hours later, cells were washed with PBS, scraped and pelleted at 4°C. Protein pellets devoid of lipid fractions were isolated according to the Bligh-Dyer method29. Briefly, pellets were resuspended in 200 μL dH20, 265 μL 100% methanol and 730 μL of chloroform. Samples were vortexed for 1 hour at 4°C. The organic phase was removed and the remaining sample washed with 1x volume of methanol and spun at 14,200g for 5 minutes. The supernatant was discarded and the pellet was resuspended in 6 M guanidine hydrochloride at 65°C for 30–45 minutes. Samples were quantified using Beckman LS 60001C instrument. Values represent the average from four wells normalized to protein of duplicate samples. Labeling experiments were performed twice.
Growth curves
ESC or EpiSCs were plated in maintenance medium at a concentration of 375,000 cells per 12-well plate. The following day cells were washed with PBS and media were changed to experimental media (for S/L conditions this included dialyzed FBS) with or without individual metabolites. Cells were counted each day using a Beckman Coulter Multisizer 4. All growth curves were performed independently at least two times.
Chromatin immunoprecipitation
Native ChIP assays (histones) were performed with approximately 6 × 106 ESCs per experiment. Cells were subject to hypotonic lysis and treated with micrococcal nuclease to recover mono- to tri-nucleosomes. Nuclei were lysed by brief sonication and dialyzed into N-ChIP buffer (10 mM Tris pH 7.6, 1 mM EDTA, 0.1% SDS, 0.1% Na-Deoxycholate, 1% Triton X-100) for 2 hr at 4°C. Soluble material was incubated overnight at 4°C following addition of 0.5–1 μg of antibody bound to 25 μL protein A Dynal magnetic beads (Invitrogen), with 5% kept as input DNA. Magnetic beads were washed, chromatin was eluted and ChIP DNA was dissolved in 10 mM Tris pH 8 for quantitative PCR reactions (see below). Three separate ChIP experiments were performed on replicate biological samples. The data shown are the average qRT-PCR values (n=3).
ChIP-qPCR
Primers are listed below. All qPCR was performed using an Applied Biosystems StepOnePlus system and Power SYBR Green PCR master mix. ChIP samples were diluted 1:100 in H20 and 5 μL used per reaction. ChIP-qPCR signals were calculated as percent input.
| Gene | Forward primer 5′ - 3′ | Reverse primer 5′ - 3′ |
|---|---|---|
| Gata6 | cgcagcacacaggtacagtt | gggatccaagcagattgaaa |
| Pax9 | aggtgtgcgacagctaaagg | atcaacccggagtgatcaag |
| Lhx1 | tgccaggcaccattacagt | aggcaaaggaaaaaccatga |
| Hoxa2 | ccaatgacaatttgggcttt | tgaggcgttcctttctgact |
| Hoxc9 | ttcttccctttggccttttt | agggtgtcttggctctctca |
| Evx1 | gccaggtgatctgggtgggga | tgagaaccggccttgtgtgct |
| Fgf5 | gggatctcctgtgcctggggt | aggcctgtactgcagccacattt |
| Ascl2 | gctccagaagcagttctcccctga | gatagagccagagcccaagcccc |
| Lrat | ccaagtccttcagtctcttgcccc | ggccacacaggctgcttcca |
| Lhx5 | aacccttaggccccagcccc | cgtgggcctggaggggagaa |
| Sox17 | gtctccccatgtagctctcctgcc | agaagagtcactgtggaggtgaggg |
| Brachyury | gccactgctttcccgagaccc | ccaggacaggcagggtagggg |
| Gata4 | acgtgtggtgttaatgtgcaagcc | tgcccacaagcctgcgatcc |
| Sox21 | aacagacatgccagtcagcagtgg | ttagcatcgcaccacccagagtc |
| Pou5f1 | gaggtcaaggctagagggtgg | agggacggtttcacctctcc |
qRT-PCR
RNA was isolated using the RNeasy kit (Qiagen). After DNase treatment, 1–2 μg RNA was used for cDNA synthesis using the First-Strand Synthesis kit (Invitrogen). Quantitative RT-PCR analysis was performed in biological triplicate using an ABI Prism 7000 (Applied Biosystems) with Platinum SYBR green. All data were generated using cDNA from three wells for each condition.
| Gene | Forward primer 5′ - 3′ | Reverse primer 5′ - 3′ |
|---|---|---|
| Pou5f1 | acatcgccaatcagcttgg | agaaccatactcgaaccacatcc |
| Nanog | aagatgcggactgtgttctc | cgcttgcacttcatcctttg |
| Esrrb | tttctggaacccatggagag | agccagcacctccttctaca |
| Klf2 | taaaggcgcatctgcgtaca | cgcacaagtggcactgaaag |
| Nr0b1 | tccaggccatcaagagtttc | atctgctgggttctccactg |
| Fgf5 | aaactccatgcaagtgccaaat | tctcggcctgtcttttcagttc |
| Zfp42 | cgagtggcagtttcttcttgg | cttcttgaacaatgcctatgactcacttcc |
| Actin | tggcgcttttgactcaggat | gggatgtttgctccaaccaa |
| Asz1 | gagtgggcttctcccagaaa | ggtcattttcccgctcattc |
| Wdfc15a | tgtgtggaaccctggacaac | gccaatgccgtcgttatttt |
| Dazl | caactgttaactaccactgcag | caagagaccactgtctgtatgc |
| Gapdh | ttcaccaccatggagaaggc | cccttttggctccaccct |
DNA methylation
Genomic DNA was extracted from ESC samples using Puregene Core Kit A (Sigma). DNA methylation was measured using the colorimetric MethylFlash Methylated DNA quantification kit (Epigentek) according to manufacturer instructions. ELISA experiments were performed independently two times.
CRISPR/Cas9 ESCs
A Cas9-2A-PURO plasmid was purchased from Addgene (Addgene plasmid 48139)30. Two gRNAs targeting exon 17 of mouse JMJD3 were designed using the online software (crispr.mit.edu) resource from the Zhang Laboratory (MIT Cambridge, MA) and were cloned into Cas9-2A-Puro using the BbsI restriction enzyme sites. ESC-1 cells cultured in 2i/L medium were transfected with either Cas9-2A-Puro control or JMJD3 gRNA-containing plasmids using Lipofectamine 2000 (Life Technologies). After 24 hours, cells were changed to fresh medium containing 1 μg/ml puromycin for 48 hours. Following selection, cells were cultured for 24 hours in 2i/L medium and then split to clonal density. After approximately 7 days, colonies were picked and expanded for analysis. Genomic DNA was purified from individual clones and used for PCR amplification of regions surrounding each gRNA target site. gRNA #1 product is 367 bp and gRNA #2 is 317 bp. Cloning of PCR products was performed using pGEM-T Easy (Promega). Mutants were identified by Sanger sequencing (Genewiz Inc.).
| gRNA oligos | Forward primer 5′ - 3′ | Reverse primer 5′ - 3′ |
|---|---|---|
| Jmjd3 gRNA #1 | cacctgtggatgttacccgcatga | aaactcatgcgggtaacatccaca |
| Jmjd3 gRNA #2 | caccgtccctggcagccgaacgcc | aaacggcgttcggctgccagggac |
| PCR primers | Forward primer 5′ - 3′ | Reverse primer 5′ - 3′ |
|---|---|---|
| Jmjd3 gRNA #1 | ggctaaggcctaagagtgcg | cggaccccaagaaccatcac |
| Jmjd3 gRNA #2 | tggcctgcagagggagatag | atttcgtcggcattcctgtg |
FACS
Nanog-GFP ESCs22 were cultured in S/L experimental medium for three passages and 2.5 × 104 cells were plated into a 6-well plate. Twenty-four hours later media was changed to S/L medium containing vehicle control or DM-αKG. Media were subsequently changed 48 hours later and cells harvested the following day. FACS analysis was performed at The Rockefeller University Flow Cytometry Resource Center using a BD LSR II. Data were generated using FlowJo. Experiments were performed two independent times and a representative experiment depicting triplicate biological wells is shown.
Western blot analysis
Lysates were extracted in 1X laemmli buffer, separated by SDS–PAGE and transferred to Immobilon PVDF (Millipore) membranes. Membranes were blocked in 5% milk prepared in phosphate-buffered saline (PBS) plus 0.1% Tween 20 (PBS-T), incubated with primary antibodies overnight at 4°C and HRP-conjugated secondary antibodies for 1_h the following day. After ECL application (Millipore), imaging was performed using Lumimager LAS-3000 (FujiFilm). The following antibodies were used for Western blotting: H3 (Abcam 1791), H3K4me3 (Active Motif 39159), H3K4me1 (Millipore 07-436), H3K9me1 (kind gift of T. Jenuwein), H3K9me3 (Active Motif 39161), H4 (Abcam 0158), H4K20me1 (Abcam 9051), H4K20me3 (Millipore 07-463), H3K27me1 (Millipore 07-448), H3K27me3 (Millipore 07-449), H3K36me3 (Abcam 9050) and H3K36me1 (Millipore 07-548). All antibodies were used at a dilution of 1:1000. H3K27me3 antibody used for ChIP-qPCR, Cell Signaling 9733BF.
Self-renewal assays
ESCs free of feeder MEFs were plated at 100 cells per well in 6-well plates coated with 20 μg/mL mouse laminin (Stemgent 06-0002) in maintenance S/L medium. The following day media was changed to S/L experimental medium containing dimethyl-α-ketoglutarate (4mM, Sigma 349631), dimethyl-succinate (4mM, Sigma W239607) or DMSO vehicle control. Four days later cells were washed with PBS and stained for alkaline phosphatase using Vector Red Alkaline Phosphatase Kit (Vector Labs) according to manufacturer’s instructions. Self-renewal assays were performed independently at least two times.
Statistics
Comparisons were made using unpaired two-tailed Student’s t-tests or 2-way ANOVA with appropriate post-test (determined using GraphPad Prism) as indicated. Experiments were performed with three or four replicates as is the standard in the field. Variation is shown as s.d., s.e.m. or 95% confidence intervals as indicated in figure legends.
Extended Data
Extended Data Figure 1. Pluripotent stem cells can proliferate in the absence of glutamine when cultured in 2i/LIF medium.

a, Doubling time of ESC-V19 cells cultured in serum/LIF (S/L) or 2i/LIF (2i/L). b, Growth curve of ESC-1 cells cultured in S/L or S/L/2i medium devoid of glucose. c, Samples of S/L (left) and 2i/L (right) media with and without glutamine were analyzed by gas chromatography-mass spectrometry. Representative chromatograms of the total ion count reveal a clear glutamine (Q) peak in +Q media (grey) and no detectable glutamine in –Q media (red). m, minutes. d, Teratoma formation from ESCs grown in 2i/L medium without glutamine for three days. Representative images of haematoxylin and eosin staining reveal neural tissue (ectoderm), hepatocytes and pancreatic acinar cells (endoderm) and smooth muscle (mesoderm). Scale bar, 200 μm. e, Growth curve of ESC-1 cells grown in glutamine-free 2i/L or 2i medium. f, Gene expression analysis confirms that EpiSCs, which represent post-implantation pluripotency, were generated from ESC-1 cells by culture with Fgf and Activin A. Transcript levels were assessed by qRT-PCR, normalized to Gapdh and expressed as a ratio of values of mESCs cultured in 2i/L medium. g, Growth curve of epiblast stem cells (EpiSCs) cultured in serum-free epiblast medium (serum-free medium containing FGF and Activin A, Fgf/ActA) with or without glutamine. h, Growth curve of an induced pluripotent (iPS) cell line derived from fibroblasts using Oct3/4 (O), Klf4 (K), and Sox2 (S) cultured in glutamine-free S/L or 2i/L medium. i, Doubling time of ESC-1 cells cultured in 2i/L medium in the presence and absence of glutamine. j, Growth curve ESC-V19 cells cultured in glutamine-free 2i/L medium in the presence or absence of 1 μM methyl-sulfoxide (MSO). k, ESC-V19 cells grown glutamine free S/L (left) or 2i/L (right) medium with or without 4 mM dimethyl-α-ketoglutarate (DM-αKG). For growth curve experiments, cells were seeded on day 0 in complete medium and then were changed to experimental medium on day 1 (indicated by red arrow). Data are presented as the mean ± s.d of triplicate wells from a representative experiment.
Extended Data Figure 2. mESCs cultured with 2i demonstrate altered glucose and glutamine utilization.
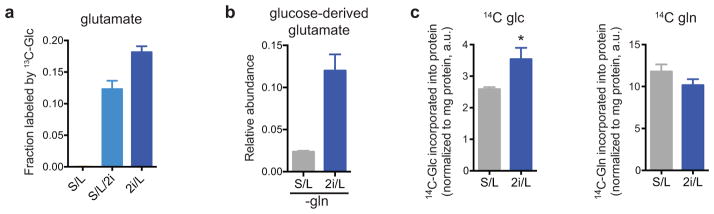
a, 2i enables glutamate synthesis from glucose-derived carbons. ESC-1 cells cultured in S/L, S/L/2i or 2i/L medium were incubated with medium containing [U-13C]glucose for four hours and the fraction of glutamate containing glucose-derived carbons is shown. b, ESC-1 cells were cultured for four hours in glutamine-free S/L or 2i/L medium containing [U-13C]glucose and the total amount of glutamate labeled by glucose-derived carbons is shown. c, Incorporation of 14C derived from [U-14C]glucose (14C-glc) (left) or derived from [U-14C]glutamine (14C-gln) (right) into total cellular protein after 48 hour incubations. p < 0.05 for 14C-glc, p = 0.1 for 14C-gln, calculated by unpaired two-tailed Student’s t-test. Data are presented as the mean ± s.d of triplicate wells (a,b) or ± s.e.m of quadruplicate wells (c) from a representative experiment.
Extended Data Figure 3. Regulation of histone methylation in 2i/LIF cells.

a, Western blot analysis of ESC-1 cells grown in glutamine-free 2i/L medium for 24 hours with supplementation as indicated (DM-αKG, dimethyl-α-ketoglutarate). b,c, H3K27me3 ChIP-PCR of ESC-1 cells cultured in S/L (b) or 2i/L (c) medium with or without 30 μM UTX/JMJD3 inhibitor GSK-J4 for five hours. Data are presented as the mean ± s.e.m. of triplicate samples. *, p < 0.05 by unpaired Student’s two-tailed t-test.
Extended Data Figure 4. Generation of JMJD3 mutant cells.

a, Schematic of targeting strategy for gRNAs to mouse Jmjd3 exon 17. gRNA sequences are highlighted in blue. b. Representative sequences from two clones used in this study. Sanger sequencing revealed indels as shown in schematic. Red dashes, deleted bases; red bases, insertions. gRNA is highlighted in blue and protospacer adjacent motif (PAM) sequences identified in green. Predicted cut site indicated by red triangle. Location of in-frame downstream stop is indicated on the right. c, An example chromatogram for clone JMJD3Δ/Δ-2 showing single base-pair insertions at predicted Cas9 cleavage site.
Extended Data Figure 5. αKG increases Tet activity in mESCs.

a, Simplified schematic of the reaction mechanism of TET1/2 enzymes. b, Relative percent 5-methylcytosine (% 5-mC) in ESC-1 cells cultured in S/L medium with or without DM-αKG for 24 hours. Each data point represents a sample from triplicate wells of a representative experiment. c, Gene expression in ESC-1 cells cultured with DM-αKG or DM-succinate for three days. d, Gene expression in ESC-1 cells cultured in S/L medium with or without DM-αKG for four passages. e, Gene expression in wild-type or Tet1/Tet2 double knock out (KO) mESCs cultured with DM-αKG or DM-succinate for 72 hours. qRT-PCR data (c–e) was normalized to Actin or Gapdh and samples were normalized to the control group. Oct3/4 is not expected to change and is included as a control. Data are presented as the mean ± s.e.m. of triplicate wells.
Extended Data Fig. 6. αKG increases Nanog expression.

a, Representative histogram of GFP intensity of Nanog-GFP cells treated with or without DM-αKG for three days. Grey represents background staining. b, ESC-1 cells were cultured in S/L medium with DM-αKG for four passages and then switched to medium containing the indicated amounts of DM-αKG (0.5 – 4 mM) or vehicle control (S/L) for three days. GFP expression (mean fluorescence intensity, M.F.I.) was determined by FACS. Data are presented as the mean ± s.d. of triplicate wells from a representative experiment.
Supplementary Material
Acknowledgments
B.C. is a HHMI fellow of the Jane Coffin Childs Memorial Research Fund. L.F. is the Jack Sorrell Fellow of the Damon Runyon Cancer Research Foundation (DRG-2144-13). This work was funded by grants from NIH/NIGMS (C.D.A) and from the NCI (C.B.T). We thank Charlie Li for assistance with FACS analysis and Meelad Dawlaty, Dina Faddah and Rudolf Jaenisch (Whitehead Institute/MIT) for sharing cell lines used in this study.
Footnotes
Author Information. C.B.T. is a co-founder of Agios Pharmaceuticals and a member of the board directors of Merck. C.D.A. is a co-founder of Chroma Therapeutics and Constellation Pharmaceuticals.
Reprints and permissions information is available at www.nature.com/reprints.
Supplementary Information is attached.
Author Contributions. B.C. and L.F. designed and performed all experiments in the study under the guidance of C.D.A and C.B.T. J.C. contributed material support. B.C., L.F., C.D.A. and C.B.T. wrote the manuscript.
References
- 1.Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. 2011;27:441–64. doi: 10.1146/annurev-cellbio-092910-154237. [DOI] [PubMed] [Google Scholar]
- 2.Eagle H, Oyama VI, Levy M, Horton CL, Fleischman R. The growth response of mammalian cells in tissue culture to L-glutamine and L-glutamic acid. J Biol Chem. 1956;218:607–16. [PubMed] [Google Scholar]
- 3.Ying QL, et al. The ground state of embryonic stem cell self-renewal. Nature. 2008;453:519–23. doi: 10.1038/nature06968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nichols J, Silva J, Roode M, Smith A. Suppression of Erk signalling promotes ground state pluripotency in the mouse embryo. Development. 2009;136:3215–22. doi: 10.1242/dev.038893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith ZD, et al. A unique regulatory phase of DNA methylation in the early mammalian embryo. Nature. 2012;484:339–44. doi: 10.1038/nature10960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wray J, Kalkan T, Smith AG. The ground state of pluripotency. Biochem Soc Trans. 2010;38:1027–32. doi: 10.1042/BST0381027. [DOI] [PubMed] [Google Scholar]
- 7.Leitch HG, et al. Naive pluripotency is associated with global DNA hypomethylation. Nat Struct Mol Biol. 2013;20:311–6. doi: 10.1038/nsmb.2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ficz G, et al. FGF signaling inhibition in ESCs drives rapid genome-wide demethylation to the epigenetic ground state of pluripotency. Cell Stem Cell. 2013;13:351–9. doi: 10.1016/j.stem.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Habibi E, et al. Whole-genome bisulfite sequencing of two distinct interconvertible DNA methylomes of mouse embryonic stem cells. Cell Stem Cell. 2013;13:360–9. doi: 10.1016/j.stem.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 10.Borgel J, et al. Targets and dynamics of promoter DNA methylation during early mouse development. Nat Genet. 2010;42:1093–100. doi: 10.1038/ng.708. [DOI] [PubMed] [Google Scholar]
- 11.Marks H, et al. The transcriptional and epigenomic foundations of ground state pluripotency. Cell. 2012;149:590–604. doi: 10.1016/j.cell.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ying QL, Nichols J, Chambers I, Smith A. BMP induction of Id proteins suppresses differentiation and sustains embryonic stem cell self-renewal in collaboration with STAT3. Cell. 2003;115:281–92. doi: 10.1016/s0092-8674(03)00847-x. [DOI] [PubMed] [Google Scholar]
- 13.Zhang J, Nuebel E, Daley GQ, Koehler CM, Teitell MA. Metabolic regulation in pluripotent stem cells during reprogramming and self-renewal. Cell Stem Cell. 2012;11:589–95. doi: 10.1016/j.stem.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaelin WG., Jr Cancer and altered metabolism: potential importance of hypoxia-inducible factor and 2-oxoglutarate-dependent dioxygenases. Cold Spring Harb Symp Quant Biol. 2011;76:335–45. doi: 10.1101/sqb.2011.76.010975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cloos PA, Christensen J, Agger K, Helin K. Erasing the methyl mark: histone demethylases at the center of cellular differentiation and disease. Genes Dev. 2008;22:1115–40. doi: 10.1101/gad.1652908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kruidenier L, et al. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature. 2012;488:404–8. doi: 10.1038/nature11262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bernstein BE, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–26. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 18.Boyer LA, et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature. 2006;441:349–53. doi: 10.1038/nature04733. [DOI] [PubMed] [Google Scholar]
- 19.Mikkelsen TS, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–60. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blaschke K, et al. Vitamin C induces Tet-dependent DNA demethylation and a blastocyst-like state in ES cells. Nature. 2013;500:222–6. doi: 10.1038/nature12362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hackett JA, et al. Synergistic Mechanisms of DNA Demethylation during Transition to Ground-State Pluripotency. Stem Cell Reports. 2013;1:518–31. doi: 10.1016/j.stemcr.2013.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Faddah DA, et al. Single-cell analysis reveals that expression of nanog is biallelic and equally variable as that of other pluripotency factors in mouse ESCs. Cell Stem Cell. 2013;13:23–9. doi: 10.1016/j.stem.2013.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shipony Z, et al. Dynamic and static maintenance of epigenetic memory in pluripotent and somatic cells. Nature. 2014;513:115–9. doi: 10.1038/nature13458. [DOI] [PubMed] [Google Scholar]
- 24.Markoulaki S, Meissner A, Jaenisch R. Somatic cell nuclear transfer and derivation of embryonic stem cells in the mouse. Methods. 2008;45:101–14. doi: 10.1016/j.ymeth.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 25.Dawlaty MM, et al. Combined deficiency of Tet1 and Tet2 causes epigenetic abnormalities but is compatible with postnatal development. Dev Cell. 2013;24:310–23. doi: 10.1016/j.devcel.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Buganim Y, et al. Single-cell expression analyses during cellular reprogramming reveal an early stochastic and a late hierarchic phase. Cell. 2012;150:1209–22. doi: 10.1016/j.cell.2012.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Millard P, Letisse F, Sokol S, Portais JC. IsoCor: correcting MS data in isotope labeling experiments. Bioinformatics. 2012;28:1294–6. doi: 10.1093/bioinformatics/bts127. [DOI] [PubMed] [Google Scholar]
- 28.Yuan J, Bennett BD, Rabinowitz JD. Kinetic flux profiling for quantitation of cellular metabolic fluxes. Nat Protoc. 2008;3:1328–40. doi: 10.1038/nprot.2008.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–7. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 30.Ran FA, et al. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281–308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.