

Abstract
In Gram-negative bacteria, lipoproteins are transported to the outer membrane by the Lol system. In this process, lipoproteins are released from the inner membrane by the ABC transporter LolCDE and passed to LolA, a diffusible periplasmic molecular chaperone. Lipoproteins are then transferred to the outer membrane receptor protein, LolB, for insertion in the outer membrane. Here we describe the discovery and characterization of novel pyridineimidazole compounds that inhibit this process. Escherichia coli mutants resistant to the pyridineimidazoles show no cross-resistance to other classes of antibiotics and map to either the LolC or LolE protein of the LolCDE transporter complex. The pyridineimidazoles were shown to inhibit the LolA-dependent release of the lipoprotein Lpp from E. coli spheroplasts. These results combined with bacterial cytological profiling are consistent with LolCDE-mediated disruption of lipoprotein targeting to the outer membrane as the mode of action of these pyridineimidazoles. The pyridineimidazoles are the first reported inhibitors of the LolCDE complex, a target which has never been exploited for therapeutic intervention. These compounds open the door to further interrogation of the outer membrane lipoprotein transport pathway as a target for antimicrobial therapy.
INTRODUCTION
The most distinguishing feature of Gram-negative bacteria is their cell envelope, which is comprised of both an inner and an outer membrane bilayer. The outer membrane has a unique composition of lipoproteins, β-barrel proteins, lipopolysaccharides, and phospholipids. Lipoproteins, membrane proteins that are covalently modified with lipids, are involved in a variety of integral cellular functions, such as the synthesis and maintenance of the cell surface and the transport of substrates (reviewed in reference 1). Lipoproteins are synthesized as precursors in the cytoplasm. Upon transit across the inner membrane by either the Sec or Tat machinery, the export signal peptide is cleaved, and attached to its amino terminus is a lipid moiety. This lipid serves as a membrane anchor for the lipoprotein. Some lipoproteins remain in the outer leaflet of the inner membrane, while others must cross the hydrophilic periplasmic space to the outer membrane. This sorting of lipoproteins to the outer membrane is achieved by the Lol system, which consists of five proteins (Fig. 1) (reviewed in reference 1). In this process, lipoproteins destined for the outer membrane are released from the inner membrane by the LolCDE complex, an inner membrane ABC transporter. LolCDE transfers the lipoproteins to LolA, a diffusible periplasmic chaperone (2, 3). LolA then transfers the lipoprotein to LolB, the outer membrane lipoprotein receptor, which incorporates these lipoproteins into the inner leaflet of the outer membrane (1, 4). This is in contrast to Gram-positive bacteria, which have a single membrane bilayer; therefore, localization of lipoproteins to the cell surface requires only export through the cytoplasmic membrane and acylation (5).
FIG 1.

Transport of lipoproteins by the Lol system in E. coli. (Top) Schematic of the localization of lipoprotein (Lol) system, which consists of five proteins and is responsible for transferring lipoproteins across the periplasm from the inner membrane to the outer membrane. (Bottom) Predicted membrane topologies of LolC and LolE (38). Mutations that confer resistance to compound 1 are shown. How LolC and LolE interact with LolD is not known.
Infections caused by multidrug-resistant Gram-negative bacteria are a growing threat to human health, and there is an urgent need for new antibacterial agents (6–8). All five of the proteins that comprise the Lol system have been shown to be essential for growth of Escherichia coli and are well conserved across the gammaproteobacteria (3, 9–11). This system is unique to Gram-negative bacteria and therefore comprises an attractive new target for antibiotics.
Using a high-throughput phenotypic screen for inhibitors of E. coli growth, we discovered a new pyridineimidazole compound with a unique mechanism of inhibition. Using resistance mutation mapping and biochemical transport assays, we found that this compound inhibits the function of the LolCDE complex, blocking the release of outer membrane-specific lipoproteins from the inner membrane, and, as such, represents the first description of inhibitors of this novel and essential Gram-negative target.
MATERIALS AND METHODS
Bacterial strain construction.
Gene deletions from the E. coli chromosome were constructed by using λ Red-mediated recombination as previously described (12). Briefly, the DNA deletion construct was created by PCR using primers that anneal to the kanamycin resistance cassette of pKD4 and that contain 36-bp regions of homology to the gene being deleted on the 5′ ends. To create the tolC deletion, the forward primer 5′-ATCGCGCTAAATACTGCTTCACCACAAGGAATGCAAGTGTAGGCTGGAGCTGCTTCG-3′ and the reverse primer 5′-TTACGTTCAGACGGGGCCGAAGCCCCGTCGTCGTCACATATGAATATCCTCCTTA-3′ were used. To create the lpp deletion, the forward primer 5′-CTGGTACTGGGCGCGGTAATCCTGGGTTCTACTCTGGTGTAGGCTGGAGCTGCTTCG-3′ and the reverse primer 5′-CTTGCGGTATTTAGTAGCCATGTTGTCCAGACGCTGCATATGAATATCCTCCTTA-3′ were used. The waaP deletion was created by using the forward primer 5′-CCAGAAAAAGCCGCGGATATCATTACAGGTGGTTTAGGTGTAGGCTGGAGCTGCTTCG-3′ and the reverse primer 5′-AATAAAGTTAGTTCCAGTACATACTAATAAATATTTCATATGAATATCCTCCTTA-3′. The resulting PCR product was purified and electroporated into an E. coli strain BW25113 containing the λ Red system on plasmid pKD46 (12). Recombinants were selected on LB agar containing 50 μg/ml kanamycin, and chromosomal deletions were verified by PCR. The tolC gene deletion was then moved by P1 phage transduction into E. coli MG1655 (13). The kanamycin resistance gene was excised from the chromosome by using the FLP recombinase expressed from pCP20, as previously described (12). The lpp deletion was subsequently moved by P1 phage transduction into this MG1655 ΔtolC strain. The waaP deletion was moved by P1 phage transduction into E. coli W3110.
High-throughput screening.
The AstraZeneca compound collection (∼1.2 million compounds) was screened at 10 μM in a 384-well plate format for inhibition of growth of an E. coli W3110 ΔwaaP strain, constructed as described above. This strain has a shortened lipopolysaccharide (LPS), rendering it more permeable to some small molecules (14). Approximately 1 × 105 cells in Mueller-Hinton broth (MHB) medium were dispensed into 384-well Greiner plates (catalog number 781165; Greiner Bio One, Frickenhausen, Germany) containing test compounds (1% dimethyl sulfoxide [DMSO] [final concentration]). Plates were incubated at 37°C with humidity for 18 h and read at an optical density at 600 nm (OD600).
Susceptibility determinations.
MICs were determined according to the guidelines of the Clinical and Laboratory Standards Institute (15). Activity against the human lung carcinoma cell line A549 was measured as described previously (16).
Inhibition of macromolecular synthesis pathways.
Inhibition of cell wall, fatty acid, DNA, RNA, and protein biosynthesis was measured as previously described, except that an E. coli W3110 ΔtolC ΔlysA strain was used and, to detect inhibition of cell wall biosynthesis, the incorporation of [3H]diaminopimelic acid ([3H]DAP) was measured (17, 18).
Isolation of resistant mutants.
Mutants resistant to compound 1 were raised against the E. coli MG1655 ΔtolC strain and isolated by using compound gradient LB plates (19). Resistant isolates were passaged onto LB plates twice without selection prior to determining their MICs to compounds 1 and 2 to ensure that they were stably resistant. To determine the frequency of resistance, E. coli ATCC 25922 ΔtolC cells (100 μl at an OD600 of ∼3) were plated in triplicate onto MHB II agar plates containing 4×, 8×, 16×, and 32× MIC of compound 1 or 2. In addition, 10-fold serial dilutions of the culture were spread onto plates without selection to determine the total number of bacteria in the sample. The plates were incubated at 37°C for 24 h, and the CFU were subsequently counted. The resistance frequency was calculated as the CFU/ml on the compound-containing plates divided by the total CFU/ml of the bacterial culture.
Whole-genome sequencing.
Genomic DNA was extracted by using the Promega Maxwell 16 instrument and Maxwell 16 Cell DNA purification kit according to procedures recommended by the manufacturer (Promega, Madison, WI). The resulting DNA was quantitated on a Qubit 2.0 fluorometer using the double-stranded DNA (dsDNA) Broad Range Assay kit (Life Technologies, Grand Island, NY). Five microliters of a 0.3-ng/μl solution was used for library generation using the Nextera XT DNA sample preparation kit, and Nextera XT index primers (Illumina, San Diego, CA) were used for library generation. The manufacturer's instructions were followed, except that quantitative PCR (qPCR) was used for library quantification using the Bio-Rad CFX96 cycler with the Kapa BioSytems Library quantification kit (Kapa BioSytems, Woburn, MA). Samples were sequenced on an Illumina MiSeq V2 instrument as 150-base paired-end reads. Assembly and analysis were performed by using CLCBio Genomics Workbench v 6.5 (CLCBio, Cambridge, MA). Detection of single nucleotide polymorphisms (SNPs)/indels was done by mapping to a parent reference assembly.
Spheroplast release assays.
E. coli MG1655 ΔtolC cells were grown in LB medium at 37°C until the culture reached an OD600 of 1.0. The cells were then converted into spheroplasts as described previously (2). Aliquots (100 μl) of suspensions containing 2 × 108 spheroplasts were incubated with or without His-tagged LolA (3.5 μg) in the presence of DMSO or compound 1 or 2 (1.4 μg) at 30°C for 1 min. LB medium (250 μl) containing 0.3 M sucrose and 10 μg/ml DNase I was then added, and the suspensions were incubated at 30°C for 30 min. After pelleting the spheroplasts by centrifugation at 16,000 × g for 2 min, the medium was diluted 3-fold with 7.15 mM MgCl2 and was ultracentrifuged at 100,000 × g for 30 min to remove membranes. The supernatants were analyzed by SDS-PAGE and subsequent immunoblotting with anti-Lpp antibody and anti-OmpA antibody.
Microscopy.
Bacterial cytological profiling was performed by Linnaeus Bioscience as previously described (20). Briefly, compounds were tested at 5× MIC in E. coli ATCC 25922 cells. Bacteria were grown at 30°C with shaking until the early log phase (OD600 of between 0.15 and 0.2). Cells were then mixed with the appropriate concentration of the compound and rolled in test tubes at 30°C for 120 min. After exposure to the compound, the cells were stained, concentrated by centrifugation, and observed by microscopy on agarose pads.
RESULTS
Pyridineimidazoles have activity against E. coli and H. influenzae.
Over 1.2 million compounds were screened for inhibition of growth of a permeabilized E. coli strain (W3110 ΔwaaP). Following a triage process to remove nonspecific inhibitors, impure compounds, and known antibacterials, a compound with a pyridineimidazole core scaffold (compound 1) was identified for further study (Fig. 2). Compound 1 has a molecular weight of 335.4, a solubility of 10.25 μM, and a measured LogD of 3.22. Compound 1 showed high human serum protein binding (4.5% free). Compound 1 was resynthesized (see the supplemental material) and profiled for its antibacterial activity against both Gram-negative and Gram-positive organisms. While compound 1 showed weak activity against wild-type E. coli ATCC 25922 (32 μg/ml), the MIC against an efflux-compromised tolC mutant of this strain was 0.25 μg/ml (Table 1). Compound 1 also showed activity against a wild-type strain of Haemophilus influenzae; however, no activity against other Gram-negative pathogens, such as Pseudomonas aeruginosa, was seen. Since disabling the efflux pump in E. coli improved the MIC by 128-fold, the activity of compound 1 against a strain of P. aeruginosa lacking five efflux pumps (ΔmexABDXY) was tested, but this strain was still not susceptible (Table 1). Compound 1 was not active against any of the Gram-positive pathogens tested (data not shown). The activity of compound 1 is specific to bacteria, as no measurable MIC was observed against the yeast Candida albicans (Table 1) and no inhibition of the proliferation of the human cell line A549 was observed at 100 μM (data not shown). The frequencies of resistance to compound 1 in an E. coli ΔtolC strain were found to be 1.2 × 10−6 at 8× MIC (2 μg/ml), 8.0 × 10−7 at 16× MIC (4 μg/ml), and 2.3 × 10−7 at 32× MIC (8 μg/ml). The kinetics of growth inhibition in the presence of compound 1 was also examined. At 8× MIC and higher, there was a 3-log decrease in CFU/ml by 4 h of incubation at 37°C; however, growth began to rebound by 6 h (data not shown).
FIG 2.
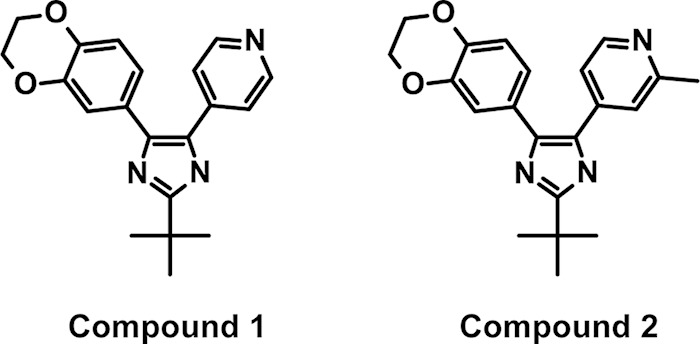
Chemical structures of compound 1 and compound 2. The structures of the high-throughput sequencing hit compound 1 and its more active analog compound 2 are shown.
TABLE 1.
Antibacterial activities of pyridineimidazole compounds
| Species and strain | Description | MIC (μg/ml) of: |
|
|---|---|---|---|
| Compound 1 | Compound 2 | ||
| Escherichia coli | |||
| ATCC 25922 | Wild type | 32 | 4 |
| ATCC 25922 ΔtolC | Efflux mutant | 0.25 | <0.06 |
| ATCC 35218 | Wild type | >64 | 16 |
| Haemophilus influenzae | Wild type | 2 | 0.25 |
| ATCC 49247 | |||
| Pseudomonas aeruginosa | |||
| PAO1 | Wild type | >64 | >64 |
| PAO1 ΔmexABDXY | Efflux mutant | >64 | >64 |
| Candida albicans | Counterscreen | >64 | >64 |
| ATCC 90028 | |||
A close analog to compound 1 with an ortho-methyl substitution off the pyridine (compound 2) showed improved activity against E. coli and H. influenzae (Fig. 2). The MIC for this analog against wild-type E. coli and H. influenzae decreased by 8-fold, while the MIC against the E. coli ΔtolC strain fell by at least 4-fold (Table 1). This analog showed no change in the spectrum of activity compared to the original hit compound (Table 1 and data not shown). Compound 2 also showed no detectable inhibition of A549 cell proliferation and had resistance frequencies in E. coli ΔtolC cells very similar to those of compound 1 (data not shown).
Mode of action of the pyridineimidazoles.
To determine which macromolecular synthesis pathway(s) is affected by the pyridineimidazoles, the inhibition of incorporation (50% inhibitory concentration [IC50]) of cellular pathway-specific radioactive precursors was measured (Table 2). Compound 1 was found to exclusively inhibit the incorporation of [3H]diaminopimelic acid, which is a component of the E. coli peptidoglycan, indicating that its activity is related to the inhibition of cell wall biogenesis.
TABLE 2.
Inhibition of radioactive precursor incorporation into E. coli macromolecular synthesis pathways
| Compoundb | Target | Incorporation IC50 (µg/ml)a for: |
||||
|---|---|---|---|---|---|---|
| Protein [14C]leucine | Cell wall [3H]DAP | Fatty acid [14C]acetic acid | RNA [3H]uridine | DNA [3H]thymidine | ||
| Erythromycin | Protein synthesis | 1.3 | >256 | >256 | >256 | >256 |
| Ampicillin | Cell wall synthesis | >256 | 49 | >256 | >256 | >256 |
| Triclosan | Fatty acid synthesis | 1.3 | 0.8 | 0.007 | 0.08 | 1.3 |
| Rifampin | Transcription | 16 | >256 | >256 | 7 | >256 |
| Ciprofloxacin | DNA replication | 0.6 | >256 | >256 | 0.2 | 0.02 |
| CCCP | Membrane potential | 0.3 | 0.04 | 0.02 | 0.03 | 0.1 |
| Compound 1 | Cell wall synthesis | >67 | 0.2 | >67 | >67 | >67 |
Incorporation of radiolabeled precursors was measured in E. coli ΔtolC ΔlysA cells.
CCCP, carbonyl cyanide m-chlorophenylhydrazone.
To further define the mode of action of the pyridineimidazoles, mutants resistant to compound 1 were selected with an E. coli MG1655 ΔtolC strain. The entire genome of 13 stably resistant isolates was sequenced by using the Illumina platform. Results from whole-genome sequencing showed that each of the resistant isolates had a single mutation that resulted in a single amino acid substitution in one of the components of the inner membrane ABC transporter LolCDE. LolCDE is responsible for releasing lipoproteins from the inner membrane that are destined for the outer membrane. There were no additional mutations identified in the reported strains compared with the parental genome. Nine mutants mapped to LolC, resulting in either a Q258L (n = 4) mutation or an N265K (n = 5) mutation. Additionally, three distinct substitutions were found in LolE: I59N (n = 2), P372L (n = 1), and L371P (n = 1). All of these mutations in LolC and LolE are predicted to be near the periplasm/inner membrane interface of these proteins (Fig. 1). Isolates with each of these LolC or LolE mutations were used to measure the MIC of compounds 1 and 2 as well as a number of other antibacterials of various classes (Table 3). Large shifts in the MIC were observed for these mutants versus both compounds 1 and 2 (32-fold and greater), which is consistent with these two compounds having the same mode of action. However, the MIC of these LolC and LolE mutant strains against antibiotics that inhibit other pathways showed no change, indicating that these mechanisms of resistance are specific to the pyridineimidazoles. In addition, the LolC and LolE mutants were not more sensitive than the parental strain to membrane-active compounds such as polymyxin, SDS, or EDTA, suggesting that the outer membrane of these mutants is largely intact. The mutants also remained resistant to vancomycin, again indicating that the outer membrane of the mutants was not affected, because E. coli strains with outer membrane defects such as changes in the lipopolysaccharide have been shown to exhibit sensitivity to vancomycin (21).
TABLE 3.
Relative sensitivities of compound 1-resistant strains to various classes of antibiotics
| Strain | MICa of: |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Compound 1 | Compound 2 | Meropenem | Doxycycline | Nalidixic acid | Polymyxin B | Rifampin | Vancomycin | SDS | EDTA | |
| E. coli ΔtolC (parent) | 0.125 | <0.06 | 0.03 | 0.5 | 1 | 2 | 8 | >256 | 0.003 | 10 |
| Δlpp mutant | 1 | 0.25 | 0.06 | 0.5 | 0.5 | 2 | 2 | >256 | 0.0015 | 5 |
| LolC(Q258L) mutant | 8 | 4 | 0.06 | 0.5 | 1 | 4 | 8 | >256 | 0.0015 | 10 |
| LolE(I59N) mutant | >64 | >64 | 0.06 | 0.5 | 1 | 4 | 8 | >256 | 0.0015 | 10 |
| LolC(N265K) mutant | >64 | >64 | 0.06 | 0.5 | 1 | 4 | 8 | >256 | 0.0015 | 10 |
| LolE(P372L) mutant | 64 | 8 | 0.06 | 0.5 | 0.5 | 2 | 8 | >256 | 0.0015 | 5 |
| LolE(L371P) mutant | >64 | >64 | 0.06 | 0.5 | 1 | 2 | 8 | >256 | 0.0015 | 5 |
MICs of SDS and EDTA are expressed as percentages and millimolar concentrations, respectively. All other MICs are expressed in micrograms per milliliter.
One of the most abundant E. coli outer membrane lipoproteins, Lpp, interacts with the peptidoglycan both covalently and noncovalently, where it contributes to the integrity of the cell surface structure (22, 23). It has been shown that when Lpp accumulates in the inner membrane, due to mutations in the Lpp protein itself, Lpp covalently binds to the peptidoglycan and is lethal for the cell (24, 25). In fact, mutations in Lpp are one route of resistance to globomycin and myxovirescin, natural-product antibiotics which inhibit signal peptidase II (LspA) that cleaves the signal peptide from lipoproteins, a step in lipoprotein processing that occurs prior to trafficking to the outer membrane (26–28). In order to confirm genetically that the pyridineimidazoles inhibit the maturation of the outer membrane, a deletion in lpp was created in an E. coli ΔtolC background for MIC testing. The MICs of both compounds 1 and 2 for the E. coli ΔtolC Δlpp strain showed at least an 8-fold increase compared to those for the E. coli ΔtolC strain (Table 3). There was no change in the MICs of antibiotics that do not inhibit lipoprotein transport to the outer membrane for the lpp deletion mutant strain, as expected. These data are consistent with compounds 1 and 2 being inhibitors of lipoprotein sorting to the outer membrane.
Pyridineimidazoles block lipoprotein release from spheroplasts.
The above-described genetic results suggest that the pyridineimidazole compounds 1 and 2 may inhibit bacterial growth by binding the LolCDE complex to block lipoprotein sorting to the outer membrane. To biochemically investigate whether the pyridineimidazoles prevent the trafficking of lipoproteins from the inner membrane, we tested these compounds for inhibition of Lpp release from spheroplasts to purified LolA protein. Spheroplasts contain the inner membrane phospholipid bilayer but lack the periplasmic and outer membrane fractions. For these studies, spheroplasts were prepared from E. coli MG1655 ΔtolC cells and incubated with purified His-tagged LolA in the presence and absence of the inhibitor compound. Following centrifugation to remove the spheroplasts, the supernatant was analyzed by SDS-PAGE and Western blotting with anti-Lpp antibody. Lipoprotein-releasing activity was measured as the amount of Lpp released from the spheroplasts. The appearance of Lpp in the supernatant was dependent on the presence of both purified LolA and spheroplasts (Fig. 3A). As shown in Fig. 3A, in the absence of the compound, Lpp was found in the supernatant, indicating that Lpp was released from the spheroplast membrane to the purified LolA protein. However, when either compound 1 or 2 was present, the amount of Lpp recovered was greatly diminished. Compound 2 appears to inhibit the release of Lpp slightly better than compound 1, which is consistent with its more potent cellular activity. Compounds 1 and 2 did not inhibit the release of OmpA from spheroplasts, which indicates that these compounds specifically inhibit the release of lipoproteins and not other outer membrane proteins. To examine the effect of the LolC and LolE mutations that confer resistance to the pyridineimidazoles, spheroplasts were also prepared from two of these resistant E. coli strains. Compounds 1 and 2 were not able to inhibit release of Lpp from spheroplasts formed from either the E. coli MG1655 ΔtolC LolE(L371P) or LolC(N265K) mutant at the same concentration used to inhibit release from spheroplasts from the susceptible strain (Fig. 3B). The amount of Lpp released from the LolC and LolE mutant spheroplasts in the presence of compound 1 or 2 was similar to the levels seen with the DMSO control. These data are consistent with compounds 1 and 2 having a mode of action through the LolCDE complex and support the genetic findings that mutations in LolCDE cause resistance to the pyridineimidazoles.
FIG 3.
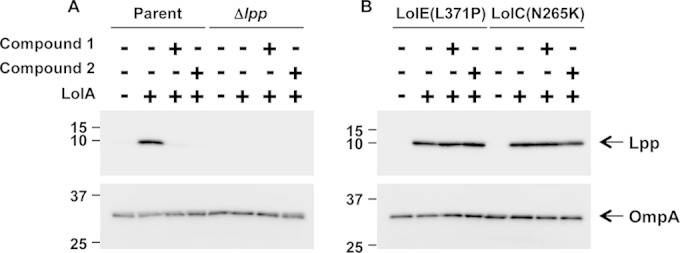
Pyridineimidazoles inhibit Lpp release from E. coli spheroplasts to purified LolA. Spheroplasts were prepared from the parental E. coli ΔtolC or E. coli ΔtolC Δlpp strain (A) or the E. coli ΔtolC strain carrying a LolE(L371P) or LolC(N265K) mutation (B) and subsequently incubated with His-tagged LolA in the presence of DMSO or pyridineimidazole compounds 1 and 2. (Top) The amount of Lpp released to LolA was detected by SDS-PAGE and immunoblotting with anti-Lpp antibodies. Detection of Lpp was dependent on the presence of LolA. (Bottom) OmpA was also detected with an anti-OmpA antibody. Detection of OmpA is independent of LolA.
Pyridineimidazoles cause morphological changes similar to those caused by globomycin.
Previous studies have shown that antibacterial compounds can cause changes in cellular morphology specific to the cellular pathways that they inhibit (20). To investigate the effects of the pyridineimidazoles on cellular morphology, E. coli cells were treated with compound 1 or 2 at 5× MIC for 120 min. These cells were then subjected to fluorescence microscopy with staining of membranes by FM4-64, 4′,6-diamidino-2-phenylindole (DAPI) to visualize the nucleoid, as well as Sytox green to detect permeabilization of the membrane. Relative to the DMSO control, cells in the presence of compound 1 or 2 were markedly swollen, and the nucleoids appeared to be less condensed (Fig. 4A to C). The presence of Sytox green staining in the interior of the cells also indicates that the membranes of some of the cells had become permeabilized in the presence of these compounds. The changes in cell morphology seen in the presence of compounds 1 and 2 are quite similar to those seen in the presence of globomycin (Fig. 4D), consistent with the fact that all these compounds appear to inhibit stages of lipoprotein transport to the outer membrane. These morphological changes are quite different from those caused by other antibiotics that inhibit cell wall biogenesis, such as amdinocillin and aztreonam (Fig. 4E and F).
FIG 4.

Morphology of E. coli in the presence of pyridineimidazoles and other cell wall inhibitors. E. coli was treated with each inhibitor at 5× MIC for 120 min and stained with FM4-64 (red), DAPI (blue), and Sytox green (green). An overlay of FM4-64 and DAPI is also shown. Bar, 1 μm.
DISCUSSION
In the work presented here, a novel pyridineimidazole compound with antibacterial activity against E. coli and H. influenzae was discovered via a high-throughput phenotypic screen. Mutants resistant to the pyridineimidazole compound map to the LolCDE complex, an inner membrane ABC transporter, which suggests that the mode of action of these compounds is to inhibit lipoprotein targeting to the outer membrane. The LolCDE transporter is responsible for releasing lipoproteins from the inner membrane to LolA, a periplasmic lipoprotein-specific molecular chaperone, for transport to the outer membrane. Biochemical assays with spheroplasts and purified LolA confirm the genetic evidence that the pyridineimidazoles directly inhibit the release of lipoproteins from the inner membrane. Experiments with spheroplasts isolated from LolC and LolE mutants resistant to the pyridineimidazoles further suggest that these compounds act through the LolCDE complex. The Gram-negative specificity of the pyridineimidazoles is consistent with this proposed mechanism of action, because Gram-positive bacteria, which lack an outer membrane, do not have such a lipoprotein-sorting pathway. This is the first reported example of an inhibitor of the LolCDE complex.
Growth inhibition by the pyridineimidazole compounds likely occurs by two mechanisms. The first mechanism is the mislocalization of the lipoprotein Lpp to the inner membrane, which is toxic to the cell (25). In fact, a deletion in lpp leads to an 8-fold increase in the MIC of these compounds. The second mechanism by which the pyridineimidazole compounds likely inhibit growth is via the prevention of essential lipoproteins from being localized to the outer membrane. E. coli encodes at least 90 lipoproteins, most of which are predicted to reside in the outer membrane. The majority of these lipoproteins are of unknown function; however, three outer membrane lipoproteins have been identified as being essential for cell viability (reviewed in reference 1). These lipoproteins are BamD, which is a component of the β-barrel assembly machine (BAM) complex and is required for the integration of β-barrel proteins into the outer membrane (29–31); LptE, which helps mediate the transport of the LPS to the outer surface of the outer membrane (32–34); and LolB, which, as mentioned above, is a component of lipoprotein transport to the outer membrane (4). Additionally, the lipoproteins LpoA and LpoB are essential for the function of PBP1A and PBP1B in peptidoglycan biogenesis (35, 36). Therefore, an interruption of the transport of lipoproteins to the outer membrane would have consequences for the structure of the cell wall. A similar mechanism of growth inhibition has been proposed for the antibiotics globomycin and myxovirescin, both of which inhibit an upstream step of lipoprotein maturation, namely, the type II signal peptidase that cleaves the signal sequence from the lipoprotein prior to transport by the Lol system to the outer membrane (26).
The LolCDE complex is composed of one copy each of LolC and LolE and two copies of the LolD ATPase subunit (Fig. 1) (3). Mutations conferring resistance to the pyridineimidazoles were mapped to both the LolC and LolE proteins. The E. coli LolC and LolE amino acid sequences are similar to one another, with 26% identity (37, 38). Cross-linking studies suggest that despite their structural similarity, LolC and LolE are functionally different, which is consistent with both proteins being essential for growth (9). These studies show that LolC interacts with LolA and that LolE interacts with the lipoprotein to mediate lipoprotein transfer from the inner membrane to LolA (39). Both LolC and LolE are predicted to have 4 transmembrane-spanning (TMS) regions, with a large periplasmic loop between TMS1 and TMS2 and a smaller periplasmic loop between TMS3 and TMS4 (38). It has been proposed that these periplasmic regions are functionally important for the transfer of lipoproteins from LolE to LolC (38, 40). All the mutations that were found in the pyridineimidazole-resistant strains are predicted to be near the periplasm/inner membrane interface of LolC and LolE. Both of the mutations found in LolC and one found in LolE are predicted to be in the base of the large periplasmic loop, near the membrane. Two of the LolE mutations (at positions 371 and 372) are predicted to lie at the beginning of TMS4 near the interface with the periplasm (Fig. 1). Since the crystal structure of the LolCDE complex is not available, it is unknown whether these amino acids constitute a distinct pocket in the quaternary structure to which the compound binds. Given that mutations in two different proteins confer resistance, the pyridineimidazoles may act to prevent crucial interactions between LolC and LolE that are required for the transfer of lipoproteins to LolA. Thus, a mutation in either protein in this interface that prevents the binding of the inhibitor could lead to resistance. It is also possible that these mutations alter the interaction of either LolC or LolE with other components of the Lol system to compensate for the pyridineimidazole binding to another site in the complex.
The data presented here indicate that the pyridineimidazoles inhibit the targeting of lipoproteins to the outer membrane. Several features of this target pathway make it attractive for antibiotic development. One feature is that it exists only in bacteria, so its inhibition would not lead to target-based mammalian toxicity. Another advantage is its largely periplasmic location, which means that inhibitors would not need to cross both membranes to reach their target. The differing physical properties of the inner and outer membranes make it challenging to discover compounds that can efficiently permeate both membranes to reach cytoplasmic targets (41, 42). Although small-molecule inhibitors of LolA have been described, whether this series is amenable for development for clinical use remains to be seen (43).
Although their target is attractive, the pyridineimidazole inhibitors require optimization in several ways, the first of which is improving the physical properties of the series. Currently, the low solubility and high protein binding of these compounds are not sufficient for dosing in mammalian systems. Additionally, the spectrum of bacteria against which these compounds are active would need to be increased. Despite the fact that the Lol system is widely conserved across the gammaproteobacteria, activity was seen in only E. coli and H. influenzae. At present, it is not clear why cellular activity was not seen against a wider range of gammaproteobacteria, as the five Lol proteins are highly conserved across species (37), with the exception of Acinetobacter baumannii, for which no clear homolog of LolC has been identified (44). Some other clinically relevant gammaproteobacteria, such as P. aeruginosa, are notorious for their resistance to antibiotics due to the impermeability of their outer membranes and active efflux systems. However, a P. aeruginosa mutant lacking 5 genes encoding components of efflux pumps was still not susceptible to the pyridineimidazoles, indicating that efflux cannot fully explain the lack of activity against this organism. Thus, it is possible that the pyridineimidazole compounds do not cross the outer membrane of P. aeruginosa. While the molecular mechanisms by which lipoproteins are targeted to the outer membrane have been extensively studied in E. coli, less is known about the Lol system in other gammaproteobacteria. For example, some work has been done to profile the proteins from P. aeruginosa, which showed that the five P. aeruginosa Lol proteins are responsible for the sorting of lipoproteins to the outer membrane, as in the case of E. coli lipoproteins (45). However, it was also shown that there are differences between E. coli and P. aeruginosa in the lipoprotein-sorting signals that dictate retention in the inner membrane or transfer to the outer membrane (45, 46). It is possible that the differences between the E. coli and P. aeruginosa Lol systems are significant enough that the pyridineimidazoles are not active against the P. aeruginosa LolCDE complex. Interestingly, homologs of the Lol proteins in spirochetes such as Borrelia burgdorferi have been described, but their susceptibilities to compounds 1 and 2 are not known (5).
Another feature limiting the potential of these compounds for therapeutic use is the high frequency (∼10−6) of resistance, which results in single-step resistant mutants displaying large shifts in MICs. Also, there was a rebound of growth during measurement of the kinetics of growth inhibition. Further investigation is required to determine whether these properties of the pyridineimidazoles are intrinsic to the compound series or to the target pathway. Nevertheless, the mechanism of action of this series is clearly an important discovery for antibacterial research, as it will guide a better definition and exploitation of lipoprotein sorting for the discovery and development of other analogs or classes of inhibitors of this pathway.
The current arsenal of antibiotics inhibits only a small number of cellular targets. Given the alarming rise in antibacterial resistance, the discovery of new compounds that inhibit novel cellular targets is crucial. Although the lipoprotein-sorting pathway is essential for growth, its potential for antibiotic therapy has never been realized. This report is the first description of inhibitors of the LolCDE transporter complex by a new lead series with a unique mechanism selective for Gram-negative pathogens. The pyridineimidazoles therefore may have significant promise as new chemical probes of lipoprotein sorting to the outer membrane.
Supplementary Material
ACKNOWLEDGMENTS
We thank the AstraZeneca Infection Bioscience Department for MIC testing, Helen Plant and colleagues at AstraZeneca in Alderley Park for high-throughput screening, and Shubha Sriram for measuring the inhibition of macromolecular synthesis pathways. We also thank Joe Pogliano and employees of Linnaeus Bioscience for their bacterial cytological profiling services and helpful discussions.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.02352-14.
REFERENCES
- 1.Okuda S, Tokuda H. 2011. Lipoprotein sorting in bacteria. Annu Rev Microbiol 65:239–259. doi: 10.1146/annurev-micro-090110-102859. [DOI] [PubMed] [Google Scholar]
- 2.Matsuyama S, Tajima T, Tokuda H. 1995. A novel periplasmic carrier protein involved in the sorting and transport of Escherichia coli lipoproteins destined for the outer membrane. EMBO J 14:3365–3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yakushi T, Masuda K, Narita S, Matsuyama S, Tokuda H. 2000. A new ABC transporter mediating the detachment of lipid-modified proteins from membranes. Nat Cell Biol 2:212–218. doi: 10.1038/35008635. [DOI] [PubMed] [Google Scholar]
- 4.Matsuyama S, Yokota N, Tokuda H. 1997. A novel outer membrane lipoprotein, LolB (HemM), involved in the LolA (p20)-dependent localization of lipoproteins to the outer membrane of Escherichia coli. EMBO J 16:6947–6955. doi: 10.1093/emboj/16.23.6947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zuckert WR. 2014. Secretion of bacterial lipoproteins: through the cytoplasmic membrane, the periplasm and beyond. Biochim Biophys Acta 1843:1509–1516. doi: 10.1016/j.bbamcr.2014.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rex JH, Eisenstein BI, Alder J, Goldberger M, Meyer R, Dane A, Friedland I, Knirsch C, Sanhai WR, Tomayko J, Lancaster C, Jackson J. 2013. A comprehensive regulatory framework to address the unmet need for new antibacterial treatments. Lancet Infect Dis 13:269–275. doi: 10.1016/S1473-3099(12)70293-1. [DOI] [PubMed] [Google Scholar]
- 7.Rice LB. 2008. Federal funding for the study of antimicrobial resistance in nosocomial pathogens: no ESKAPE. J Infect Dis 197:1079–1081. doi: 10.1086/533452. [DOI] [PubMed] [Google Scholar]
- 8.Spellberg B, Guidos R, Gilbert D, Bradley J, Boucher HW, Scheld WM, Bartlett JG, Edwards J Jr. 2008. The epidemic of antibiotic-resistant infections: a call to action for the medical community from the Infectious Diseases Society of America. Clin Infect Dis 46:155–164. doi: 10.1086/524891. [DOI] [PubMed] [Google Scholar]
- 9.Narita S, Tanaka K, Matsuyama S, Tokuda H. 2002. Disruption of lolCDE, encoding an ATP-binding cassette transporter, is lethal for Escherichia coli and prevents release of lipoproteins from the inner membrane. J Bacteriol 184:1417–1422. doi: 10.1128/JB.184.5.1417-1422.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tajima T, Yokota N, Matsuyama S, Tokuda H. 1998. Genetic analyses of the in vivo function of LolA, a periplasmic chaperone involved in the outer membrane localization of Escherichia coli lipoproteins. FEBS Lett 439:51–54. doi: 10.1016/S0014-5793(98)01334-9. [DOI] [PubMed] [Google Scholar]
- 11.Tanaka K, Matsuyama SI, Tokuda H. 2001. Deletion of lolB, encoding an outer membrane lipoprotein, is lethal for Escherichia coli and causes accumulation of lipoprotein localization intermediates in the periplasm. J Bacteriol 183:6538–6542. doi: 10.1128/JB.183.22.6538-6542.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thomason LC, Costantino N, Court DL. 2007. E. coli genome manipulation by P1 transduction. Curr Protoc Mol Biol Chapter 1:Unit 1.17. doi: 10.1002/0471142727.mb0117s79. [DOI] [PubMed] [Google Scholar]
- 14.Raetz CR, Whitfield C. 2002. Lipopolysaccharide endotoxins. Annu Rev Biochem 71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clinical and Laboratory Standards Institute. 2009. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically: approved standard, 9th ed, M07-A8, vol 29, no 2 Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 16.de Jonge BL, Walkup GK, Lahiri SD, Huynh H, Neckermann G, Utley L, Nash TJ, Brock J, San Martin M, Kutschke A, Johnstone M, Laganas V, Hajec L, Gu RF, Ni H, Chen B, Hutchings K, Holt E, McKinney D, Gao N, Livchak S, Thresher J. 2013. Discovery of inhibitors of 4′-phosphopantetheine adenylyltransferase (PPAT) to validate PPAT as a target for antibacterial therapy. Antimicrob Agents Chemother 57:6005–6015. doi: 10.1128/AAC.01661-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Buurman ET, Johnson KD, Kelly RK, MacCormack K. 2006. Different modes of action of naphthyridones in Gram-positive and Gram-negative bacteria. Antimicrob Agents Chemother 50:385–387. doi: 10.1128/AAC.50.1.385-387.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wientjes FB, Pas E, Taschner PE, Woldringh CL. 1985. Kinetics of uptake and incorporation of meso-diaminopimelic acid in different Escherichia coli strains. J Bacteriol 164:331–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hunt DE, Sandham HJ. 1969. Improved agar gradient-plate technique. Appl Microbiol 17:329–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nonejuie P, Burkart M, Pogliano K, Pogliano J. 2013. Bacterial cytological profiling rapidly identifies the cellular pathways targeted by antibacterial molecules. Proc Natl Acad Sci U S A 110:16169–16174. doi: 10.1073/pnas.1311066110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shlaes DM, Shlaes JH, Davies J, Williamson R. 1989. Escherichia coli susceptible to glycopeptide antibiotics. Antimicrob Agents Chemother 33:192–197. doi: 10.1128/AAC.33.2.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Choi DS, Yamada H, Mizuno T, Mizushima S. 1986. Trimeric structure and localization of the major lipoprotein in the cell surface of Escherichia coli. J Biol Chem 261:8953–8957. [PubMed] [Google Scholar]
- 23.Inouye M, Shaw J, Shen C. 1972. The assembly of a structural lipoprotein in the envelope of Escherichia coli. J Biol Chem 247:8154–8159. [PubMed] [Google Scholar]
- 24.Inukai M, Takeuchi M, Shimizu K, Arai M. 1979. Existence of the bound form of prolipoprotein in Escherichia coli B cells treated with globomycin. J Bacteriol 140:1098–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yakushi T, Tajima T, Matsuyama S, Tokuda H. 1997. Lethality of the covalent linkage between mislocalized major outer membrane lipoprotein and the peptidoglycan of Escherichia coli. J Bacteriol 179:2857–2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xiao Y, Gerth K, Muller R, Wall D. 2012. Myxobacterium-produced antibiotic TA (myxovirescin) inhibits type II signal peptidase. Antimicrob Agents Chemother 56:2014–2021. doi: 10.1128/AAC.06148-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zwiebel LJ, Inukai M, Nakamura K, Inouye M. 1981. Preferential selection of deletion mutations of the outer membrane lipoprotein gene of Escherichia coli by globomycin. J Bacteriol 145:654–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Inukai M, Takeuchi M, Shimizu K, Arai M. 1978. Mechanism of action of globomycin. J Antibiot (Tokyo) 31:1203–1205. doi: 10.7164/antibiotics.31.1203. [DOI] [PubMed] [Google Scholar]
- 29.Hagan CL, Kim S, Kahne D. 2010. Reconstitution of outer membrane protein assembly from purified components. Science 328:890–892. doi: 10.1126/science.1188919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Malinverni JC, Werner J, Kim S, Sklar JG, Kahne D, Misra R, Silhavy TJ. 2006. YfiO stabilizes the YaeT complex and is essential for outer membrane protein assembly in Escherichia coli. Mol Microbiol 61:151–164. doi: 10.1111/j.1365-2958.2006.05211.x. [DOI] [PubMed] [Google Scholar]
- 31.Wu T, Malinverni J, Ruiz N, Kim S, Silhavy TJ, Kahne D. 2005. Identification of a multicomponent complex required for outer membrane biogenesis in Escherichia coli. Cell 121:235–245. doi: 10.1016/j.cell.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 32.Chng SS, Ruiz N, Chimalakonda G, Silhavy TJ, Kahne D. 2010. Characterization of the two-protein complex in Escherichia coli responsible for lipopolysaccharide assembly at the outer membrane. Proc Natl Acad Sci U S A 107:5363–5368. doi: 10.1073/pnas.0912872107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ruiz N, Kahne D, Silhavy TJ. 2009. Transport of lipopolysaccharide across the cell envelope: the long road of discovery. Nat Rev Microbiol 7:677–683. doi: 10.1038/nrmicro2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu T, McCandlish AC, Gronenberg LS, Chng SS, Silhavy TJ, Kahne D. 2006. Identification of a protein complex that assembles lipopolysaccharide in the outer membrane of Escherichia coli. Proc Natl Acad Sci U S A 103:11754–11759. doi: 10.1073/pnas.0604744103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paradis-Bleau C, Markovski M, Uehara T, Lupoli TJ, Walker S, Kahne DE, Bernhardt TG. 2010. Lipoprotein cofactors located in the outer membrane activate bacterial cell wall polymerases. Cell 143:1110–1120. doi: 10.1016/j.cell.2010.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Typas A, Banzhaf M, van den Berg van Saparoea B, Verheul J, Biboy J, Nichols RJ, Zietek M, Beilharz K, Kannenberg K, von Rechenberg M, Breukink E, den Blaauwen T, Gross CA, Vollmer W. 2010. Regulation of peptidoglycan synthesis by outer-membrane proteins. Cell 143:1097–1109. doi: 10.1016/j.cell.2010.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Narita S. 2011. ABC transporters involved in the biogenesis of the outer membrane in Gram-negative bacteria. Biosci Biotechnol Biochem 75:1044–1054. doi: 10.1271/bbb.110115. [DOI] [PubMed] [Google Scholar]
- 38.Yasuda M, Iguchi-Yokoyama A, Matsuyama S, Tokuda H, Narita S. 2009. Membrane topology and functional importance of the periplasmic region of ABC transporter LolCDE. Biosci Biotechnol Biochem 73:2310–2316. doi: 10.1271/bbb.90451. [DOI] [PubMed] [Google Scholar]
- 39.Mizutani M, Mukaiyama K, Xiao J, Mori M, Satou R, Narita S, Okuda S, Tokuda H. 2013. Functional differentiation of structurally similar membrane subunits of the ABC transporter LolCDE complex. FEBS Lett 587:23–29. doi: 10.1016/j.febslet.2012.11.009. [DOI] [PubMed] [Google Scholar]
- 40.Okuda S, Tokuda H. 2009. Model of mouth-to-mouth transfer of bacterial lipoproteins through inner membrane LolC, periplasmic LolA, and outer membrane LolB. Proc Natl Acad Sci U S A 106:5877–5882. doi: 10.1073/pnas.0900896106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nikaido H. 2001. Preventing drug access to targets: cell surface permeability barriers and active efflux in bacteria. Semin Cell Dev Biol 12:215–223. doi: 10.1006/scdb.2000.0247. [DOI] [PubMed] [Google Scholar]
- 42.Silhavy TJ, Kahne D, Walker S. 2010. The bacterial cell envelope. Cold Spring Harb Perspect Biol 2:a000414. doi: 10.1101/cshperspect.a000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pathania R, Zlitni S, Barker C, Das R, Gerritsma DA, Lebert J, Awuah E, Melacini G, Capretta FA, Brown ED. 2009. Chemical genomics in Escherichia coli identifies an inhibitor of bacterial lipoprotein targeting. Nat Chem Biol 5:849–856. doi: 10.1038/nchembio.221. [DOI] [PubMed] [Google Scholar]
- 44.Henry R, Vithanage N, Harrison P, Seemann T, Coutts S, Moffatt JH, Nation RL, Li J, Harper M, Adler B, Boyce JD. 2012. Colistin-resistant, lipopolysaccharide-deficient Acinetobacter baumannii responds to lipopolysaccharide loss through increased expression of genes involved in the synthesis and transport of lipoproteins, phospholipids, and poly-beta-1,6-N-acetylglucosamine. Antimicrob Agents Chemother 56:59–69. doi: 10.1128/AAC.05191-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tanaka SY, Narita S, Tokuda H. 2007. Characterization of the Pseudomonas aeruginosa Lol system as a lipoprotein sorting mechanism. J Biol Chem 282:13379–13384. doi: 10.1074/jbc.M611840200. [DOI] [PubMed] [Google Scholar]
- 46.Narita S, Tokuda H. 2007. Amino acids at positions 3 and 4 determine the membrane specificity of Pseudomonas aeruginosa lipoproteins. J Biol Chem 282:13372–13378. doi: 10.1074/jbc.M611839200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.