

Abstract
The presence of multidrug-tolerant persister cells within microbial populations has been implicated in the resiliency of bacterial survival against antibiotic treatments and is a major contributing factor in chronic infections. The mechanisms by which these phenotypic variants are formed have been linked to stress response pathways in various bacterial species, but many of these mechanisms remain unclear. We have previously shown that in the cariogenic organism Streptococcus mutans, the quorum-sensing peptide CSP (competence-stimulating peptide) pheromone was a stress-inducible alarmone that triggered an increased formation of multidrug-tolerant persisters. In this study, we characterized SMU.2027, a CSP-inducible gene encoding a LexA ortholog. We showed that in addition to exogenous CSP exposure, stressors, including heat shock, oxidative stress, and ofloxacin antibiotic, were capable of triggering expression of lexA in an autoregulatory manner akin to that of LexA-like transcriptional regulators. We demonstrated the role of LexA and its importance in regulating tolerance toward DNA damage in a noncanonical SOS mechanism. We showed its involvement and regulatory role in the formation of persisters induced by the CSP-ComDE quorum-sensing regulatory system. We further identified key genes involved in sugar and amino acid metabolism, the clustered regularly interspaced short palindromic repeat (CRISPR) system, and autolysin from transcriptomic analyses that contribute to the formation of quorum-sensing-induced persister cells.
INTRODUCTION
The classical view of bacterial survival against antibiotic killing has usually been seen as the expression of genetic resistance mechanisms that arise from mutations or gained through horizontal gene transfer. These resistance mechanisms include host target modification, degradation or modification of the antibiotic itself, and reduction in the permeability or increase in the efflux of the drug (1). A major survival mechanism of bacteria is antibiotic tolerance, whereby bacterial cells that have a slower or reduced growth rate become more tolerant toward antibiotic killing (2, 3). This reduction in bacterial growth is prominently perceived as one of the main survival mechanisms elicited in bacterial biofilms, by which the slower growth of biofilm cells contributes toward the highly recalcitrant nature of biofilm infections, even in biofilm populations that lack genetically encoded antibiotic resistance markers (4, 5).
Formation of persister cells is the main factor responsible for the tolerance of pathogens to antibiotics. Persisters are nongrowing dormant cells that are produced in a clonal population of genetically identical cells. They constitute a small fraction of the bacterial population. Persisters are not mutants but phenotypic variants of the wild-type population (6). In contrast to the case with the aforementioned drug resistance mechanisms, which allow for bacterial cells to actively grow and divide unimpeded in the presence of specific antimicrobials, persisters are capable of surviving lethal doses of multiple classes of bactericidal antibiotics at the expense of cellular growth in both planktonic and biofilm populations (7).
Persister formation is governed by both stochastic and deterministic mechanisms (8, 9). Persister cells are formed through random intrinsic fluctuations of protein levels, such as the well-studied stable toxins expressed by chromosomal toxin-antitoxin (TA) modules (10–13). Spontaneous gene expression and noise during transcription and translation, amplified by feedback regulatory processes, also give rise to fluctuating protein levels of persister genes that result in a dormant multidrug-tolerant state (3, 14). Since the formation of persister cells is a result of a variety of mechanisms, the subpopulation of persisters is heterogenous. The redundancy in the formation of persisters hampers efforts in preventing tolerance toward antimicrobial drugs through the targeting of a single mechanism (14). For example, it requires genetic knockouts of at least 10 TA modules to significantly reduce persister levels in Escherichia coli (15). Recent studies have shown that the formation of dormant persister cells can be induced by environmental and external stimuli (deterministic mechanisms), including various stressors and signaling molecules. Studies done with E. coli have shown that DNA damage, as a result of ciprofloxacin challenge, induced persisters through SOS-dependent expression of the TisB toxin from the tisAB-istR TA locus (16). Another study showed that indole, an intercellular signaling molecule expressed during stationary growth phase, was capable of inducing the formation of persisters through the activation of stress response pathways (17).
While little work has been performed with streptococci, our laboratory showed—using Streptococcus mutans as a model organism—that persisters were formed stochastically through mild overexpression of chromosomal MazEF and RelBE TA modules (18). S. mutans is the main causative agent of human dental caries (tooth decay) and a resident of the complex dental plaque microflora (19, 20). As it inhabits and colonizes the dental biofilm, S. mutans continuously confronts a variety of nutritional, chemical, and physical stresses (21, 22). We recently demonstrated that challenge with an incoming stressor produced increased numbers of multidrug-tolerant persisters (18). More importantly, we discovered that S. mutans used its competence-stimulating peptide (CSP)–ComDE quorum-sensing system governing the development of genetic competence to produce stress-inducible persisters (18). The CSP-ComDE system is composed of the CSP pheromone acting through a membrane-bound histidine kinase receptor (ComD) and its cognate cytoplasmic response regulator (ComE) (23). When the extracellular level of CSP reaches a critical threshold, CSP interacts with the ComD sensor, resulting in its autophosphorylation. This event initiates a phosphorelay cascade that results in the activation of ComE. The phosphorylated form of ComE regulates transcription of several genes (24, 25). Among the early genes is cipB, encoding a small antimicrobial peptide that also functions as a peptide regulator for the transcriptional control of the competence regulon via the alternative sigma factor SigX (26). Recently, it has been demonstrated that the ComR/ComS system is the proximal regulator of SigX in S. mutans (27, 28).
Many environmental stresses are capable of causing damage to the chromosomal DNA, leading to potential mutations and even the formation of DNA strand breaks that are lethal during transcription and DNA replication (29–31). Streptococci lack a canonical SOS response pathway to repair DNA damage, and the induction of the SigX regulon has been proposed to act as a general stress response (32). In this study, we continued our investigation of the CSP-ComDE pathway in regulating bacterial persistence in S. mutans. In particular, we characterized the CSP-inducible gene SMU.2027, encoding a LexA-like transcriptional regulator. We showed that this regulator was involved in initiating tolerance toward DNA-damaging agents through a nonclassical SOS pathway. More importantly, we demonstrated that the LexA pathway affected the formation of CSP-induced persisters.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
A summary of the bacterial strains and plasmids used in this study is provided in Table 1. Nonpolar insertion-deletion mutants were constructed in the S. mutans UA159 wild-type (WT) strain by PCR ligation mutagenesis (33). Primers used for the generation of PCR products are listed in Table S1 in the supplemental material. S. mutans strains were grown in Todd-Hewitt medium supplemented with 0.3% yeast extract (THYE) and incubated statically at 37°C in air with 5% CO2. E. coli strains were cultivated aerobically in Luria-Bertani (LB) medium at 37°C. Plasmids were introduced into E. coli by transformation using electroporation or chemical transformation. Plasmids were transferred to S. mutans by natural transformation as described previously (26). When needed, antibiotics were added as follows: erythromycin (150 μg/ml) or kanamycin (50 μg/ml) for E. coli and kanamycin (300 μg/ml), spectinomycin (1 mg/ml), or erythromycin (10 μg/ml) for S. mutans. Cell growth was monitored by determining the optical density at 600 nm (OD600). Cell viability was assessed by counting CFU on replica agar plates. The MIC test was performed according to the broth microdilution method using THYE broth as described previously (34).
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Relevant characteristic(s)a | Source or reference |
|---|---|---|
| Strains | ||
| S. mutans UA159 | Wild-type reference strain | Laboratory stock |
| E. coli DH10B | Host strain for cloning and plasmid production | Laboratory stock |
| E. coli LMG194 | Host strain for pBAD expression | Invitrogen |
| E. coli XL10-Gold | Host strain for site-directed mutagenesis | Agilent Technologies |
| ΔlexA mutant | In-frame SMU.2027 deletion mutant derived from S. mutans UA159; Spr | This study |
| UA159(pIB184) | UA159 harboring pIB184; Emr | This study |
| ΔlexA::lexA A115D mutant | ΔlexA mutant harboring pVL6 chromosomally integrated; Spr Kmr | This study |
| ΔlexA(pVL2) mutant | ΔlexA mutant harboring pVL2; Spr Emr | This study |
| ΔlexA(pVL3) mutant | ΔlexA mutant harboring pVL3; Spr Emr | This study |
| LMG194(pVL4) | LMG194 containing pVL4; Kmr | This study |
| LMG194(pVL5) | LMG194 containing pVL5; Kmr | This study |
| ΔumuC mutant | In-frame SMU.403 deletion mutant derived from S. mutans UA159; Emr | This study |
| ΔclpP mutant | In-frame SMU.1672 deletion mutant derived from S. mutans UA159; Emr | This study |
| ΔcomE mutant | In-frame SMU.1917 deletion mutant derived from S. mutans UA159; Emr | 25 |
| ΔcipB mutant | In-frame SMU.1914 deletion mutant derived from S. mutans UA159; Emr | 25 |
| ΔcomS mutant | In-frame comS deletion mutant derived from S. mutans UA159; Emr | This study |
| ΔcomR mutant | In-frame SMU.61 deletion mutant derived from S. mutans UA159; Emr | D. Morrison, UICb |
| ΔsigX mutant | In-frame SMU.1997 deletion mutant derived from S. mutans UA159; Emr | 25 |
| ΔscrA mutant | In-frame SMU.1841 deletion mutant derived from S. mutans UA159; Emr | This study |
| Δcsn2 mutant | In-frame SMU.1402 deletion mutant derived from S. mutans UA159; Emr | This study |
| Δ63 mutant | In-frame SMU.63 deletion mutant derived from S. mutans UA159; Emr | This study |
| Δ984 mutant | In-frame SMU.984 deletion mutant derived from S. mutans UA159; Spr | This study |
| Plasmids | ||
| pBAD202/D-TOPO | Expression vector linearized and topoisomerase activated; Kmr | Invitrogen |
| pIB184 | Shuttle plasmid containing the constitutive P23 lactococcal promoter; Emr | 35 |
| pIB107 | Plasmid for chromosomal integration into S. mutans; Kmr | 35 |
| pVL2 | lexA cloned into pIB184 under the control of its own promoter; Emr | This study |
| pVL3 | lexA A115D mutation into pVL2; Emr | This study |
| pVL4 | lexA cloned under the control of the araBAD promoter into pBAD202/D-TOPO vector; Kmr | This study |
| pVL5 | lexA A115D mutation cloned under the control of the araBAD promoter into pBAD202/D-TOPO vector; Kmr | This study |
| pVL6 | lexA A115D mutation cloned under the control of its own promoter into pIB107; Kmr | This study |
Emr, erythromycin resistance; Kmr, kanamycin resistance; Spr, spectinomycin resistance.
UIC, University of Illinois at Chicago.
Construction of a noncleavable LexA regulator.
A noncleavable LexA regulator was generated by site-directed mutagenesis using the QuikChange II site-directed mutagenesis kit (Agilent Technologies) according to the manufacturer's recommendations. Briefly, the full-length coding region and promoter region of the lexA gene (SMU.2027) were PCR amplified using UA159 genomic DNA as the template and the primer pair CMT-669/CMT-700. The PCR product was purified, doubly digested with ApaI/BamHI, and then cloned onto the shuttle vector pIB184 (35) precut by the same enzymes. The recombinant plasmid pVL2 was confirmed by restriction digestion and sequencing. Plasmid pVL2 was then used as the template for the replacement of A115D using the two mutagenic PCR primers CMT-728 and CMT-729. The clone used to produce LexA A115D mutant was designated pVL3 and confirmed by sequencing. The recombinant plasmid pVL3 was next used as a template for cloning into plasmid pIB107 (35) for integration into the chromosome of S. mutans UA159, and the resulting plasmid was designated pVL6.
Construction of expression vectors for induction in E. coli.
To generate inducible expression constructs for induction of LexA and LexA A115D mutation, a fragment containing the open reading frame for the lexA gene was PCR amplified using UA159 genomic DNA as the template and the primer pair CMT-718/CMT-719. The PCR product was purified and cloned in-frame upstream from the His6 sequence under the control of the araBAD promoter into the pBAD202/D-TOPO vector (Invitrogen). The recombinant plasmid pVL4 was transferred into E. coli DH10B and sequenced on both strands for confirmation. The LexA A115D mutation was achieved via site-directed mutagenesis as described above using the primer pair CMT-728/CMT-729 and pVL4 as the template. The mutation was confirmed by sequencing. The plasmids designated pVL4 (lexA in pBAD) and pVL5 (lexA A115D in pBAD) were then transferred into electrocompetent E. coli LMG194 cells for induction by arabinose.
Production and purification of recombinant fusion proteins.
Overnight E. coli LMG194(pVL4) and E. coli LMG194(pVL5) cultures were diluted (1:100) into fresh LB medium supplemented with kanamycin and grown aerobically at 37°C until an OD600 of ∼0.5 was reached. Arabinose was then added at a final concentration of 0.2% (wt/vol) to induce the expression of recombinant fusion proteins, and the incubation was continued for another 3 h at 37°C, with agitation. Protein expression was verified by SDS-PAGE using the Laemmli buffer system, and the protein bands were visualized by staining with Coomassie brilliant blue. In addition, the expression of recombinant proteins was confirmed with anti-His antibodies (Sigma). The cells were collected by centrifugation, resuspended in 1× binding buffer (Novagen), and disrupted on ice by sonication. The LexA-His6 and LexA A115D-His6 recombinant proteins were then purified by affinity chromatography on Ni2+-nitrilotriacetic acid (Ni-NTA) resin (Novagen) as described by the manufacturer. The Bradford protein assay was used to determine the protein concentrations of the purified samples.
Self-cleavage assay.
Aliquots of purified recombinant LexA proteins (1 μg) were incubated in 50 mM Tris-HCl buffer adjusted to pH values ranging from 6.0 to 10.0. LexA autodigestion was performed at 37°C for 16 h, and aliquots were stopped by adding SDS-PAGE loading buffer. Samples were analyzed by SDS-PAGE using the buffer system of Laemmli at a constant voltage (200 V) with gels containing 12% polyacrylamide in the separating gel and 4.5% polyacrylamide in the stacking gel. The protein bands were visualized by staining with Coomassie brilliant blue.
Gene expression analysis following heat and DNA damage induction.
Overnight cultures of S. mutans strains were diluted (1:100) into fresh THYE broth and incubated at 37°C until an OD600 of ∼0.4 to 0.5 was reached. Cells were then exposed for 2 h at 37°C (except for heat shock) to the following stresses: antibiotic stress (20 μg/ml of ofloxacin), DNA cross-linker (0.5 μg/ml of mitomycin C), oxidative stress (0.5 mM hydrogen peroxide), and heat shock (30 min at 50°C). Cells were processed with the Bio101 FastPrep system (Qbiogene), and total RNA was extracted using TRIzol reagent (Invitrogen). Samples were treated with RQ1 DNase (Promega) and converted to cDNA using a RevertAid H Minus First-Strand cDNA synthesis kit (Fermentas). Negative controls without reverse transcriptase were included in all experiments. Real-time quantitative PCR (qPCR) analysis was performed using the SsoFast EvaGreen Supermix (Bio-Rad) and the CFX96 real-time PCR detection system (Bio-Rad). qPCR assays were performed in triplicate with RNA isolated from three independent experiments. Statistical significance was determined by using Student's t test with the parametric P value cutoff set at <0.01.
Persistence assays.
Overnight cultures of S. mutans strains were diluted (1:20) into fresh THYE broth containing 20 μg/ml of ofloxacin (DNA damage), 15 μg/ml of oxacillin (inhibition of cell wall synthesis), 50 μg/ml of rifampin (inhibition of transcription), or 20 μg/ml of vancomycin (inhibition of cell wall synthesis), followed by incubation for 24 h at 37°C. For the quorum-sensing peptide pheromone assays, diluted S. mutans cells were incubated with 2 μM synthetic competence-stimulating peptide (CSP; Advanced Protein Technology Centre, Hospital for Sick Children, Toronto, Ontario, Canada) pheromone for 2 h at 37°C before exposure to the stresses (18). Samples were withdrawn at the times indicated below, serially diluted, and plated on THYE agar plates. The colonies were counted after 48 h of incubation. All assays were performed in triplicate from three independent cultures. The statistical significance was determined by using Student's t test and a P value of <0.01.
DNA microarrays.
DNA microarrays were performed as previously described (25). Briefly, the S. mutans WT strain and ΔlexA mutant cells were grown with 2 μM CSP (CSP induced) or without CSP (uninduced) to mid-log phase. Cells were then collected by centrifugation, resuspended in ice-cold RNAwiz (Ambion), and disrupted using 0.1-mm zirconia beads. Total RNA was extracted using the Ambion RiboPure-Bacteria kit according to the provided protocol. Isolated RNA was treated twice with DNase I (Ambion) to remove traces of genomic DNA. After the treatment, RNA samples were cleaned with a Qiagen RNeasy MinElute cleanup kit. Aliquots of the extracted total RNA were tested for quantity and quality. RNA concentration was determined spectrophotometrically using a NanoDrop spectrophotometer, and RNA integrity was assessed with the Agilent Bioanalyzer 6000 (ProkaryoteTotal RNA Nano assay). Inclusion criteria considered total RNA with a ratio (A260/A280) ideally of >1.8 and absence of signs of RNA degradation on the Agilent BioAnalyzer.
For cDNA synthesis, 15 μg of RNA was mixed with 1.25 μg of random primers and incubated at 70°C for 10 min and then at 25°C for 10 min. Reverse transcription was performed in 1× First Strand buffer supplemented with 10 mM dithiothreitol, 0.5 mM deoxynucleoside triphosphate mix, 60 U of RNAseOut (Invitrogen), and 1,500 U of SuperScript II reverse transcriptase (Invitrogen). The reaction mixture was incubated at 25°C for 10 min, 37°C for 1 h, 42°C for 1 h, and 70°C for 10 min. To remove residual RNA, the cDNA samples were treated with 0.2 N NaOH at 65°C for 30 min and then neutralized with 0.2 N HCl before being purified with the Qiagen MinElute PCR purification kit. The cDNA was fragmented using Roche DNase I (0.06 U per μg of cDNA) at 37°C for 10 min. Inactivation of the enzyme was performed at 98°C for 10 min. Synthesized cDNA was quantified spectrophotometrically and the integrity assessed with the Agilent Bioanalyzer 6000. These processes were performed before and after fragmentation. Fragmented samples represented by a smear of products falling between 50 and 200 bases were included and labeled with biotin-ddUTP using the BioArray terminal labeling kit (Enzo). Microarrays were run using a whole-genome custom GeneChips antisense expression microarray previously designed in collaboration with Affymetrix (Santa Clara, CA) (36). Hybridization, washing, and scanning of the microarray chips were performed at the Princess Margaret Genomics Centre (Toronto, Ontario, Canada) using the Affymetrix protocol. Results were validated by qPCR.
DNA transformation assays.
Overnight cultures of the S. mutans WT strain and its deletion mutants were diluted (1:20) with fresh THYE broth and incubated at 37°C until an OD600 of 0.1 was reached. To test the effect of CSP on genetic transformation (CSP-induced conditions), synthetic CSP was added at a final concentration of 0.2 μM (26). Ten micrograms of UA159 genomic DNA containing the spectinomycin resistance marker inserted into the rgp locus of the UA159 strain, as inactivation of this locus has no effect on cell viability and transformation efficiency (25), was added to the cultures (0.5-ml aliquots), which were grown for a further 2.5 h at 37°C before differential plating. The transformation efficiency was calculated as the percentage of spectinomycin-resistant transformants divided by the total number of recipient cells, which was determined by the number of CFU on antibiotic-free THYE agar plates. All assays were performed in triplicate from three independent experiments.
RESULTS
An intact CSP-dependent competence pathway is necessary for CSP-inducible formation of persisters.
In the oral cavity, bacteria are constantly challenged by a wide variety of stressful environmental conditions. We previously demonstrated that particular stresses activated CSP production (25) and cells responded to high levels of CSP by inducing the formation of multidrug-tolerant persisters (18). In pursuit of determining the mechanism by which the CSP-ComDE pathway regulates the inducible formation of persisters, several components of the competence regulon necessary for the development of genetic competence in S. mutans were investigated. Individual mutants in which comE, cipB, comR, comS, and sigX genes were knocked out were constructed and tested for the CSP-inducible formation of persisters tolerant to ofloxacin. We used exogenously added synthetic CSP to artificially mimic the high CSP concentrations induced by stress. Our results showed that whereas the WT strain produced an increased number of persisters tolerant to ofloxacin (∼4-fold) in the presence of the CSP pheromone, the CSP-inducible persistence phenotype was abolished for all mutants tested (Fig. 1). These results signified the importance of an intact competence pathway initiating from CSP binding to the ComD membrane sensor to the eventual activation of SigX and suggest that putative persister genes are regulated by this alternative sigma factor.
FIG 1.

Role of essential competence genes in the formation of CSP-induced persister cells. Overnight cultures of the S. mutans WT strain and its ΔcomD, ΔcomE, ΔcipB, ΔcomR, ΔcomS, and ΔsigX mutants were diluted (1:100) into fresh THYE broth in the absence or presence of the CSP pheromone and incubated at 37°C for 2 h before being challenged with ofloxacin. Aliquots of cells were removed at the introduction of the antibiotic and after the antibiotic treatment (h 24) to determine cell survival by spot plating onto THYE agar plates. Results are expressed as the log fold change in cell survival normalized to the non-CSP prestress value. The data are the averages and standard errors of results from three independent cultures. The average value for each strain is indicated.
SMU.2027 gene induced by the quorum-sensing CSP pheromone possesses characteristics of LexA-like transcriptional regulators.
Work performed by some members of our group showed that S. mutans employs its quorum-sensing CSP pheromone as a stress-inducible “alarmone” to give an early warning signal of hostile conditions and mounts a specific response to adapt and survive environmental assaults (reviewed in reference 37). SMU.2027, encoding a putative LexA-like (or HdiR-like) regulator, was found to be upregulated (∼5-fold) under CSP conditions used to mimic the high concentrations of CSP induced by stress (25). Although the SMU.2027 gene is induced by the CSP pheromone, it is unlikely that SigX directly regulates the SMU.2027 gene, since no conserved CIN (competence-induced) box consensus sequence (23, 38) has been found in its promoter region. A perfect inverted repeat (IR; 15 bp) was found in the promoter region of SMU.2027 (see Fig. S2 in the supplemental material), suggesting a putative binding site for autoregulatory activity as observed in other LexA-like regulators (39). Bioinformatic analysis of S. mutans SMU.2027 showed that it possesses the two conserved domains found in LexA-like regulators: (i) a helix-turn-helix motif involved in sequence-specific DNA binding and (ii) a signal peptidase-like serine peptidase domain. A multiple-sequence alignment of the product of S. mutans SMU.2027 with the amino acid sequences of various LexA and HdiR regulators revealed that the Ala-Gly bond, which is involved in RecA-dependent autocleavage, was conserved in all sequences aligned (Fig. 2).
FIG 2.

Primary structures of LexA-like regulators. Shown is a multisequence alignment of the amino acid sequence of the S. mutans SMU.2027 product with those of LexA and HdiR of E. coli, Lactococcus lactis, Staphylococcus aureus, Streptococcus uberis, Rhodobacter sphaeroides, and Bifidobacterium longum. The helix-turn-helix motif is shown as a gray box at the N-terminal end of the SMU.2027 sequence. The C-terminal serine-peptidase domain (white box) is also shown. Asterisks indicate conserved amino acid residues involved in LexA autocleavage. The amino acid mutated in the present study is in bold and underlined.
Under heat and DNA damage conditions, LexA regulators are cleaved and derepress expression of genes involved in DNA damage tolerance and repair and the lexA gene itself (i.e., it is autoregulatory) (39). The expression of the S. mutans SMU.2027 gene in response to heat and DNA damage conditions was then investigated. Our results showed that treatment with ofloxacin antibiotic and heat shock, two stresses known to initiate self-cleavage of LexA repressors (39, 40), led to activation of the S. mutans SMU.2027 gene (Table 2). Oxidative stress was also investigated since reactive oxygen species (ROS) are known to cause DNA damage via oxidation of nucleotide bases. Our results showed that hydrogen peroxide stress resulted in an increase in SMU.2027 gene expression (Table 2). Consequently, we can hypothesize that the SMU.2027 product (called LexA here) belongs to the LexA-like (or HdiR-like) family based on domain organization, sequence similarity, and induction in response to heat and DNA damage conditions.
TABLE 2.
Effects of heat and DNA damage stresses on lexA (SMU.2027) gene expression
| Stress | Change in S. mutansa |
||
|---|---|---|---|
| WT | ΔlexA::lexA A115D mutant | ΔclpP mutant | |
| 50°C | +2.84 ± 0.55 | +1.18 ± 0.17* | −1.31 ± 0.14* |
| Antibiotic ofloxacin | +5.22 ± 3.03 | +1.09 ± 0.11* | +1.29 ± 0.45* |
| Hydrogen peroxide | +5.04 ± 3.93 | +1.32 ± 0.25* | +1.28 ± 0.54* |
qPCR was performed to analyze lexA gene expression. The expression is presented as the average fold change ± standard deviation compared with lexA gene expression in the absence of stress. Statistical significance was determined by using Student's t test with the parametric P value cutoff set at <0.01 (*, mutant versus WT).
LexA undergoes self-cleavage and requires the ClpP proteolytic subunit for its autoregulation.
To further characterize the S. mutans LexA regulator, we produced a recombinant LexA protein by cloning the full-length coding region of SMU.2027 into the pBAD expression system for induction of gene expression using araBAD promoter dose-dependent regulation. Purified recombinant protein was then utilized for determining if LexA undergoes self-cleavage in vitro at alkaline pH, a characteristic feature of LexA-like repressors (41). In vitro cleavage requires RecA at neutral pH, but at alkaline pH, a spontaneous cleavage takes place. This RecA-independent reaction is called autodigestion. As the purified recombinant LexA protein was incubated in increasing pHs, spontaneous self-cleavage occurred which resulted in two cleavage products at a pH of ≥8.0 (Fig. 3A). A recombinant LexA regulator with a point mutation at the conserved alanine residue at position 115 of the Ala-Gly bond to an aspartic acid abolished the alkaline-induced self-cleavage (Fig. 3B). These results confirm that LexA is capable of self-cleavage, specifically at the conserved A115 cleavage site.
FIG 3.
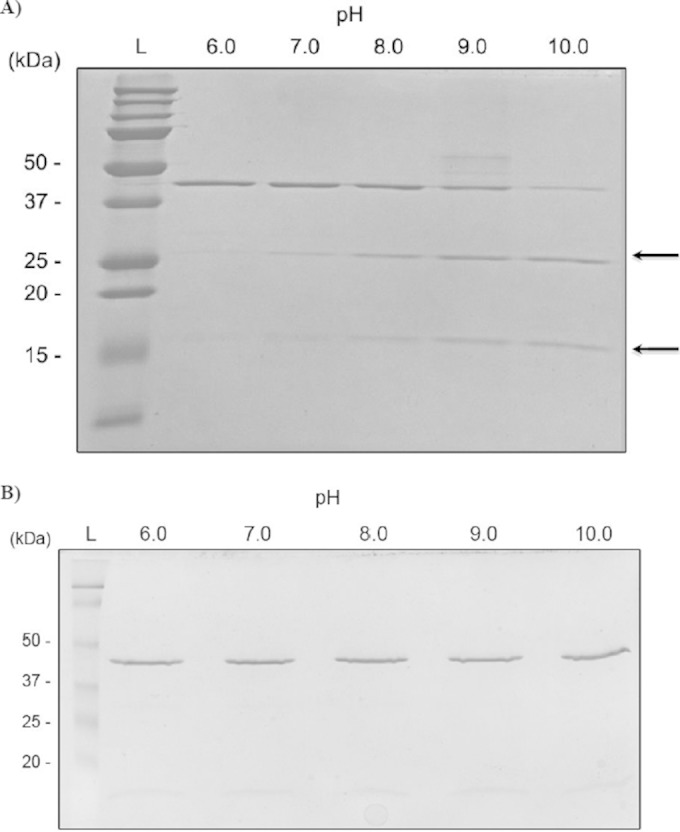
LexA self-cleavage. LexA (A) and LexA A115D (B) recombinant proteins were incubated in Tris-HCl buffer at pH values varying from 6.0 to 10.0. Autocleavage assay results were visualized by SDS-PAGE with staining with Coomassie brilliant blue. Sizes of Precision Plus Protein Dual Color Standards (Bio-Rad) are on the left. Arrows indicate the positions of breakdown products of LexA.
After cleavage, the N-terminal domain of LexA-like repressors can remain bound to the DNA, which can still repress gene expression (42). For this reason, the N-terminal cleavage products are usually removed by Clp proteolytic complexes prior to derepression of target gene expression. qPCR analysis of the lexA gene was performed using the WT strain and a mutant deficient in the ClpP protease catalytic domain (ΔclpP). Results showed that derepression of lexA did not occur in the ΔclpP mutant upon heat or DNA damage (Table 2). Together, these results suggest that in S. mutans, Clp protease is required to degrade the N-terminal helix-turn-helix motif of LexA that is predicted to remain bound to the DNA upon heat and DNA damage, similar to the case with other LexA-like repressors.
To confirm the role of S. mutans LexA in its autoregulation, we constructed a strain expressing a mutated LexA protein unable to undergo autocleavage. A single-site mutation of amino acid residue was introduced by PCR in the Ala-Gly bond of the S. mutans LexA regulator. The conserved alanine at position 115 was changed to aspartic acid (Fig. 2). The uncleavable form of S. mutans lexA was then cloned under the control of its own promoter into pIB107 vector (pVL6) and integrated into the chromosome of an S. mutans ΔlexA mutant. The new mutant was designated ΔlexA::lexA A115D and was confirmed by PCR. The disruption of lexA had no effect upon S. mutans cell growth under normal growth conditions (data not shown). Moreover, no discernible growth difference was observed between the WT strain, the ΔlexA mutant, and the ΔlexA::lexA A115D mutant under nonstress conditions (data not shown). As expected, no upregulation of lexA was observed in the S. mutans ΔlexA::lexA A115D uncleavable strain tested under the same stress conditions (Table 2). Our results indicate that induction of lexA gene expression by heat and DNA damage is controlled by LexA cleavage-mediated derepression of lexA. Altogether, these results confirm that SMU.2027 encodes a LexA-like repressor.
S. mutans LexA regulates tolerance toward DNA damage through a mechanism that is different from the SOS-like response pathway in streptococci.
In order to test the role of the LexA regulator in tolerance to DNA damage, S. mutans cells were incubated in the presence of a lethal concentration of the fluoroquinolone ofloxacin, a broad-spectrum antibiotic. Fluoroquinolones induce DNA damage by preventing the ligation reactions of DNA gyrase and topoisomerase, resulting in double-strand breaks (43). Cell survival assays showed that the ΔlexA(pIB184) mutant had a significant decrease (3.7-fold) in survival after 24 h compared with that of the WT strain expressing the empty plasmid (Table 3). A more severe reduction (22.9-fold) was observed with the ΔlexA(pVL3) strain expressing the uncleavable form of LexA. Similar results were obtained with mitomycin C, a potent DNA cross-linker. Cell survival of exposure to antibiotic oxacillin exhibiting a different mechanism of action remained unaffected, suggesting that LexA does not affect tolerance toward non-DNA-damaging agents (Table 3). Together, these results further indicate that the S. mutans lexA gene encodes a regulator involved in tolerance toward DNA damage.
TABLE 3.
S. mutans cell survival of exposure to different drugs
| Drugb | % cell survival in S. mutans straina: |
||
|---|---|---|---|
| WT(pIB184) | ΔlexA(pIB184) mutant | ΔlexA(pVL3) mutant | |
| Ofloxacin | (110 ± 5.0) × 10−3 | (30 ± 20) × 10−3* | (4.8 ± 1.2) × 10−3* |
| Mitomycin C | (67 ± 1.0) × 10−4 | (7 ± 2.1) × 10−4* | (1.6 ± 1.3) × 10−4* |
| Oxacillin | 2.2 ± 1.0 | 1.6 ± 0.8 | 1.6 ± 0.7 |
Aliquots of cells were removed at the introduction of the drug and after the treatment (h 24) to determine cell survival by spot plating onto THYE agar plates. The data are the averages and standard errors of results from three independent cultures. Statistical significance was determined by using Student's t test with the parametric P value cutoff set at <0.01 [*, mutant versus WT(pIB184)].
Lethal concentrations of drugs were used.
In determining if the route by which LexA initiates a DNA damage tolerance response via an SOS-like pathway, we examined the role of a UmuC homolog in S. mutans. UmuC is a subunit of the error-prone DNA polymerase V found in bacteria and is involved in bypassing DNA lesions during DNA replication. In streptococci, this gene was found to be regulated by LexA-like repressors in Streptococcus thermophilus (44) and Streptococcus uberis (45). In S. mutans strain UA159, SMU.403 encodes a UmuC homolog exhibiting more than 85% amino acid sequence similarity to UmuC of S. thermophilus and S. uberis. A strain disrupted in SMU.403 (ΔumuC mutant) was constructed from the UA159 WT strain. The gene disruption had no effect upon cell growth under normal growth conditions (data not shown). We then tested the effect of umuC mutation on tolerance of S. mutans to DNA damage. Our results showed that in S. mutans, the UmuC homolog was dispensable in the tolerance and survival toward both ofloxacin (concentrations varying between 2 μg/ml and 20 μg/ml) and mitomycin C (concentrations varying between 0.18 μg/ml and 0.25 μg/ml). Furthermore, qPCR analysis showed that S. mutans umuC was not differentially expressed in a ΔlexA mutant background (data not shown). We conducted BLAST searches for homologous sequences using SMU.403 (umuC) as a query sequence. The SMU.403 gene is highly conserved in all S. mutans strains, but the SMU.403 sequence does not share homology with any other sequences in S. mutans. Since knocking out SMU.403 failed to cause a change in the phenotype, our results suggest that another gene(s) can compensate for the loss of SMU.403. Altogether, these results suggest that LexA regulates tolerance and survival of DNA damage through a mechanism that differs from the SOS-like response pathway as described for S. thermophilus and S. uberis.
CSP-inducible formation of persisters in S. mutans is affected by the LexA pathway.
Persisters are dormant cells within an isogenic bacterial population that can tolerate lethal concentrations of antibiotics (6, 46). The presence of dormant persisters has no effect on the MICs of antibiotics, since persisters are not growing and lack antibiotic resistance mechanisms. The results presented above showed that although cell survival is decreased in the ΔlexA mutant and the ΔlexA(pVL3) strain expressing the uncleavable form of LexA (Table 3), antibiotic sensitivity remained the same, as the MICs of ofloxacin against the mutants were not different from that for the WT strain (data not shown). Consequently, we hypothesized that LexA could promote the formation of persister cells. Our previous work showed that environmental stresses, such as heat and DNA damage, induce the formation of persister cells (18), reinforcing our hypothesis. Moreover, we demonstrated that S. mutans integrates its response to specific environmental stresses with its quorum-sensing system, the CSP-ComDE regulatory circuit, by positively influencing the persister levels (18). Given the above, we tested the direct impact of S. mutans LexA in the CSP-inducible development of persisters. The S. mutans WT strain and its ΔlexA mutant were preexposed to the quorum-sensing CSP pheromone before being treated with different drugs for 24 h. As expected, our data showed that the CSP pheromone significantly increased the numbers of persister cells tolerant to ofloxacin, oxacillin, rifampin, and vancomycin for the WT strain (Fig. 4). Interestingly, the CSP-inducible persistence phenotype was abolished in the ΔlexA mutant for all classes of antibiotics tested except vancomycin, suggesting that the LexA pathway affects the formation of CSP-inducible persisters toward several different classes of antibiotics, not just toward antibiotics inducing DNA damage.
FIG 4.

Effects of S. mutans LexA on the development of CSP-induced persister cells. Overnight cultures of the S. mutans WT strain and its ΔlexA mutant were diluted (1:100) into fresh THYE broth in the absence or presence of CSP pheromone and incubated at 37°C for 2 h before being challenged with ofloxacin, oxacillin, rifampin, or vancomycin. Aliquots of cells were removed at the introduction of the antibiotic and after the antibiotic treatment (h 24) to determine cell survival by spot plating onto THYE agar plates. Results are expressed as the log fold change in cell survival normalized to the non-CSP prestress value. The data are the averages and standard errors of results from three independent cultures. The average value for each treatment is indicated.
It is most likely that the CSP conditions induce SigX factor to upregulate genes involved in DNA repair and recombination leading to formation of single-stranded DNA (ssDNA) intermediates. Based on work done with E. coli, we can hypothesize that the highly expressed RecA proteins (recA is a SigX-regulated gene in S. mutans and showed an ∼7-fold increase under CSP conditions [25]) probably bind to any exposed ssDNA within the cell to form RecA filaments, and this phenomenon is capable of serving as an SOS-inducing signal that leads to the autocleavage of LexA and subsequent gene derepression (47).
The CSP-inducible LexA regulator is not necessary for the development of genetic competence.
It was recently shown that competence development and the SOS pathway in S. thermophilus were antagonistic to one another (44). Since inactivation of lexA gene abolished the formation of CSP-induced persister cells in S. mutans, whether competence is also hindered was investigated. The transformation efficiency of the ΔlexA mutant was evaluated under CSP-induced and uninduced conditions. Our results showed that inactivation of the lexA gene, and thus constant activation of the LexA-regulated pathway, had no significant effect on transformation efficiency under both conditions (Fig. 5). These results suggest that the S. mutans LexA pathway, while preventing CSP-inducible formation of persisters, does not interfere with the development of genetic competence.
FIG 5.
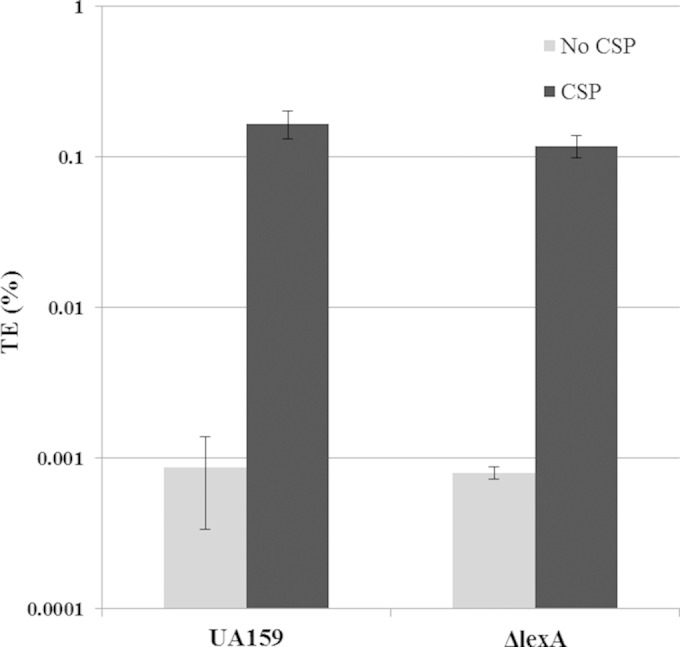
Effect of inactivation of the lexA gene on the development of genetic competence. Overnight cultures of the WT strain and its ΔlexA mutant were diluted (1:20) into fresh THYE broth and grown until an OD600 of 0.1 was reached. Aliquoted cultures were cultivated in the absence and presence of CSP and 20 μg/ml of UA159 genomic DNA carrying the kanamycin resistance marker. Cells were incubated at 37°C for 2.5 h before differential plating. The transformation efficiency (TE) was expressed as the percentage of kanamycin-resistant transformants divided by the total number of recipient cells. The data are the averages and standard errors of results from three independent cultures.
Comparison of the gene expression profile between the WT and the ΔlexA mutant.
To determine the potential genes regulated by the LexA transcriptional regulator, transcriptional analysis of both the WT and its ΔlexA mutant were compared. RNA was extracted from cultures of both the WT and the deletion mutant grown to mid-exponential phase under nonstress conditions (see Materials and Methods for microarray experiment details). The results from the microarray identified scrA, coding for enzyme II of the phosphoenolpyruvate-dependent sucrose phosphotransferase system (48), as the only gene differentially regulated (3-fold reduction in the ΔlexA mutant). Confirming our earlier results, the transcriptome analyses showed that the genes involved in DNA repair and recombination, including umuC, did not appear to be differentially expressed when LexA was inactivated (see Table S2 in the supplemental material). These results confirmed that LexA regulates tolerance toward DNA damage in a manner different from the classical SOS response.
Genome-wide expression response to CSP in the absence of LexA.
Since our results demonstrated that the CSP-inducible formation of persisters was affected by the LexA pathway, we performed a comparison of the gene expression profiles between the WT strain and its ΔlexA mutant in the presence of the CSP pheromone (see Materials and Methods for microarray experiment details). Our results showed that LexA regulated 11 genes (Table 4). Four genes were selected for further analysis. Individual deletion mutants were constructed in the WT strain, and each mutant was tested for its ability to produce CSP-induced persister cells. SMU.63 and SMU.1402, two genes known to be differentially regulated under CSP conditions (25), were first tested. SMU.1402 encodes a CRISPR-associated Csn2 protein belonging to the CRISPR-1 region of S. mutans (49). CRISPR arrays with CRISPR-associated proteins are involved in resistance to bacteriophage, and resistance specificity is determined by the insertion of CRISPR spacer-phage sequence similarity (50). Interestingly, inactivation of the csn2 gene in S. mutans completely abolished the increase in persister numbers observed for the WT strain following pretreatment with CSP (Fig. 6). S. mutans Csn2 protein showed high similarity (81%; 154/187 amino acids) to Csn2 protein from S. thermophilus. In S. thermophilus, CRISPR systems provide resistance against phages, and Csn2 protein is involved in spacer integration (50, 51). For S. mutans, work previously done by van der Ploeg using strain OMZ 381, one of a few strains sensitive to M102 phage attacking S. mutans, showed that deletion of the CRISPR-1 region is insufficient to render sensitivity to infection by the phage M102 (49). Natural phage resistance mechanisms in S. mutans have not been previously described, mainly because phages and sensitive strains are actually quite rare in S. mutans (52).
TABLE 4.
DNA microarray data obtained by comparing the expression profiles of the WT strain and ΔlexA mutant in the presence of the CSP pheromone
| Locusa | Common name and/or putative function of protein | Fold changeb in ΔlexA mutant |
|---|---|---|
| SMU.63 | Conserved membrane protein | −1.8 |
| SMU.503 | Hypothetical protein | −1.5 |
| SMU.563 | Ornithine carbamoyltransferase | −1.7 |
| SMU.663-666 | Glutamate family amino acid synthesis operon | −1.5 |
| SMU.984 | Hypothetical protein; CHAP domain-containing protein | −1.5 |
| SMU.1402 | CRISPR-associated protein | −1.5 |
| SMU.1841 | ScrA; sucrose-specific IIABC component | −2.2 |
| SMU.1907 | Hypothetical protein; bacteriocin-related genomic island | −1.7 |
Results for selected genes were ordered based on their position in the UA159 chromosome.
Differential gene expression was based on a postnormalization cutoff ± ≥1.5-fold.
FIG 6.

Role of selected LexA-regulated genes in the formation of CSP-induced persister cells. Overnight cultures of the S. mutans WT strain and its Δ63, Δcsn2, ΔscrA, and Δ984 mutants were diluted (1:100) into fresh THYE broth in the absence or presence of CSP pheromone and incubated at 37°C for 2 h before being challenged with ofloxacin. Aliquots of cells were removed at the introduction of the antibiotic and after the antibiotic treatment (h 24) to determine cell survival by spot plating onto THYE agar plates. Results are expressed as the log fold change in cell survival normalized to the non-CSP prestress value. The data are the averages and standard errors of results from three independent cultures. The average value for each strain is indicated.
The SMU.63 gene encodes a conserved membrane protein of 613 amino acid residues of unknown function. This gene is located immediately downstream of the comRS locus, involved in CSP-inducible persister formation (Fig. 1) and genetic competence (27, 28). Our results showed that the Δ63 mutant was still able to increase the number of ofloxacin-tolerant persister cells following treatment with the CSP pheromone (Fig. 6). Moreover, inactivation of SMU.63 did not alter the number of persisters under no-CSP conditions (see Fig. S1 in the supplemental material), suggesting that SMU.63 is not involved in the formation of persister cells.
The SMU.984 gene encodes a protein of 166 amino acid residues with no known function. The protein possesses a C-terminal CHAP (cysteine- and histidine-dependent aminohydrolase/peptidase) domain that functions in peptidoglycan hydrolysis. Prestressing cells with the CSP pheromone followed by ofloxacin treatment revealed that a lack of SMU.984 abolished the CSP-inducible persistence phenotype (Fig. 6). Global transcriptome analysis performed by Senadheera and colleagues showed that the SMU.984 gene was regulated by the two-component system VicRK (D. Senadheera, personal communication). In S. mutans, the VicRK system modulates stress tolerance by direct modulation of the CSP pheromone (53). Altogether, these results suggest that SMU.984 could be involved in stress tolerance by modulating the CSP-inducible persistence phenotype.
The scrA gene (SMU.1841) is part of the scr regulon composed of the three genes scrA, scrB, and scrR, coding for a sucrose-specific IIABC phosphoenolpyruvate:carbohydrate phosphate transferase system (PTS) component, a sucrose-6-phosphate hydrolase, and a sucrose operon repressor, respectively (54). The scrA gene was found to be downregulated in both transcriptome profile comparisons (CSP induced and uninduced). The ΔscrA mutant was unable to increase the number of ofloxacin-tolerant persister cells following treatment with the CSP pheromone (Fig. 6). Moreover, a ΔscrA mutant had an approximately 4-fold decrease in persister level after 24 h of ofloxacin treatment under the no-CSP condition (see Fig. S1 in the supplemental material), suggesting that ScrA also participates in the formation of persisters through a quorum-sensing-independent mechanism(s).
Using the online RSAT bioinformatic tool (http://rsat.ulb.ac.be/rsat/), we searched the promoter regions of SMU.984, SMU.1402 (csn2), and SMU.1841 (scrA) with the IR identified in the promoter region of lexA (see Fig. S2 in the supplemental material). No IR sequences were found, suggesting that these persister genes are most probably indirectly regulated by LexA.
DISCUSSION
In this study, we have identified and characterized a LexA transcriptional regulator in S. mutans. This LexA ortholog has many of the notable hallmarks of a typical LexA regulator, such as autoregulatory expression upon heat shock and DNA damage and a conserved Ala-Gly self-cleavage site. The observed full autoregulatory expression is contingent on a functional ClpP subunit, which is most likely required for the degradation of the N-terminal DNA binding domain after LexA autocleavage to allow complete derepression of its own gene expression. Furthermore, the S. mutans LexA regulator plays a role in a DNA damage response with regulation of the formation of persisters tolerant toward DNA-damaging agents, such as mitomycin C and fluoroquinolones. Indeed, a ΔlexA(pVL3) strain overexpressing the mutated and uncleavable LexA A115D protein showed a significantly decreased rate of survival of exposure to ofloxacin and mitomycin C. However, the LexA-regulated pathway in S. mutans differs greatly from the canonical SOS response seen in other bacteria and even from other streptococcal species that have been identified with an SOS-like pathway via the LexA-like HdiR regulator. In S. thermophilus and S. uberis, HdiR functions similarly to LexA in regulating an SOS-like response by directly upregulating the expression of umuC upon DNA damage to bypass DNA lesions during DNA replication (44, 45). However, the umuC gene and other genes with known or putative roles in DNA repair were not found to be regulated by LexA in S. mutans (see Table S2 in the supplemental material). In fact, a ΔumuC mutant did not affect tolerance toward or survival of DNA damage.
Most interestingly, S. mutans LexA was also identified as playing a significant role in the formation of quorum-sensing-induced multidrug-tolerant persisters, in addition to its role in forming persisters tolerant toward DNA-damaging agents. A lack of LexA completely abolished the formation of CSP-induced persisters tolerant to multiple antibiotics, suggesting that LexA influences the deterministic formation of persisters mediated by the stress-inducible alarmone. It is interesting that a lack of LexA does not greatly affect survival of exposure to or tolerance toward the antibiotic oxacillin, while under CSP conditions the lack of the LexA regulator prevents CSP-inducible persistence. This suggests that there are at least two mechanisms regulated by LexA by which S. mutans can respond to and tolerate DNA-damaging stress. One mechanism occurs under normal growth, in which LexA functions to provide an immediate response for tolerating DNA damage conditions. The other mechanism occurs through activation of the CSP-ComDE quorum-sensing pathway, most like responding to various environmental stressors. In this case, activation of LexA regulator indirectly alters the expression profile of a group of genes that are required for the formation of CSP-induced persisters tolerant not only to DNA damage but also to several classes of antibiotics.
In S. thermophilus, it was recently shown that the HdiR pathway functioned antagonistically toward its own competence development (44). Activation of the SOS-like response via HdiR led to the downregulation of competence genes and to the decrease in transformation efficiency, while a SigX-deficient strain had an increased survivability toward both mitomycin C and norfloxacin. Our results showed that the continuous activation of the LexA pathway in a LexA-deficient mutant acts negatively toward CSP-induced persisters, where the formation requires the intact competence pathway from ComE to SigX. This may suggest that competence is also negatively impacted in S. mutans, but this was not the case, as we observed that the transformation efficiency (Fig. 5) and the expression of competence genes (data not shown) were not affected in a ΔlexA mutant treated or not with exogenous CSP. This difference is most likely due to differing mechanism(s) and genes regulated by LexA in S. mutans compared to those regulated by HdiR in S. thermophilus.
Our transcriptomic analyses identified a number of genes with various cellular roles that appear to contribute toward the formation of CSP-induced persisters. A few of these identified genes are involved with metabolism, specifically amino acid biosynthesis. Although we did not test individual knockout mutants for SMU.563, encoding the ornithine carbamoyltransferase enzyme involved in arginine biosynthesis, or the genes SMU.663 to SMU.666, belonging to the glutamate family amino acid synthesis operon, it is likely that affecting amino acid biosynthesis would greatly influence persister formation, as has been recently shown in E. coli through the use of a transposon library (55).
Our study led us to identify new genes involved in the formation of quorum-sensing-induced persisters. These genes are most likely indirectly regulated through LexA. They include SMU.984, encoding a hypothetical CHAP-containing protein. CHAP domains are typically associated with lytic proteins, such as autolysins, involved in peptidoglycan hydrolysis (56). Our group had previously identified and characterized LytF-Sm, a SigX-regulated autolysin involved in cell death induced by the CSP pheromone (57). Preliminary work done in our laboratory showed that SMU.984 encodes a functional murein hydrolase but is not involved in the CSP-induced cell death pathway. As similarities go, SMU.984 may be a lytic protein that potentially functions with reduced efficiency or potency in altering the cell wall integrity, leading to CSP-inducible persister formation.
The CRISPR-associated csn2 gene was found to be important in the CSP-inducible persistence phenotype. To the best of our knowledge, this is the first time a link is proposed between a CRISPR-associated protein and persister formation. The CRISPR system is not well characterized in S. mutans, and the csn2 gene is found only in the type II-A systems. The Csn2 protein was recently characterized as forming a ring-shaped tetramer complex that binds to double-strand DNA ends with the proposed function of assisting in the integration of new spacer DNA fragments by stabilizing the DNA ends together and as a potential accessory protein recruiting DNA repair proteins (58). Perhaps increased csn2 gene expression contributes to ofloxacin tolerance by recruiting DNA repair proteins at the posttranscriptional level not detected in our microarrays. How exactly Csn2 protein plays a role in the formation of CSP-induced persisters remains to be determined.
The scrA gene, encoding a PTS sucrose-specific component, was the only gene found to be significantly differentially regulated in a ΔlexA mutant cultivated under both CSP-induced and uninduced conditions. The role of carbohydrate metabolism has been previously associated with the formation of persisters (59, 60). Based on our results, we can hypothesize that a lack of LexA derepresses a gene(s) involved in the downregulation of the scrA gene contributing to the reduced persister levels. Interestingly, inactivation of scrA led to decrease persister formation under both CSP-induced and uninduced conditions, suggesting that scrA is involved in the formation of persisters governed by both stochastic and deterministic mechanisms. ScrA is a sucrose permease that is heavily involved in sucrose uptake. However, there are other systems that contribute to uptake and/or catabolism of sucrose in S. mutans, specifically the glucosyltransferases (GtfB, GtfC, and GtfD), the fructosyltransferase (Ftf), and the multiple-sugar-metabolism pathway (61). This would likely eliminate the notion of sucrose metabolism being the mechanism by which scrA contributes to persister formation. Additionally, our persistence assays were performed in a nutrient-rich complex medium that contains glucose as the major carbohydrate source. Interestingly, we have shown from an overexpression genomic library that deletion of the sucrose operon repressor ScrR, which normally represses the expression of scrA and scrB genes in S. mutans, led to an ∼10-fold increase in persister formation (18), suggesting that a derepression of scrA may lead to an increase of persister formation. These results also suggest that there are multiple layers of regulation of scrA. Regulation of scrA could also occur from the IGR1445 intergenic region corresponding to the scrA-scrB promoter region. Unfortunately, efforts in identifying a regulatory antisense RNA in this intergenic region have been unsuccessful thus far.
It is important to note that significantly upregulated genes were expected to be detected in our comparative microarray analysis. Detection of upregulated genes would signify that these genes are most likely directly regulated by the LexA regulator in S. mutans. In contrast, all significantly differentially regulated genes were downregulated. Our results suggest two possible mechanisms by which LexA contributes to the persister phenotype in S. mutans. In the first model, LexA acts as a transcriptional activator following its autocleavage. Under these conditions, deletion of the lexA gene would cause downregulation of genes and the loss of the CSP-inducible persistence phenotype. However, this would suggest that the LexA regulator in S. mutans possesses a different transcriptional function following its autocleavage, which would be in stark contrast to the well-characterized LexA family of transcriptional regulators described for other bacteria. In the second scenario, LexA acts as a repressor inhibiting another repressor of persister genes, where LexA autocleavage leads to the indirect repression of these genes. The fact that the identified IR located in the promoter region of the S. mutans lexA gene was not found upstream of the downregulated genes identified in our arrays strongly supports this model.
It is also possible that upregulated genes were not detected because the binding affinity of LexA toward the promoter regions of potential LexA-regulated genes is heterogenous depending on the strength and conservation of the consensus LexA box motif, their relative location in the target promoter, and even the promoter strength of the target gene itself, as has been well characterized in E. coli (62). This variation in LexA binding results in a difference in the expression level, timing, and duration for different LexA-regulated genes. Some SOS-regulated genes in E. coli have low promoter activity in the absence of DNA-damaging agents (63). Furthermore, genomic analyses with E. coli have shown that LexA is capable of binding and regulating genes that do not contain a canonical LexA box motif in their promoter regions (64). All these factors make it challenging to identify genes that are directly regulated by LexA. Moreover, it is possible that LexA-regulated genes are non-protein-encoding regulatory RNAs that would not be detected in our microarrays. In this case, transcriptome analyses using high-throughput RNA sequencing (RNA-seq) of S. mutans in the presence of DNA-damaging agents would need to be performed.
The results of this study present two roles by which the LexA transcriptional regulator participates in regulating bacterial survival toward antibiotic challenge in S. mutans. One pathway involves DNA-damaging agents initiating LexA self-cleavage that indirectly leads to tolerance through the regulation of scrA. The second pathway involves the stress-inducible CSP alarmone that activates the competence regulon. Our results suggest that the lexA gene is part of the sigX regulon acting as a general stress response in streptococci. Once activated, the lexA pathway leads to the expression of a group of genes, including csn2, scrA, and SMU.984, that collectively contribute to the stress-inducible multidrug-tolerant persister state. Although CSP-inducible persister genes have been identified, their exact roles are unknown. Further experiments would need to be performed to elucidate the exact mechanisms by which each identified gene contributes toward persister formation. Nonetheless, disrupting the inducible persister pathway at any point between the components of the bacterial quorum-sensing system to the effector genes to prevent stress-inducible persister formation could lead to innovative drug designs and strategies.
Supplementary Material
ACKNOWLEDGMENTS
We thank Delphine Dufour for careful reading of the manuscript. We thank Indranil Biswas for the pIB107 and pIB184 plasmids.
This study was supported by Canadian Institutes of Health Research (CIHR) grant MOP-93555 to C.M.L. and Natural Sciences and Engineering Research Council of Canada (NSERC) grant RGPIN 355968 to C.M.L. C.M.L. is a recipient of a Canada Research Chair.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.02496-14.
REFERENCES
- 1.Alekshun MN, Levy SB. 2007. Molecular mechanisms of antibacterial resistance. Cell 128:1037–1050. doi: 10.1016/j.cell.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 2.Levin BR, Rozen DE. 2006. Non-inherited antibiotic resistance. Nat Rev Microbiol 4:556–562. doi: 10.1038/nrmicro1445. [DOI] [PubMed] [Google Scholar]
- 3.Wiuff C, Zappala RM, Regoes RR, Garner KN, Baquero F, Levin BR. 2005. Phenotypic tolerance: antibiotic enrichment of noninherited resistance in bacterial populations. Antimicrob Agents Chemother 49:1483–1494. doi: 10.1128/AAC.49.4.1483-1494.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dufour D, Leung V, Lévesque CM. 2012. Bacterial biofilm: structure, function, and antimicrobial resistance. Endod Topics 22:2–16. doi: 10.1111/j.1601-1546.2012.00277.x. [DOI] [Google Scholar]
- 5.Lewis K. 2008. Multidrug tolerance of biofilms and persister cells. Curr Top Microbiol Immunol 322:107–131. doi: 10.1007/978-3-540-75418-3_6. [DOI] [PubMed] [Google Scholar]
- 6.Lewis K. 2010. Persister cells. Annu Rev Microbiol 64:357–372. doi: 10.1146/annurev.micro.112408.134306. [DOI] [PubMed] [Google Scholar]
- 7.Lewis K. 2012. Persister cells: molecular mechanisms related to antibiotic tolerance. Handb Exp Pharmacol 211:121–133. doi: 10.1007/978-3-642-28951-4_8. [DOI] [PubMed] [Google Scholar]
- 8.Jayaraman R. 2008. Bacterial persistence: some new insights into an old phenomenon. J Biosci 33:795–805. doi: 10.1007/s12038-008-0099-3. [DOI] [PubMed] [Google Scholar]
- 9.Veening JW, Smits WK, Kuipers OP. 2008. Bistability, epigenetics, and bet-hedging in bacteria. Annu Rev Microbiol 62:193–201. doi: 10.1146/annurev.micro.62.081307.163002. [DOI] [PubMed] [Google Scholar]
- 10.Yamaguchi Y, Park JH, Inouye M. 2011. Toxin-antitoxin systems in bacteria and archae. Annu Rev Genet 45:61–79. doi: 10.1146/annurev-genet-110410-132412. [DOI] [PubMed] [Google Scholar]
- 11.Gerdes K, Maisonneuve E. 2012. Bacterial persistence and toxin-antitoxin loci. Annu Rev Microbiol 66:103–123. doi: 10.1146/annurev-micro-092611-150159. [DOI] [PubMed] [Google Scholar]
- 12.Schuster CF, Bertram R. 2013. Toxin-antitoxin systems are ubiquitous and versatile modulators of prokaryotic fate. FEMS Microbiol Lett 340:73–85. doi: 10.1111/1574-6968.12074. [DOI] [PubMed] [Google Scholar]
- 13.Van Melderen L, Saavedra de Bast M. 2009. Bacterial toxin-antitoxin systems: more than selfish entities? PLoS Genet 5:e1000437. doi: 10.1371/journal.pgen.1000437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kint CI, Verstraeten N, Fauvart M, Michiels J. 2012. New-found fundamentals of bacterial persistence. Trends Microbiol 20:577–585. doi: 10.1016/j.tim.2012.08.009. [DOI] [PubMed] [Google Scholar]
- 15.Maisonneuve E, Shakespeare LJ, Jorgensen MG, Gerdes K. 2011. Bacterial persistence by RNA endonucleases. Proc Natl Acad Sci U S A 108:13206–13211. doi: 10.1073/pnas.1100186108. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 16.Dörr T, Vulić M, Lewis K. 2010. Ciprofloxacin causes persister formation by inducing the TisB toxin in Escherichia coli. PLoS Biol 8:e1000317. doi: 10.1371/journal.pbio.1000317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vega NM, Allison KR, Khalil AS, Collins JJ. 2012. Signaling-mediated bacterial persister formation. Nat Chem Biol 8:431–433. doi: 10.1038/nchembio.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leung V, Lévesque CM. 2012. A stress-inducible quorum-sensing peptide mediates the formation of persister cells with noninherited multidrug tolerance. J Bacteriol 194:2265–2274. doi: 10.1128/JB.06707-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Loesche WJ. 1986. Role of Streptococcus mutans in tooth decay. Microbiol Rev 50:353–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marsh PD. 2006. Dental plaque as a biofilm and a microbial community: implications for health and disease. BMC Oral Health 6:S14. doi: 10.1186/1472-6831-6-S1-S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lemos JA, Burne RA. 2008. A model of efficiency: stress tolerance by Streptococcus mutans. Microbiology 154:3247–3255. doi: 10.1099/mic.0.2008/023770-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith EG, Spatafora GA. 2012. Gene regulation in S. mutans: complex control in a complex environment. J Dent Res 91:133–141. doi: 10.1177/0022034511415415. [DOI] [PubMed] [Google Scholar]
- 23.Oggioni MR, Morrison DA. 2008. Cooperative regulation of competence development in Streptococcus pneumoniae: cell-to-cell signaling via a peptide pheromone and an alternative sigma factor, p 345–362. In Winans SC, Bassler BL (ed), Chemical communication among bacteria. ASM Press, Washington, DC. [Google Scholar]
- 24.Kreth J, Merritt J, Shi W, Qi F. 2005. Co-ordinated bacteriocin production and competence development: a possible mechanism for taking up DNA from neighbouring species. Mol Microbiol 57:392–404. doi: 10.1111/j.1365-2958.2005.04695.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perry JA, Jones MB, Peterson SN, Cvitkovich DG, Lévesque CM. 2009. Peptide alarmone signalling triggers an auto-active bacteriocin necessary for genetic competence. Mol Microbiol 72:905–917. doi: 10.1111/j.1365-2958.2009.06693.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dufour D, Cordova M, Cvitkovitch DG, Lévesque CM. 2011. Regulation of the competence pathway as a novel role associated with a streptococcal bacteriocin. J Bacteriol 193:6552–6559. doi: 10.1128/JB.05968-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mashburn-Warren L, Morrison DA, Federle MJ. 2010. A novel double-tryptophan peptide pheromone controls competence in Streptococcus spp. via an Rgg regulator. Mol Microbiol 78:589–606. doi: 10.1111/j.1365-2958.2010.07361.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fontaine L, Goffin P, Dubout H, Delplace B, Baulard A, Lecat-Guillet N, Chambellon E, Gardan P, Hols P. 2013. Mechanism of competence activation by the ComRS signalling system in streptococci. Mol Microbiol 87:1113–1132. doi: 10.1111/mmi.12157. [DOI] [PubMed] [Google Scholar]
- 29.Miller RA, Britigan BE. 1997. Role of oxidants in microbial pathophysiology. Clin Microbiol Rev 10:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cooke MS, Evans MD, Dizdaroglu M, Lunec J. 2003. Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J 17:1195–1214. doi: 10.1096/fj.02-0752rev. [DOI] [PubMed] [Google Scholar]
- 31.Gonzalez K, Faustoferri RC, Quivey RG Jr. 2012. Role of DNA base excision repair in the mutability and virulence of Streptococcus mutans. Mol Microbiol 85:361–377. doi: 10.1111/j.1365-2958.2012.08116.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Claverys JP, Prudhomme M, Martin B. 2006. Induction of competence regulon as a general response to stress in Gram-positive bacteria. Annu Rev Microbiol 60:451–475. doi: 10.1146/annurev.micro.60.080805.142139. [DOI] [PubMed] [Google Scholar]
- 33.Lau PC, Sung CK, Lee JH, Morrison DA, Cvitkovitch DG. 2002. PCR ligation mutagenesis in transformable streptococci: application and efficiency. J Microbiol Methods 49:193–205. doi: 10.1016/S0167-7012(01)00369-4. [DOI] [PubMed] [Google Scholar]
- 34.Suntharalingam P, Senadheera MD, Mair RW, Lévesque CM, Cvitkovitch DG. 2009. The LiaFSR system regulates the cell envelope stress response in Streptococcus mutans. J Bacteriol 191:2973–2984. doi: 10.1128/JB.01563-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Biswas I, Jha JK, Fromm N. 2008. Shuttle expression plasmids for genetic studies in Streptococcus mutans. Microbiology 154:2275–2282. doi: 10.1099/mic.0.2008/019265-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ajdić D, Pham VT. 2007. Global transcriptional analysis of Streptococcus mutans sugar transporters using microarrays. J Bacteriol 189:5049–5059. doi: 10.1128/JB.00338-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dufour D, Lévesque CM. 2013. Bacterial behaviors associated with the quorum-sensing peptide pheromone (‘alarmone') in streptococci. Future Microbiol 8:593–605. doi: 10.2217/fmb.13.23. [DOI] [PubMed] [Google Scholar]
- 38.Lee MS, Morrison DA. 1999. Identification of a new regulator in Streptococcus pneumoniae linking quorum sensing to competence for genetic transformation. J Bacteriol 181:5004–5016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van der Veen S, Hain T, Wonters JA, Hossain H, de Vos WM, Abee T, Chakraborty T, Wells-Bennik MH. 2007. The heat-shock response of Listeria monocytogenes comprises genes involved in heat shock, cell division, cell wall synthesis, and the SOS response. Microbiology 153:3593–3607. doi: 10.1099/mic.0.2007/006361-0. [DOI] [PubMed] [Google Scholar]
- 40.Savijoki K, Ingmer H, Frees D, Vogensen FK, Palva A, Varmanen P. 2003. Heat and DNA damage induction of the LexA-like regulator HdiR from Lactococcus lactis is mediated by RecA and ClpP. Mol Microbiol 50:609–621. doi: 10.1046/j.1365-2958.2003.03713.x. [DOI] [PubMed] [Google Scholar]
- 41.Little JW. 1984. Autodigestion of lexA and phage lambda repressors. Proc Natl Acad Sci U S A 81:1375–1379. doi: 10.1073/pnas.81.5.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Neher SB, Flynn JM, Sauer RT, Baker TA. 2003. Latent ClpX-recognition signals ensure LexA destruction after DNA damage. Genes Dev 17:1084–1089. doi: 10.1101/gad.1078003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Drlica K, Zhao X. 1997. DNA gyrase, topoisomerase IV, and the 4-quinolones. Microbiol Mol Biol Rev 61:377–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Boutry C, Delplace B, Clippe A, Fontaine L, Hols P. 2013. SOS response activation and competence development are antagonistic mechanisms in Streptococcus thermophilus. J Bacteriol 195:696–707. doi: 10.1128/JB.01605-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Varhimo E, Savijoki K, Jalava J, Kuipers OP, Varmanen P. 2007. Identification of a noval streptococcal gene cassette mediating SOS mutagenesis in Streptococcus uberis. J Bacteriol 189:5210–5222. doi: 10.1128/JB.00473-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wood TK, Knabel SJ, Kwan BW. 2013. Bacterial persister cell formation and dormancy. Appl Environ Microbiol 79:7116–7121. doi: 10.1128/AEM.02636-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kuzminov A. 1999. Recombinational repair of DNA damage in Escherichia coli and bacteriophage lambda. Microbiol Mol Biol Rev 63:751–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sato Y, Poy F, Jacobson GR, Kuramitsu HK. 1989. Characterization and sequence analysis of the scrA gene encoding enzyme IIScr of the Streptococcus mutans phosphoenolpyruvate-dependent sucrose phosphotransferase system. J Bacteriol 171:263–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.van der Ploeg JR. 2009. Analysis of CRISPR in Streptococcus mutans suggests frequent occurrence of acquired immunity against infection by M102-like bacteriophages. Microbiology 155:1966–1976. doi: 10.1099/mic.0.027508-0. [DOI] [PubMed] [Google Scholar]
- 50.Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, Horvath P. 2007. CRISPR provides acquired resistance against viruses in prokaryotes. Science 315:1709–1712. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- 51.Deveau H, Barrangou R, Garneau JE, Labonte J, Fremaux C, Boyaval P, Romero DA, Horvath P, Moineau S. 2008. Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J Bacteriol 190:1390–1400. doi: 10.1128/JB.01412-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Maruyama F, Kobata M, Kurokawa K, Nishida K, Sakurai A, Nakano K, Nomura R, Kawabata S, Ooshima T, Nakai K, Hattori M, Hamada S, Nakagawa I. 2009. Comparative genomic analyses of Streptococcus mutans provide insight into chromosomal shuffling and species-specific content. BMC Genomics 10:358. doi: 10.1186/1471-2164-10-358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Senadheera DB, Cordova M, Ayala EA, Chávez de Paz LE, Singh K, Downey JS, Svensäter G, Goodman SD, Cvitkovitch DG. 2012. Regulation of bacteriocin production and cell death by the VicRK signaling system in Streptococcus mutans. J Bacteriol 194:1307–1316. doi: 10.1128/JB.06071-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang B, Kuramitsu HK. 2003. Control of enzyme IIscr and sucrose-6-phosphate hydrolase activities in Streptococcus mutans by transcriptional repressor ScrR binding to the cis-active determinants of the scr regulon. J Bacteriol 185:5791–5799. doi: 10.1128/JB.185.19.5791-5799.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bernier SP, Lebeaux D, DeFrancesco AS, Valomon A, Soubigou G, Coppee JY, Ghigo JM, Beloin C. 2013. Starvation, together with the SOS response, mediates high biofilm-specific tolerance to the fluoroquinolone ofloxacin. PLoS Genet 9:e1003144. doi: 10.1371/journal.pgen.1003144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vollmer W, Joris B, Charlier P, Foster S. 2008. Bacterial peptidoglycan (murein) hydrolases. FEMS Microbiol Rev 32:259–286. doi: 10.1111/j.1574-6976.2007.00099.x. [DOI] [PubMed] [Google Scholar]
- 57.Dufour D, Lévesque CM. 2013. Cell death of Streptococcus mutans induced by quorum-sensing peptide occurs via a conserved streptococcal autolysin. J Bacteriol 195:105–114. doi: 10.1128/JB.00926-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Arslan Z, Wurm R, Brener O, Ellinger P, Nagel-Steger L, Oesterhelt F, Schmitt L, Willbold D, Wagner R, Gohlke H, Smits SH, Pul U. 2013. Double-strand DNA end-binding and sliding of the toroidal CRISPR-associated protein Csn2. Nucleic Acids Res 41:6347–6359. doi: 10.1093/nar/gkt315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Spoering AL, Vulic M, Lewis K. 2006. GlpD and PlsB participate in persister cell formation in Escherichia coli. J Bacteriol 188:5136–5144. doi: 10.1128/JB.00369-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Girgis HS, Harris K, Tavazoie S. 2012. Large mutational target size for rapid emergence of bacterial persistence. Proc Natl Acad Sci U S A 109:12740–12745. doi: 10.1073/pnas.1205124109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zeng L, Choi SC, Danko CG, Siepel A, Stanhope MJ, Burne RA. 2013. Gene regulation by CcpA and catabolite repression explored by RNA-Seq in Streptococcus mutans. PLoS One 8:e60465. doi: 10.1371/journal.pone.0060465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Butala M, Zgur-Bertok Busby SJW. 2009. The bacterial LexA transcriptional repressor. Cell Mol Life Sci 66:82–93. doi: 10.1007/s00018-008-8378-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kamenšek S, Podlesek Z, Gillor O, Zgur-Bertok D. 2010. Genes regulated by the Escherichia coli SOS repressor LexA exhibit heterogenous expression. BMC Microbiol 10:283. doi: 10.1186/1471-2180-10-283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wade JT, Reppas NB, Church GM, Struhl K. 2005. Genomic analysis of LexA binding reveals the permissive nature of the Escherichia coli genome and identifies unconventional target sites. Genes Dev 19:2619–2630. doi: 10.1101/gad.1355605. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.