

Abstract
Bacterial strain variation exists in natural populations of bacteria and can be generated experimentally through directed or random mutation. The advent of rapid and cost-efficient whole-genome sequencing has facilitated strain-level genotyping. Even with modern tools, however, it often remains a challenge to map specific traits to individual genetic loci, especially for traits that cannot be selected under culture conditions (e.g., colonization level or pathogenicity). Using a combination of classical and modern approaches, we analyzed strain-level variation in Vibrio fischeri and identified the basis by which some strains lack the ability to utilize glycerol as a carbon source. We proceeded to reconstruct the lineage of the commonly used V. fischeri laboratory strains. Compared to the wild-type ES114 strain, we identify in ES114-L a 9.9-kb deletion with endpoints in tadB2 and glpF; restoration of the missing portion of glpF restores the wild-type phenotype. The widely used strains ESR1, JRM100, and JRM200 contain the same deletion, and ES114-L is likely a previously unrecognized intermediate strain in the construction of many ES114 derivatives. ES114-L does not exhibit a defect in competitive squid colonization but ESR1 does, demonstrating that glycerol utilization is not required for early squid colonization. Our genetic mapping approach capitalizes on the recently discovered chitin-based transformation pathway, which is conserved in the Vibrionaceae; therefore, the specific approach used is likely to be useful for mapping genetic traits in other Vibrio species.
INTRODUCTION
Identifying relevant differences in bacterial strains is fundamental to determining the genetic basis of microbial phenotypes. In many cases, the number of polymorphisms between strains is so high that elucidating which locus or loci contribute to specific phenotypes cannot be achieved simply by determining the genome sequence of the isolates. This challenge is especially pronounced in identifying loci that contribute to colonization and/or pathogenicity phenotypes. The study of genomic islands has made it clear that the acquisition of large regions of DNA can profoundly influence a bacterium's ability to engage with a eukaryotic host (1, 2). Recently, it has become increasingly apparent that defined genetic changes in bacteria at individual loci, single genes, or even nucleotide changes have led to dramatic effects in the evolution of colonizing bacteria. As some examples, the acquisition of the nil locus in Xenorhabdus nematophila contributed to the species-specific association with the worm host Steinernema carpocapsae, inactivation of the RscA biofilm regulator was critical in the evolution of Yersinia pestis from Yersinia pseudotuberculosis, and acquisition of the biofilm regulation of RscS facilitated colonization of north Pacific squid by Vibrio fischeri (3–8).
In most cases, identification of factors that contribute to host colonization specificity has relied first on identifying the factor as being necessary for host colonization by standard genetic methods and then subsequent characterization of its role in host specificity by testing candidate factors to determine whether they are sufficient to confer colonization ability on a naive strain. We sought a direct approach to identify colonization specificity factors in V. fischeri. Instead of first needing to conduct a genetic screen in the context of host colonization, we reasoned that if we could mobilize DNA from a colonization-competent donor strain into a strain that is unable to colonize, then we could identify those regions of the donor that confer a gain-of-function phenotype in the context of the recipient genome. As a proof of concept to establish such a genetic system in V. fischeri, we took advantage of strain ES114-L, which cannot use glycerol and which presented a simplified case in which to develop genetic mapping in V. fischeri.
The Gram-negative bacterium V. fischeri is an important model system for studies of symbiosis, colonization, biofilm formation, quorum sensing, and luminescence. V. fischeri is the light-organ symbiont of Euprymna scolopes squid, in which bacterially produced luminescence is proposed to camouflage the silhouette generated by the nocturnally foraging animal (9, 10). Studies of V. fischeri have focused on a natural squid isolate, ES114, to understand the basis by which symbiotic bacteria colonize animal hosts (11). The use of this system has presented challenges for molecular genetics, notably in counterselection against E. coli donor strains in the conjugation of mutagenic plasmids into recipient V. fischeri. One solution that was employed in approximately 40 studies was a spontaneous rifampin-resistant V. fischeri recipient, termed ESR1 (12). It was subsequently shown that this strain exhibited a defect in a competitive squid colonization assay against the wild-type ES114 strain (13), and it has been used only minimally since that time. In addition to the wild-type strain ES114 and the rifampin-resistant derivative ESR1, recent work has identified a cryptic strain, ES114-L (14) (M. J. Mandel, unpublished data). ES114-L is thought to be a laboratory derivative of ES114, yet the former cannot utilize glycerol as the sole carbon source; the genetic basis of this deficiency is unknown.
Genetic mapping is a decades-old approach that has been invaluable to identifying the basis of specific phenotypic differences in strains. Griffith's experiment involved transformation in Streptococcus pneumoniae, and the development of prokaryotic genetics relied on experiments from Lederberg and coworkers that employed linkage analysis in Bacillus subtilis (15–17). In Gram-negative bacteria, linkage is commonly determined using generalized transducing phages that package a defined length of host DNA, yet in Gram-positive bacteria transformation still is commonly employed to introduce marked donor DNA for genetic mapping (18–20). Currently there is no generalized transduction system in V. fischeri, and as such it resembles many environmental and host-associated bacteria for which there exists only genome information and a small toolbox of techniques. The last decade has revealed that Vibrio spp. are naturally transformable in the presence of chitin, and that large fragments of DNA (>20 kb) can be exchanged in nature or in the laboratory upon induction of the transformation regulatory cascade (21–25). The availability of transformation in V. fischeri, coupled with other genetic tools under development in the laboratory, presented an opportunity to develop a generalized system for genetic mapping in V. fischeri. Accordingly, the goals of this work were 3-fold: (i) to improve genetic tools in V. fischeri, (ii) to apply those tools to map the functional basis that distinguishes V. fischeri strains, and (iii) to establish a system to use genetics to map strain-level traits that then can be applied in the future to identify factors that influence host colonization specificity. In this report, our results provide a proof of concept for a genetic approach to the study of host colonization specificity factors. Furthermore, they have revealed an unexpected unification of the lineage of V. fischeri laboratory strains and clarified the requirement for glycerol utilization during juvenile squid colonization.
MATERIALS AND METHODS
Bacterial strains, plasmids, and oligonucleotides.
V. fischeri and Escherichia coli strains used in this study are listed in Table 1. Standard microbial techniques were used to mobilize plasmids into V. fischeri strains and to integrate suicide vectors into the chromosome (26). DNA oligonucleotide primer sequences are listed in Table 2. Primers were synthesized by Integrated DNA Technologies (Coralville, IA).
TABLE 1.
Strains and plasmids
| Strain or plasmid | Genotype/description | Source or reference(s) |
|---|---|---|
| Vibrio fischeri | ||
| MJM1100 (ES114) | Wild-type Euprymna scolopes light-organ isolate | 11, 35 |
| MJM1101 (ES114 alt) | Wild-type Euprymna scolopes light-organ isolate (alternate isolate) | |
| MJM1108 | MJM1100/pVSV208 | This work |
| MJM1538 | MJM1100/pLostfoX | This work |
| MJM1000 (ES114-L) | ES114 Δ(tadB2′-′glpF) gltA VF_B0014-5 VNTRa | 14 |
| MJM1007 | MJM1000/pVSV102 | This work |
| MJM1683 | MJM1000/pLostfoX | This work |
| MJM1071 (ESR1) | ES114-L fadB lptB rpoB VF_B0014-21 VNTRa | 12 |
| MJM1002 (JRM100) | ES114-L Tn7::erm | 13 |
| MJM1003 (JRM200) | ES114-L Tn7::cam | 13 |
| MJM1125 (SR5) | Wild-type Sepiola robusta light-organ isolate | 46, 47 |
| MJM1652 | MJM1100 glpF::Tnerm | This work |
| MJM1687 | MJM1100 glpK::Tnerm | This work |
| MJM1700 | MJM1100 iucA::Tnerm | This work |
| MJM2129 | MJM1000 glpF::pM2130 | This work |
| MJM2131 | Chimera 1-1; MJM1000 transformed with MJM1100 tadA2::Tnerm | This work |
| MJM2132 | Chimera 2-4; MJM1000 transformed with intergenic MJM1100 (VF_A0234-glpF)::Tnerm | This work |
| MJM2133 | Chimera 2-9; MJM1000 transformed with MJM1100 tadB2::Tnerm | This work |
| MJM2134 | Chimera 3-1; MJM1000 transformed with MJM1100 glpC::Tnerm | This work |
| MJM2135 | Chimera 3-2; MJM1000 transformed with MJM1100 rscS::Tnerm | This work |
| MJM2136 | Chimera 3-4; MJM1000 transformed with MJM1100 VF_A0219::Tnerm | This work |
| MJM2137 | Chimera 3-5; MJM1000 transformed with MJM1100 VF_A0233::Tnerm | This work |
| Escherichia coli | ||
| MJM537 (DH5α λpir) | Cloning host strain | Laboratory stock |
| MJM661 (S17-1 λpir) | Conjugation donor strain | 48 |
| MJM1538 | DH5α/pLostfoX | 24 |
| MJM534 | CC118 λpir/pEVS122 | 49 |
| MJM2130 | MJM537/pM2130 | This work |
| MJM684 | MJM661/pMarVF1 | This work |
| Plasmids | ||
| pLostfoX | Arabinose-inducible tfoX to facilitate transformation; oriRColE1 Camr | 24 |
| pEVS122 | Suicide vector in V. fischeri; oriRR6Kɣ Ermr | 26 |
| pM2130 | pEVS122::glpF+ (including promoter); oriRR6Kɣ Ermr | This work |
| pMarVF1 | V. fischeri mariner delivery vector (Ampr backbone, Ermr transposon) | 28 |
| pVSV102 | Constitutive green fluorescent protein; oriRpES213 Kanr | 50 |
| pVSV208 | Constitutive red fluorescent protein; oriRpES213 Camr | 50 |
Genotype determined in this study.
TABLE 2.
DNA oligonucleotide primers
| Name | Sequence (5′–3′) | Note(s)a |
|---|---|---|
| MJM-615F | GATCAAGAGCATAAAGAAGCTGAA | Strain verification (Rxn 1) |
| MJM-616R | TCGCTTTTCTTGTCGTACTACCTA | Strain verification (Rxn 1) |
| MJM-617F | AATCACATCGTGATGATACAAACC | Strain verification (Rxn 2) |
| MJM-618R | ATCACAATATCTTCTCGGCTGAA | Strain verification (Rxn 2) |
| MJM-619F | ATCGGTAAATACCATGATAAGATCG | Strain verification (Rxn 3) |
| MJM-620R | AGCAAGCAGAAAAAGCTAATATCAA | Strain verification (Rxn 3) |
| MJM-621F | GCACTGAAATTAGAAGCCATACCT | Strain verification (Rxn 4) |
| MJM-622R | ATGCCTGTAATGGACGGTATTTTA | Strain verification (Rxn 4) |
| MJM-623F | AAGTTTGATGAGGCGATATGATTT | Strain verification (Rxn 5), sequencing |
| MJM-624R | ACCACCAATAACAGCGATTAAAATA | Strain verification (Rxn 5), sequencing |
| MJM127 | ACAAGCATAAAGCTTGCTCAATCAATCACC | Verification of transposon transformation, Tn(pEVS170) anchored |
| glpFTn | CAACAGCAAAAGCACCAAGTATCG | Verification of glpF::Tnerm transformation |
| MCG-17F | GCGGTACCGATAGCATATTTAACTCGGCCTTTTA | pM2130 cloning |
| MCG-18R | GCGGTACCATTTAATTGTAGGGTGTTATTTGTTAGA | pM2130 cloning |
| MJM-633F | GTTTAATGCAACGATCGGTATTAG | pM2130 integration confirmation |
| M13 Rev (−48) | AGCGGATAACAATTTCACACAGG | pM2130 integration confirmation |
| Arb1 | GGCCACGCGTCGACTAGTACNNNNNNNNNNGATAT | Arbitrarily primed PCR |
| MJM-440 | TCAACACACTCTTAAGTTTGCTTC | Arbitrarily primed PCR |
| Arb2 | GGCCACGCGTCGACTAGTAC | Arbitrarily primed PCR |
| MJM-477 | TTCCATAACTTCTTTTACGTTTCC | Arbitrarily primed PCR |
| gltA-F | ATAACAGACTTCAAGGTAATCTGCATT | PCR, sequencing |
| gltA-R | TGTGAAACATCTTATTACAAACCCATA | PCR, sequencing |
| fadB-F | AGTCATTGATGACAATAGGTGTTTTTC | PCR, sequencing |
| fadB-R | TAATAAACAATTAGAACGAGGACGTTT | PCR, sequencing |
| lptB-F | AGCGATACCTTTTGTACATTACAACTT | PCR, sequencing |
| lptB-R | AAAATTGTCGGTATTTGACAATATCAT | PCR, sequencing |
| rpoB-F | GATAGTGAGTTAATCAGACCGATGTTT | PCR, sequencing |
| rpoB-R | TAACGGTATTGGTGAAGTCGATGATA | PCR, sequencing |
| VF_B0014-F | AATAAAGAGTTGATGTACCTTGTGGAT | PCR, sequencing |
| VF_B0014-R | TGGTGCATTATACATTTATTTTTAGCA | PCR, sequencing |
Rxn, reaction.
Bacterial growth media.
V. fischeri strains were grown at 25°C in Luria-Bertani salt (LBS) medium (per liter, 10 g Bacto tryptone, 5 g yeast extract, 20 g NaCl, 50 ml 1 M Tris buffer, pH 7.5, in distilled water), in saltwater tryptone (SWT) medium (11), or in a Tris minimal medium (MM) (27) with one or more of the following carbon and/or nitrogen sources: 0.2% N-acetylglucosamine (GlcNAc), 0.2% glycerol, 0.2% glucose, 0.2% galactose, 0.2% mannitol, and 0.1% NH4Cl. E. coli strains, used for cloning and conjugation, were grown at 37°C in Luria-Bertani (LB) medium (18) or brain heart infusion (BHI) medium (Sigma-Aldrich). When appropriate, antibiotics or supplements were added to media at the following concentrations: erythromycin, 5 μg/ml for V. fischeri in rich medium, 15 μg/ml for V. fischeri in Tris MM containing glycerol and NH4Cl, and 150 μg/ml for E. coli; kanamycin, 100 μg/ml for V. fischeri and 50 μg/ml for E. coli; chloramphenicol, 5 μg/ml for V. fischeri in rich medium, 2.5 μg/ml for V. fischeri in minimal medium, and 25 μg/ml for E. coli; carbenicillin, 100 μg/ml for E. coli; rifampin, 100 μg/ml for V. fischeri. Growth media were solidified with 1.5% agar on 100-mm diameter plates as needed.
DNA isolation and PCR.
Genomic DNA was isolated from bacterial cells using the DNeasy blood and tissue kit Gram-negative cell protocol (Qiagen). Plasmid DNA was isolated using the Wizard plus midiprep kit (Promega). Cloning and amplification was conducted with the Pfx50 proofreading polymerase (Invitrogen). PCR to examine VF_B0014 was conducted with Pfx50; another diagnostic PCR to confirm transposon insertions was conducted with GoTaq (Promega). Sanger DNA sequencing was conducted at the Northwestern University Center for Genetic Medicine. Design and analysis of cloning constructs and analysis of sequence results were conducted with DNASTAR Lasergene software.
Transformation under tfoX induction.
An overnight LBS-chloramphenicol culture of the recipient strain harboring the pLostfoX plasmid was subcultured 1:100 into 3 ml Tris MM–N-acetylglucosamine containing 2.5 μg/ml chloramphenicol. After 14 h of growth with aeration, the recipient was subcultured 1:50 into 20 ml of Tris MM–N-acetylglucosamine with chloramphenicol and grown with aeration to an optical density at 600 nm (OD600) of 0.25 to 0.30. Five hundred microliters of prepared recipient was incubated with 2.4 μg genomic DNA from a library of MJM1100::Tnerm (28) (genomic DNA was prepared with the Qiagen DNeasy kit). Following a brief vortex, the samples were incubated statically at room temperature for 30 min. LBS (1 ml) was added and the samples then recovered for either 1 h (quantification of cotransformation frequencies) or overnight (strain construction and initial chimera generation). One hundred microliters of each sample was plated onto selective medium (typically LBS-erythromycin to identify transformants). In all cases, control samples that did not have added DNA did not yield any transformant colonies. Candidate colonies were restreaked on the selective medium and then patched onto medium to determine if the transformation plasmid was retained (LBS-chloramphenicol). Strains that retained the selectable marker but did not retain the transformation plasmid (Ermr Cams) were retained; when relevant, a PCR assay was conducted to ensure that they incorporated the marker of interest by amplifying from the transposon to the flanking chromosome in the recipient strain (negative control) and in the resulting transformant.
Transformation additionally was used to move transposon insertions from mutant collections into clean backgrounds in a manner similar to that described above. The recipient strain was MJM1100/pLostfoX (MJM1683). After transformation, loss of the pLostfoX plasmid was confirmed by ensuring that the resulting strain is sensitive to chloramphenicol. The glpK::Tnerm allele was transformed from MB08328 (29). The glpF::Tnerm and iucA::Tnerm alleles were transformed from VFS17B11 and VFS14G09, which were kindly provided by C. Whistler.
Identification of transposon insertion junctions.
Arbitrarily primed PCR-based insertion site mapping was conducted on the strains to identify the exact transposon junction site in each case (30, 31). PCR primers for round 1 (Arb1/MJM-440), round 2 (Arb2/MJM-477), and DNA sequencing (MJM-477) are listed in Table 2. The resulting sequence reads were 121 bp of transposon DNA and then the flanking chromosomal DNA. The orientation of the read is in the opposite direction relative to the erm open reading frame.
Whole-genome sequencing.
Paired-end Illumina (300-bp inserts) libraries of ES114, ES114-L, and ESR1 were constructed and sequenced to depths of approximately 400-fold on the Illumina HiSeq 2000, 2000, and 2500, respectively, at the Tufts University Core Facility for Genomics. Assembly was conducted with Seqman NGen 3.0 (DNASTAR, Madison, WI): de novo assembly of ES114 was conducted, and then the reference-based assembly of each of ES114-L and ESR1 were compared to both the published ES114 sequence and the de novo Illumina assembly with Seqman NGen 3.0. Single-nucleotide polymorphisms (SNPs) were called in Seqman, and sites for which the “P not ref” was 100% were analyzed further. Indels were analyzed manually. Mutations were confirmed by Sanger sequencing of PCR products.
Repair of the glpF 5′ end and promoter.
PCR was performed on MJM1100 genomic DNA with the primers MCG-17F and MCG-18R. These primers amplified the entire glpF (VF_A0235) open reading frame in addition to 323 bp upstream and 46 bp downstream, adding KpnI restriction sites on both primers. After digestion with KpnI, the PCR product was introduced into the KpnI site of pEVS122 (32) to generate plasmid pM2130. The sequence of the insert was confirmed by Sanger sequencing. Plasmid pM2130 was introduced into ES114-L by conjugation from E. coli and selection for erythromycin resistance. PCR amplification to confirm integration at the glpF locus in strain MJM2129 was conducted using GoTaq (Promega, Madison, WI) with the manufacturer's recommended protocol and the primers MJM-633F and M13 Rev (−48).
Squid colonization.
Newly hatched squid were colonized by exposure to a total of approximately 3,000 CFU/ml of the indicated strains in 40 ml filter-sterilized Instant Ocean (FSIO) for 3 h. Squid then were transferred to 40 ml uninoculated FSIO for an additional 45 h (water was changed at 24 h postinoculation), at which point they were euthanized and surface sterilized by storage at −80°C. Individual squid then were homogenized, and each homogenate was diluted and plated for colony counts on LBS agar using standard methods (33). One hundred colonies from each squid then were patched onto minimal medium-glycerol and minimal medium-GlcNAc plates to determine the ratio of glycerol-positive CFU (ES114) to glycerol-negative CFU (ES114-L or ESR1). The competitive index (CI) for each individual squid is calculated as log10[(homogenate ES114-L CFU/ES114 CFU)/(inoculum ES114-L CFU/ES114 CFU)]; it was calculated similarly for ESR1 in place of ES114-L. Single-sample t tests (two tailed) were conducted to identify significant deviations from neutral for each strain (GraphPad Prism).
Survival assay on SWT agar.
An overnight LBS culture of MJM1100 or LBS-chloramphenicol culture of MJM1007 (MJM1000/pVSV208) was subcultured into LBS medium (37.5 μl into 3 ml) and grown at 25°C for 1 h with aeration. Approximately 25 μl of bacteria was introduced into 40 ml FSIO, and 50 μl was plated onto SWT agar at 25°C. The resulting plates contained 2,000 to 4,000 CFU/plate, a density that ensured that colonies were in close proximity to each other, yet distinct colonies could be assessed for 76 h. MJM1100 alone, MJM1007 alone, and a mixture of MJM1100 (90%) and MJM1007 (10%) were plated. To assess viability at each time point, strains were identified by epifluorescence microscopy on a Nikon Eclipse 90i with tetramethyl rhodamine isocyanate (TRITC) filters. Individual colonies then were patched onto LBS agar and viability was assessed after incubation at 25°C for 48 h. Replicates were conducted as follows. For each single strain, 4 colonies were assessed per plate per time point from 2 to 6 separate groups of 3 plates. For mixed-strain platings, all colonies assessed were from instances in which the rare MJM1007 colony was adjacent to and/or surrounded by MJM1100 colonies. Adjacent pairs of MJM1007 and MJM1100 were selected in every case to control for spatial variation on the plate. Two such pairs were examined from each plate per time point from 3 to 8 separately plated groups of 4 plates. Only those plates that fell within the 2,000- to 4,000-CFU/plate range were examined (80% of the total plates). Single-strain analysis additionally was performed at 72 h for MJM1108 (MJM1100/pVSV208) and for MJM1000, and no difference was observed compared to MJM1100 and MJM1007, respectively, demonstrating that the marker plasmid did not influence the phenotype. Data represent the means and standard errors of the means for separately plated replicate groups. Statistical analysis to determine if MJM1007 exhibited increased survival compared to MJM1100 at each time point was conducted by a one-tailed Fischer's exact test (GraphPad Prism).
Sequence read accession number.
Illumina sequencing data for ES114, ES114-L, and ESR1 were deposited in the Sequence Read Archive under accession number SRP045732.
RESULTS
ES114-L represents a valuable test case for transformation-mediated genetic mapping in V. fischeri.
The V. fischeri ES114-L strain was discovered in a laboratory freezer collection, was believed to be a laboratory derivative of strain ES114, and shares most phenotypic characteristics with its presumed parent strain, including the robust capacity to colonize E. scolopes squid (14). ES114-L is unable to grow on glycerol, but the genetic basis of this was unknown. This difference provided a discrete system through which to develop a genetic mapping approach in V. fischeri. Therefore, we proceeded to define a genetic mapping strategy in V. fischeri using the following approach.
(i) Generation of genetic markers in the donor strain.
Our laboratory is developing techniques to conduct global genetic analysis of V. fischeri during colonization. For that purpose, we constructed a mariner transposition vector, termed pMarVF1, that transposes efficiently to generate single stable insertions in V. fischeri ES114 and other V. fischeri strains. We generated a library of over 40,000 unique erythromycin-resistant insertions (Tnerm) in strain ES114 (28), and this served as the donor library in the mapping.
(ii) Introduction of the marked donor DNA fragments (and flanking DNA) into the unmarked recipient strain.
There is no generalized transducing phage available for V. fischeri, so this approach was facilitated by the recently described chitin-induced transformation pathway (21, 24). In V. fischeri, transformation is optimally induced by overexpression of the TfoX transcriptional regulator in the chitin transformation pathway. We employed the pLostfoX plasmid described previously for this purpose (24), and TfoX induction in ES114-L allowed for the introduction of erythromycin-marked ES114 fragments.
(iii) Selection for the fragments from the donor that confer the phenotype of interest.
Strains typically have multiple genomic polymorphisms, and among them we sought to identify the genetic change that underlies the differential glycerol utilization phenotype. We selected for incorporation of erythromycin-resistance-marked ES114 DNA that carried the trait of interest by selecting on Tris MM-glycerol-NH4Cl-erythromycin plates. The only ES114-L derivatives that could grow on this medium were those that incorporated ES114 DNA that conferred the ability to utilize glycerol and that also contained a linked transposon marker. The transformation yielded colonies reliably, whereas control reaction mixtures lacking donor DNA did not. We performed three replicate transformations and selected 15 candidates to pursue further. To identify the locus that was transformed for each sample, we performed arbitrarily primed PCR on the linked transposon for each candidate strain. Twelve independent hits in 10 genes were scored, all of which represented insertions in a 32-kb region of chromosome II (Fig. 1A). The remaining 3 hits were identical to characterized insertions and likely represent sibling colonies that arose during the overnight recovery step.
FIG 1.

Mapping of the ES114-L defect to a narrow region of chromosome II. The region of chromosome II from VF_A0219 through VF_A0248 is shown. Transposon insertions in ES114 that were transformed into ES114-L, conferring a glycerol+ phenotype, are denoted by the anchored triangles. In some cases the transformed transposons subsequently were backcrossed into ES114-L, and the resulting cotransformation frequency of the erythromycin resistance (transposon) and the glycerol+ phenotypes are shown as a percentage above the transposon. The gray box indicates the 9.9 kb that is absent from ES114-L relative to ES114. (B) PCR confirmation of the deletion. PCR primers are as diagrammed in panel A. (C) Quantitative mapping of the ES114-L phenotypes.
To further narrow the possibilities as to what functionally differentiated ES114 from ES114-L, we selected seven transformants that were distributed across the region to quantitatively map the location of the affected locus (Fig. 1A). We backcrossed from the hybrid recipient strain into ES114-L and again selected for erythromycin resistance. Among the new recipient strains, we screened 100 for their glycerol utilization phenotype by patching onto the appropriate media. On our first attempts, we observed either 0% or 100% linkage in each case; in some cases a given transposon would yield 0% in one experiment and 100% in the next. We reasoned that this result was due to a “jackpot” effect following the overnight recovery stage, consistent with the siblings we identified in the previous step, so we proceeded to optimize the recovery period and determined that a 1-h recovery led to reproducible quantification of mapping percentiles. Four of the transposons had a linkage of 8 to 81% to the glycerol utilization phenotype, and three of the test transposons still yielded 100% linkage; in each case the linkage was reproducible. The 100% linkage for three adjacent insertions suggested a chromosomal disruption, so we proceeded to sequence ES114 and ES114-L by Illumina paired-end sequencing.
ES114-L contains a 9.9-kb deletion (tadB2′ to ′glpF).
We sequenced the ES114-L genome and performed a reference-guided assembly against the published ES114 sequence (34, 35). The region of chromosome II that we mapped (described above) was found to contain a 9,911-bp deletion in ES114-L that spanned from tadB2 to glpF (Fig. 1A). Many polymorphisms were identified that distinguished ES114-L from the published ES114 model; however, subsequent Illumina sequencing of ES114 revealed that those differences were present in ES114 as well, with the only exceptions being a point mutation in citrate synthase (gltA; encoded on chromosome I) and a variable-number tandem repeat (VNTR) expansion on plasmid pES100 (Table 3). Owing to our mapping results, we focused on the chromosome II deletion.
TABLE 3.
Polymorphisms identified by sequencing ES114-L and ESR1 and mapping each relative to the reference strain ES114
| ES114 replicona | Start (bp) | End (bp) | Nucleotide(s) | ES114-L |
ESR1 |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Nucleotide | % matching readsb | Depthc | Nucleotide | % matching readsb | Depthc | Protein change(s) | Feature(s) | ||||
| CP000020.2 | 27442 | 27442 | C | C | 99.9 | 858 | Ad | 100.0 | 114 | A437S | fadB |
| CP000020.2 | 422616 | 422616 | G | G | 100.0 | 925 | A | 98.9 | 94 | I171 (silent) | lptB |
| CP000020.2 | 898242 | 898242 | C | Ad | 99.7 | 756 | A | 83.7 | 123 | M18I | gltA |
| CP000020.2 | 2702527 | 2702527 | T | T | 99.9 | 1150 | A | 99.0 | 100 | Q513L | rpoB |
| CP000021.2 | 248836 | 258746 | 9,911 bpf | Deletionf | 100.0 | 0 | Deletionf | 100.0 | 0 | Multiple | tadB2′ to ′glpF |
| CP000022.1 | 8417 | 8417 | G | G | 99.8 | 1161 | C | 55.0 | 20 | VNTRe | VF_B0014 |
| CP000022.1 | 8423 | 8423 | C | C | 99.9 | 1118 | T | 67.9 | 28 | VNTRe | VF_B0014 |
NCBI accession number.
Percentage of reads with the indicated base pair.
Number of reads (depth of coverage) at the indicated base pair.
Changes relative to ES114 are shown in bold.
Polymorphisms identified for VF_B0014 in ESR1 reflect the VNTR described in the text.
ES114-L and ESR1 have a deletion of 9,911 bp relative to ES114.
The tadB2′-to-′glpF deletion overlapped all three transposon-mapped insertions that yielded 100% linkage on backcross. Through diagnostic PCR, we confirmed the absence of the deleted region in ES114-L (Fig. 1B). The high level of identity between ES114 and ES114-L supported a model in which ES114-L was derived from ES114. We examined the margins of the 9,911 ES114 unique regions and identified a direct repeat (5′-ACTCTCTA) that was duplicated in ES114 but was present as a single copy in ES114-L following excision of the intervening DNA. The chimeric strains generated by the transformation of ES114 DNA into the ES114-L genome were glycerol+ by the genetic selection imposed, and in each case the 9.9-kb fragment was cotransformed into ES114-L with the transposon marker (Fig. 1B). We plotted the cotransformation frequency based on the minimum length of donor DNA incorporated and determined that TfoX-based transformation in V. fischeri can transfer large chromosomal fragments: 10 kb with high efficiency and up to 26 kb (Fig. 1A and C).
GlpF truncation explains the glycerol utilization defect in multiple V. fischeri strains.
We reasoned that if an ES114-specific locus was responsible for the phenotype(s) that distinguished the two strains, then the candidate gene(s) was likely to lie in or adjacent to the 9.9-kb deletion that is unique to ES114. At the same time, the gene(s) itself would not be interrupted by a transposon insertion that would impair its function. One deletion endpoint was in the glpF open reading frame (ORF), which likely is encoded in an operon upstream of glpK. GlpF is an inner membrane glycerol importer of the major intrinsic protein (MIP) family, and GlpK is the glycerol kinase (36). To test if the glpF truncation was the basis for the ES114-L phenotypes observed in culture, we first asked whether glpF is required for glycerol utilization in V. fischeri. We determined that a glpF::Tnerm transposon insertion was unable to utilize glycerol, so we proceeded to determine if repair of the glpF truncation in ES114-L would restore glycerol utilization. At the ES114-L chromosomal locus, we restored the promoter and 5′ portion of glpF. The resulting strain, MJM2129, phenocopied ES114 in its ability to utilize glycerol, confirming that restoration of the glpF ORF and proper glpFK regulation is sufficient to confer positive glycerol utilization (Fig. 2).
FIG 2.
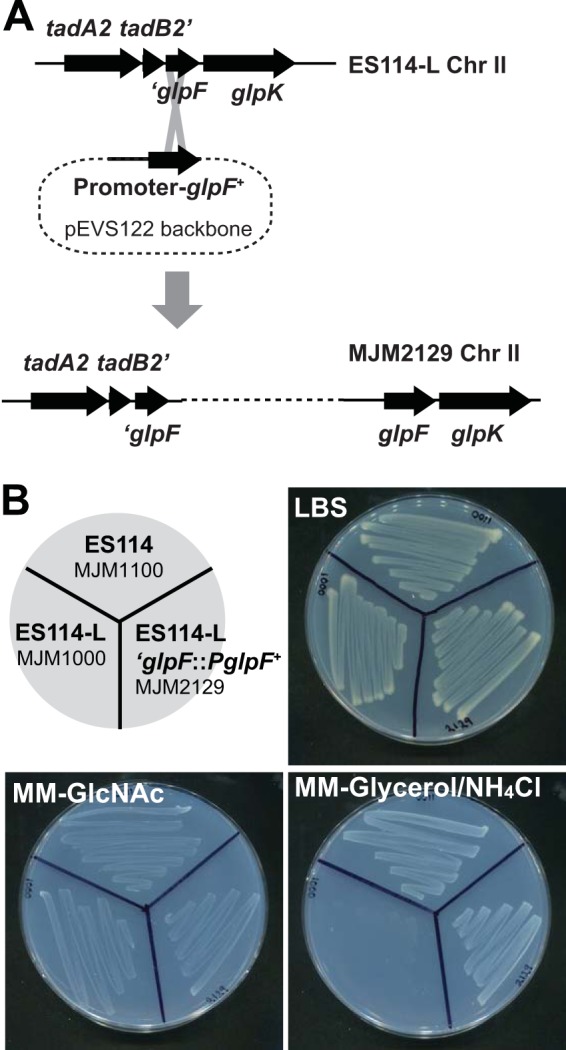
Repair of the glpF 5′ end and promoter in ES114-L phenocopies ES114. (A) Mechanism of repair at the glpF locus. The promoter and glpF open reading frame were cloned into the suicide vector pEVS122 and incorporated into the ES114-L chromosome as a single copy to generate strain MJM2129 as shown. (B) Growth phenotypes on LBS rich medium or on Tris minimal medium (MM) with the sole carbon and nitrogen sources as indicated and as described in the text. The indicated strains were streaked onto the agar shown, allowed to grow for 24 h at 25°C, and then imaged. The strains are oriented according to the key in the top left. ES114-L fails to grow on glycerol as a sole carbon source, but repair of the glpF locus restores growth on glycerol.
ESR1 is a derivative of ES114-L.
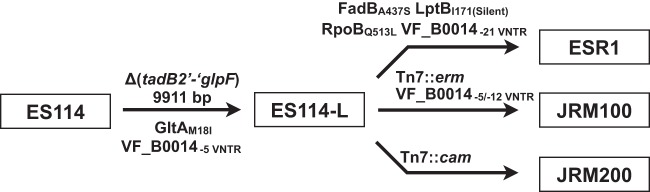
Genetic manipulation in V. fischeri relies on conjugation of mutagenesis plasmids from donor E. coli into the recipient V. fischeri cells, and counterselection against the donor was a significant challenge. For many years this challenge was solved by the use of a rifampin-resistant derivative of ES114, termed ESR1. ESR1 enabled facile genetic analysis in V. fischeri, but it was discovered that the strain did not compete well against wild-type ES114 in squid competition experiments (13). Over 40 publications used ESR1 for its counterselection advantage, but as a result of its defect in squid colonization, a key focus of symbiotic V. fischeri studies, it now is rarely used as a parent strain. It also had been appreciated that ESR1 exhibited a defect in glycerol utilization (37). ESR1 was distinguishable from ES114-L, as only the former was resistant to rifampin (Table 4); however, due to the common inability to utilize glycerol, we asked whether ESR1 and ES114-L shared a common lineage. Using diagnostic PCR and Sanger sequencing, we determined that the ESR1 chromosome similarly was missing the 9.9-kb (tadB2′-to-′glpF) fragment (Fig. 1B). We proceeded to sequence ESR1 by Illumina paired-end sequencing and performed a reference-guided assembly against ES114. Mutations at six loci were noted (Table 3). The tadB2′-to-′glpF deletion and the gltA point mutation identified in ES114-L both were present in the ESR1 assembly. Additional point mutations in fadB, lptB, and rpoB were noted in ESR1 but not in ES114-L. The mutation in rpoB was expected, as rifampin resistance mutations typically map to rpoB. Like ES114-L, ESR1 exhibited a reduction in repeats in the VNTR-containing gene VF_B0014 (Fig. 3). We discovered the unusual nature of this gene when we followed up on the Illumina sequencing results (Table 3). Up to 93% of the open reading frame is comprised of repeat sequences, and ES114-L exhibits fewer repeats than ES114 and more repeats than ESR1. The phenotypic, SNP, deletion, and VNTR data together strongly support a lineage in which the ESR1 strain was derived from ES114-L, which itself was derived from ES114 (Fig. 4).
TABLE 4.
Phenotypic characterization of V. fischeri strains
| Medium | Growth ofa: |
||
|---|---|---|---|
| ES114 | ES114-L | ESR1 | |
| LBS | + | + | + |
| LBS-rifampin | − | − | + |
| Tris MM-GlcNAc | + | + | + |
| Tris MM-GlcNAc + NH4Cl | + | + | + |
| Tris MM-glycerol + NH4Cl | + | – | – |
| Tris MM-glucose + NH4Cl | + | + | + |
| Tris MM-galactose + NH4Cl | + | + | + |
| Tris MM-mannitol + NH4Cl | + | + | + |
A plus sign indicates that the given medium supports growth.
FIG 3.
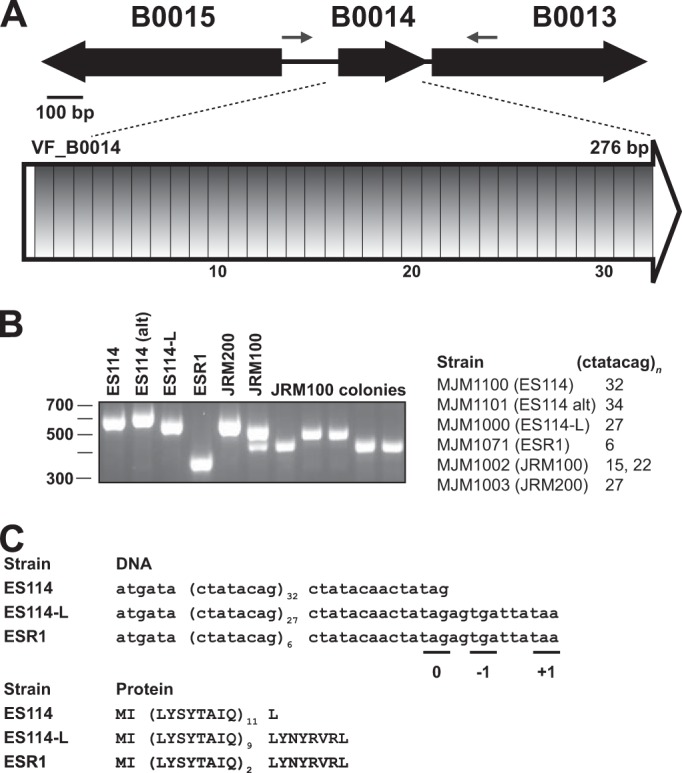
VF_B0014 is a predicted protein-coding gene that evolves rapidly, and it is composed almost entirely of 8-bp repeats. (A) In ES114, 93% of the predicted open reading frame is composed of identical direct 8-bp repeats. (B) The number of repeats is different in most strains examined, as assayed by PCR with the flanking primers shown in panel A and by Sanger sequencing reads that span the repeat region. Strain JRM100 revealed two different sizes for the overnight culture that were resolved from the analysis of plated colonies. The marker for the gel is in bp. (C) Predicted ORF and resulting protein sequence from three of the repeats. Stop codons are present in all three reading frames downstream of the repeat region.
FIG 4.
Proposed lineage for V. fischeri strains. The most parsimonious lineage of the strains under study is presented. Polymorphisms are detailed in Table 3.
It had been noted previously that two other common V. fischeri laboratory strains, JRM100 and JRM200, also failed to grow on glycerol minimal medium (13, 14). We identified the 9.9-kb Δ(tadB2′-′glpF) deletion in each of these strains. These strains are rifampin sensitive yet are tagged with antibiotic resistance cassettes, erythromycin and chloramphenicol for JRM100 and JRM200, respectively; therefore, they too likely are derived from ES114-L (Fig. 4). Compared to ES114-L, JRM200 encodes the same number of DNA repeats at VF_B0014, and JRM100 encodes fewer (Fig. 3). Our isolate of JRM100 revealed a doublet at the locus and resulting single colonies that were analyzed and sequenced to reveal strains with different numbers of repeats that had arisen in a single passage (Fig. 3). Previous work additionally attributed a siderophore production defect to ES114-L, but we have since demonstrated that this was secondary to growth on medium in which glycerol was provided as the carbon source (14) (J.F.B. and M.J.M., unpublished).
ESR1, but not ES114-L, is defective for squid colonization.
In a previous study, ESR1 exhibited a defect in colonization of squid when inoculated in competition with ES114 (13). We repeated the squid colonization assay and in each case coinoculated either ES114-L or ESR1 with the ES114 wild-type strain. Inoculation proceeded for 3 h, and we assayed fitness at 48 h. We observed no competitive defect of the ES114-L strain, but the ESR1 strain exhibited a significant defect in competition to ES114 (Fig. 5). This result is consistent with the previous report that demonstrated no significant colonization defect for JRM100 or JRM200, which we show here to be highly similar to ES114-L. Therefore, it appears that the ESR1 colonization defect arose during the generation of that strain from ES114-L.
FIG 5.
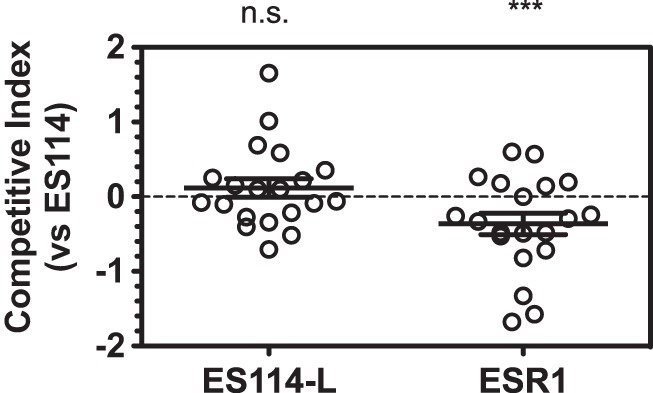
ESR1 competitive colonization defect is not observed in ES114-L. Squid competition experiments in which ES114 and the indicated strain were coinoculated with newly hatched E. scolopes squid for 3 h, with a total colonization time of 48 h. Shown are animals from two replicates for each experiment. Single-sample t tests were conducted to identify significant deviations from neutral. The ES114-L results were nonsignificant (n.s.) (P = 0.36), whereas ESR1 exhibited significant results (P = 0.001 [***]).
ES114-L exhibits a fitness advantage on SWT agar.
The origin of the ES114-L strain is not clear. ESR1 was generated for work that was published in 1994 (12); based on the model that ES114-L is the parent strain for ESR1 (Fig. 4), it seems likely that ES114-L has existed undetected for approximately 20 years. One possibility is that growth in SWT, a glycerol-containing rich medium known to cause medium acidification in overnight ES114 but not ES114-L cultures (14, 38), contributed to a fitness advantage for ES114-L. This proposal is consistent with culture practices from the time (11), but it would require that a rare mutant that does not utilize glycerol exhibits a selective advantage in the context of the majority of bacteria on the plate that utilize the carbon source and acidify the medium. We confirmed that ES114 colonies are not viable after 52 h on SWT agar, whereas ES114-L remains viable through at least 76 h (Fig. 6A). In SWT liquid culture of the two strains with ES114 at a 9-fold excess, the acidification from the ES114 cells kills the entire culture after overnight growth (data not shown). Therefore, we turned to solid medium to determine whether ES114-L had a selective advantage on SWT. We plated the two strains at high density, with ES114 present at 9-fold excess and with the rare ES114-L colonies marked by fluorescence. We proceeded to examine the viability of each strain over 76 h. At multiple time points over an 8-h window, ES114-L exhibited a significant fitness advantage, even in the context of the dense ES114 plating (Fig. 6B). Therefore, we conclude that once arisen, ES114-L would have exhibited a selective advantage under such conditions.
FIG 6.
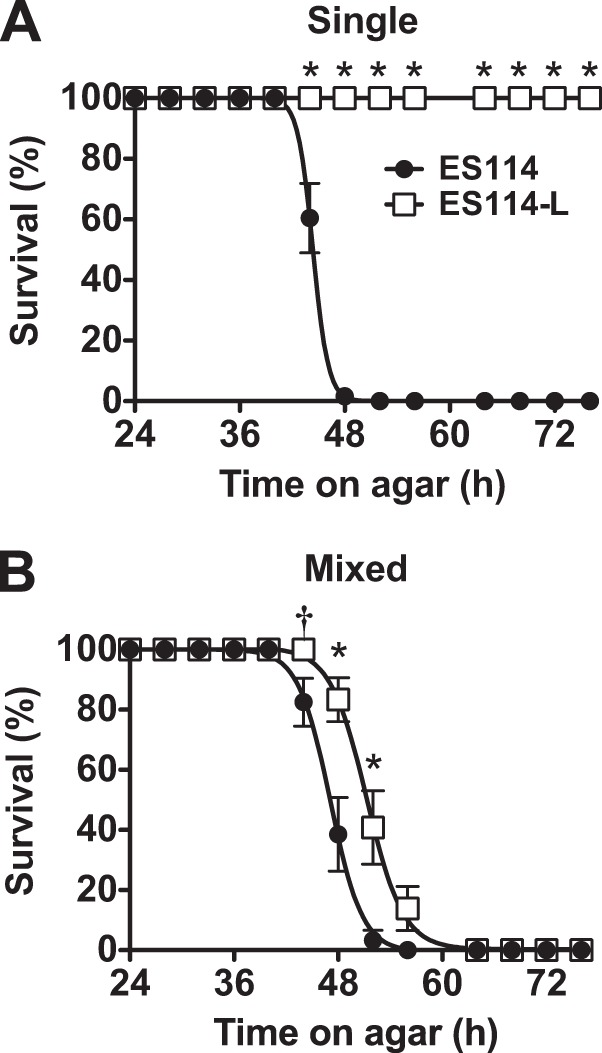
ES114-L exhibits a fitness advantage over ES114 when the strains are plated together on SWT agar. Survival of ES114 (MJM1100) and ES114-L (MJM1007) was monitored for 76 h. Individual colonies were distinguished by epifluorescence microscopy, and viability was assessed by patching onto LBS. Data points represent the percentage of viable colonies at each time point. The means and standard errors of the means from replicate platings (minimum n = 3 of 8 colonies each) are indicated as described in Materials and Methods. Fisher's exact test was conducted to compare the survival of the two strains; significant differences at P < 0.05 (†) and P < 0.001 (*) are noted. The strains were either plated separately (A) or coplated as a mixture of 90% ES114 and 10% ES114-L (B).
DISCUSSION
In this report, we discovered the lineage by which the most commonly used V. fischeri strains are related and identified a rapidly evolving repeat locus in the organism. To accomplish this, we developed a genetic mapping approach in V. fischeri that allowed us to characterize the basis by which strain ES114-L fails to utilize glycerol. Improved transposon mutagenesis, the use of TfoX induction for quantitative genetic linkage analysis, and integration with whole-genome sequencing data and complementation studies allowed us to define ES114-L as an intermediate strain that has played a cryptic yet important role in the origins of V. fischeri laboratory strains.
Chitin-based transformation was discovered in V. cholerae, and active work has elucidated the circuitry by which the TfoX pathway is induced in response to various nutritional and density cues (21, 39, 40). A previous use of the pathway has been to facilitate defined mutagenesis in multiple Vibrio species, and it was shown that in the presence of chitin V. cholerae strains could undergo a serogroup conversion, which requires the exchange of an ∼42-kb fragment of DNA (22–24, 41). In another study it was shown that chitin-based transformation led to transmission of large genomic fragments in V. cholerae, with an average size of 22.7 kb (25). We observe similarly large fragments in V. fischeri, and we demonstrate the novel application of the use of TfoX-induced transformation for genetic mapping. The mapping approach described is limited only by the selection that can be imposed. Chimeric strains that survive the selection then can be analyzed individually or en masse. Cotransformation was used for the early study of Gram-positive bacteria, including Bacillus subtilis and Streptococcus pneumoniae (16, 42). The availability of generalized transducing phage for model Gram-negative bacteria provided an equivalent tool (18). To our knowledge, this is the first time that transformation has been applied in the Vibrionaceae as a means to map genetic traits. With increased interest in studying natural and diverse isolates, many of which do not have characterized phage or sophisticated molecular genetics, the ability to cotransform markers to map a trait of interest is a powerful and simple technique that should be widely applicable. The techniques presented here were developed for V. fischeri, but the general strategy of generating a marked donor strain (e.g., high-density transposon insertion), introducing marked donor fragments into a recipient (e.g., transformation), and selecting for the donor-specific trait generally can be applied across many organisms for which specialized genetic tools have not yet been developed. The in vitro Tn5 transposons available from Epicentre (Madison, WI) have been reported to work in many genera of bacteria. Because this system requires both transposition and transformation (typically electroporation), the general mapping approach employed here can be applied immediately in a large number of microorganisms using a commercially available product. Thus, it is feasible to coopt available transposition technology to map traits of interest in environmental microorganisms and to identify the key traits that distinguish bacterial strains of interest.
Glycerol utilization has been shown to be critical for V. fischeri organisms when they inhabit adult E. scolopes, and GlpF is predicted to be a major route by which symbiotic V. fischeri acquires glycerol catabolized from host phospholipids (43). In contrast, we demonstrated that strains unable to utilize glycerol exhibit no colonization defect in the juvenile squid, arguing that glycerol catabolism is not required for symbiosis development in the first 48 h (Fig. 5). Therefore, we find that the proposed requirement for glycerol exchange in adult symbiosis is not relevant in the juvenile stage of the interaction.
The strain that does exhibit a colonization defect, both in our experiments and in a previous study, is ESR1. The ES114-L control limits the loci that may contribute to this defect. Predicted amino acid differences between the two strains are encoded by the rpoB, lptB, and VF_B0014 loci. Therefore, one or several of these mutations are the likely basis for the colonization defect in ESR1. RpoB is the β subunit of RNA polymerase, and mutations at this locus have been shown in other organisms to be pleiotropic (44). LptB is an inner membrane ATPase involved in LPS transport, so bacterial envelope alterations could underlie the ESR1 defect (45). The divergence in VF_B0014 is particularly intriguing. The changes at this locus seem to represent a rapidly evolving V. fischeri repeat region. If this gene/locus contributes to the fitness of the strain in culture or during colonization, then monitoring this locus will be particularly important during strain construction and after the extended passage of strains. Notably, contractions (or, possibly, expansions) at this locus may not be readily apparent by high-throughput sequencing technologies. We detected the contraction in ES114-L only after observing the anomalies in ESR1 and then proceeding to verify the locus in all of the strains under study. This locus appears to mutate so rapidly that two isolates of the same wild-type strain exhibited different numbers of repeats (MJM1100 with 32 repeats and MJM1101 with 34 repeats), and that an overnight passage of JRM100 led to a mixed culture (some harboring 22 repeats and others with 15) (Fig. 3).
The discovery of chitin-based transformation in vibrios has yielded numerous insights into bacterial physiology in animal hosts, in the environment, and in culture. We have extended the tools for the molecular genetic determination of strain-level differences and established a method that will be valuable to directly interrogate host colonization specificity.
ACKNOWLEDGMENTS
We acknowledge C. Brennan and S. Studer for early characterization of the ES114-L phenotype; K. Visick for noting the similarities to ESR1; M. Hadfield and the Kewalo Marine Laboratory for Euprymna field resources; and K. Bodi, D. Burnside, R. Foxall, J. Foster, A. Goodman, T. Miyashiro, V. Ray, A. Tai, K. Visick, and C. Whistler for strains, reagents, instrumentation, and advice.
This work was supported by an NSF grant to M.J.M. (IOS-0843633) and an NIGMS Cell and Developmental Basis of Disease T32 training grant (GM08061) award to J.F.B. Reagents provided by J. Foster were supported by NASA Space Biology grant NNX13AM44G.
REFERENCES
- 1.Rajashekara G, Covert J, Petersen E, Eskra L, Splitter G. 2008. Genomic island 2 of Brucella melitensis is a major virulence determinant: functional analyses of genomic islands. J Bacteriol 190:6243–6252. doi: 10.1128/JB.00520-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kung VL, Ozer EA, Hauser AR. 2010. The accessory genome of Pseudomonas aeruginosa. Microbiol Mol Biol Rev 74:621–641. doi: 10.1128/MMBR.00027-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sun Y-C, Hinnebusch BJ, Darby C. 2008. Experimental evidence for negative selection in the evolution of a Yersinia pestis pseudogene. Proc Natl Acad Sci U S A 105:8097–8101. doi: 10.1073/pnas.0803525105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cowles CE, Goodrich-Blair H. 2008. The Xenorhabdus nematophila nilABC genes confer the ability of Xenorhabdus spp. to colonize Steinernema carpocapsae nematodes. J Bacteriol 190:4121–4128. doi: 10.1128/JB.00123-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chaston J, Goodrich-Blair H. 2010. Common trends in mutualism revealed by model associations between invertebrates and bacteria. FEMS Microbiol Rev 34:41–58. doi: 10.1111/j.1574-6976.2009.00193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mandel MJ, Wollenberg MS, Stabb EV, Visick KL, Ruby EG. 2009. A single regulatory gene is sufficient to alter bacterial host range. Nature 458:215–218. doi: 10.1038/nature07660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mandel MJ. 2010. Models and approaches to dissect host-symbiont specificity. Trends Microbiol 18:504–511. doi: 10.1016/j.tim.2010.07.005. [DOI] [PubMed] [Google Scholar]
- 8.Kirzinger MWB, Stavrinides J. 2012. Host specificity determinants as a genetic continuum. Trends Microbiol 20:88–93. doi: 10.1016/j.tim.2011.11.006. [DOI] [PubMed] [Google Scholar]
- 9.Jones B, Nishiguchi M. 2004. Counterillumination in the Hawaiian bobtail squid, Euprymna scolopes Berry (Mollusca: Cephalopoda). Mar Biol 144:1151–1155. doi: 10.1007/s00227-003-1285-3. [DOI] [Google Scholar]
- 10.Ruby EG, McFall-Ngai MJ. 1992. A squid that glows in the night: development of an animal-bacterial mutualism. J Bacteriol 174:4865–4870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boettcher KJ, Ruby EG. 1990. Depressed light emission by symbiotic Vibrio fischeri of the sepiolid squid Euprymna scolopes. J Bacteriol 172:3701–3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Graf J, Dunlap PV, Ruby EG. 1994. Effect of transposon-induced motility mutations on colonization of the host light organ by Vibrio fischeri. J Bacteriol 176:6986–6991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McCann J, Stabb EV, Millikan DS, Ruby EG. 2003. Population dynamics of Vibrio fischeri during infection of Euprymna scolopes. Appl Environ Microbiol 69:5928–5934. doi: 10.1128/AEM.69.10.5928-5934.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Studer SV. 2010. Identification of AinSR-controlled activities and their role in the Vibrio fischeri-Euprymna scolopes symbiosis. Ph.D. dissertation University of Wisconsin—Madison, Madison, WI. [Google Scholar]
- 15.Griffith F. 1928. The significance of pneumococcal types. J Hyg 27:113–159. doi: 10.1017/S0022172400031879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nester EW, Lederberg J. 1961. Linkage of genetic units of Bacillus subtilis in DNA transformation. Proc Natl Acad Sci U S A 47:52–55. doi: 10.1073/pnas.47.1.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nester EW, Schafer M, Lederberg J. 1963. Gene linkage in DNA transfer: a cluster of genes concerned with aromatic biosynthesis in Bacillus subtilis. Genetics 48:529–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Silhavy TJ, Berman ML, Enquist LW. 1984. Experiments with gene fusions. Cold Spring Harbor Laboratory Press, Plainview, NY. [Google Scholar]
- 19.Button JE, Silhavy TJ, Ruiz N. 2007. A suppressor of cell death caused by the loss of σE downregulates extracytoplasmic stress responses and outer membrane vesicle production in Escherichia coli. J Bacteriol 189:1523–1530. doi: 10.1128/JB.01534-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stanley NR, Lazazzera BA. 2005. Defining the genetic differences between wild and domestic strains of Bacillus subtilis that affect poly-gamma-dl-glutamic acid production and biofilm formation. Mol Microbiol 57:1143–1158. doi: 10.1111/j.1365-2958.2005.04746.x. [DOI] [PubMed] [Google Scholar]
- 21.Meibom KL, Blokesch M, Dolganov NA, Wu C-Y, Schoolnik GK. 2005. Chitin induces natural competence in Vibrio cholerae. Science 310:1824–1827. doi: 10.1126/science.1120096. [DOI] [PubMed] [Google Scholar]
- 22.Blokesch M, Schoolnik GK. 2007. Serogroup conversion of Vibrio cholerae in aquatic reservoirs. PLoS Pathog 3:e81. doi: 10.1371/journal.ppat.0030081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marvig RL, Blokesch M. 2010. Natural transformation of Vibrio cholerae as a tool–optimizing the procedure. BMC Microbiol 10:155. doi: 10.1186/1471-2180-10-155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pollack-Berti A, Wollenberg MS, Ruby EG. 2010. Natural transformation of Vibrio fischeri requires tfoX and tfoY. Environ Microbiol 12:2302–2311. doi: 10.1111/j.1462-2920.2010.02250.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miller MC, Keymer DP, Avelar A, Boehm AB, Schoolnik GK. 2007. Detection and transformation of genome segments that differ within a coastal population of Vibrio cholerae strains. Appl Environ Microbiol 73:3695–3704. doi: 10.1128/AEM.02735-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stabb EV, Ruby EG. 2002. RP4-based plasmids for conjugation between Escherichia coli and members of the Vibrionaceae. Methods Enzymol 358:413–426. doi: 10.1016/S0076-6879(02)58106-4. [DOI] [PubMed] [Google Scholar]
- 27.Lupp C, Ruby EG. 2005. Vibrio fischeri uses two quorum-sensing systems for the regulation of early and late colonization factors. J Bacteriol 187:3620–3629. doi: 10.1128/JB.187.11.3620-3629.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brooks JF, Gyllborg MC, Cronin DC, Quillin SJ, Mallama CA, Foxall R, Whistler C, Goodman AL, Mandel MJ. 2014. Global discovery of colonization determinants in the squid symbiont Vibrio fischeri. Proc Natl Acad Sci U S A 111:17284–17289. doi: 10.1073/pnas.1415957111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brennan CA, Mandel MJ, Gyllborg MC, Thomasgard KA, Ruby EG. 2013. Genetic determinants of swimming motility in the squid light-organ symbiont Vibrio fischeri. Microbiologyopen 2:576–594. doi: 10.1002/mbo3.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Caetano-Anollés G. 1993. Amplifying DNA with arbitrary oligonucleotide primers. PCR Methods Appl 3:85–94. doi: 10.1101/gr.3.2.85. [DOI] [PubMed] [Google Scholar]
- 31.Danese PN, Pratt LA, Kolter R. 2001. Biofilm formation as a developmental process. Methods Enzymol 336:19–26. doi: 10.1016/S0076-6879(01)36574-6. [DOI] [PubMed] [Google Scholar]
- 32.Dunn AK, Martin MO, Stabb EV. 2005. Characterization of pES213, a small mobilizable plasmid from Vibrio fischeri. Plasmid 54:114–134. doi: 10.1016/j.plasmid.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 33.Naughton LM, Mandel MJ. 2012. Colonization of Euprymna scolopes squid by Vibrio fischeri. J Vis Exp 2012:e3758. doi: 10.3791/3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ruby EG, Urbanowski M, Campbell J, Dunn A, Faini M, Gunsalus R, Lostroh P, Lupp C, McCann J, Millikan D, Schaefer A, Stabb E, Stevens A, Visick K, Whistler C, Greenberg EP. 2005. Complete genome sequence of Vibrio fischeri: a symbiotic bacterium with pathogenic congeners. Proc Natl Acad Sci U S A 102:3004–3009. doi: 10.1073/pnas.0409900102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mandel MJ, Stabb EV, Ruby EG. 2008. Comparative genomics-based investigation of resequencing targets in Vibrio fischeri: focus on point miscalls and artefactual expansions. BMC Genomics 9:138. doi: 10.1186/1471-2164-9-138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin EC. 1976. Glycerol dissimilation and its regulation in bacteria. Annu Rev Microbiol 30:535–578. doi: 10.1146/annurev.mi.30.100176.002535. [DOI] [PubMed] [Google Scholar]
- 37.Visick KL, Skoufos LM. 2001. Two-component sensor required for normal symbiotic colonization of Euprymna scolopes by Vibrio fischeri. J Bacteriol 183:835–842. doi: 10.1128/JB.183.3.835-842.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Studer SV, Mandel MJ, Ruby EG. 2008. AinS quorum sensing regulates the Vibrio fischeri acetate switch. J Bacteriol 190:5915–5923. doi: 10.1128/JB.00148-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Blokesch M. 2012. Chitin colonization, chitin degradation and chitin-induced natural competence of Vibrio cholerae are subject to catabolite repression. Environ Microbiol 14:1898–1912. doi: 10.1111/j.1462-2920.2011.02689.x. [DOI] [PubMed] [Google Scholar]
- 40.Sun Y, Bernardy EE, Hammer BK, Miyashiro T. 2013. Competence and natural transformation in vibrios. Mol Microbiol 89:583–595. doi: 10.1111/mmi.12307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gulig PA, Tucker MS, Thiaville PC, Joseph JL, Brown RN. 2009. USER friendly cloning coupled with chitin-based natural transformation enables rapid mutagenesis of Vibrio vulnificus. Appl Environ Microbiol 75:4936–4949. doi: 10.1128/AEM.02564-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hotchkiss RD. 1955. Bacterial transformation. J Cell Physiol Suppl 45:1–22. [DOI] [PubMed] [Google Scholar]
- 43.Wier AM, Nyholm SV, Mandel MJ, Massengo-Tiassé RP, Schaefer AL, Koroleva I, Splinter-Bondurant S, Brown B, Manzella L, Snir E, Almabrazi H, Scheetz TE, de Bonaldo M, Casavant FTL, Soares MB, Cronan JE, Reed JL, Ruby EG, McFall-Ngai MJ. 2010. Transcriptional patterns in both host and bacterium underlie a daily rhythm of anatomical and metabolic change in a beneficial symbiosis. Proc Natl Acad Sci U S A 107:2259–2264. doi: 10.1073/pnas.0909712107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jin DJ, Gross CA. 1989. Characterization of the pleiotropic phenotypes of rifampin-resistant rpoB mutants of Escherichia coli. J Bacteriol 171:5229–5231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sherman DJ, Lazarus MB, Murphy L, Liu C, Walker S, Ruiz N, Kahne D. 2014. Decoupling catalytic activity from biological function of the ATPase that powers lipopolysaccharide transport. Proc Natl Acad Sci U S A 111:4982–4987. doi: 10.1073/pnas.1323516111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fidopiastis PM, von Boletzky S, Ruby EG. 1998. A new niche for Vibrio logei, the predominant light organ symbiont of squids in the genus Sepiola. J Bacteriol 180:59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gyllborg MC, Sahl JW, Cronin DC, Rasko DA, Mandel MJ. 2012. Draft genome sequence of Vibrio fischeri SR5, a strain isolated from the light organ of the Mediterranean squid Sepiola robusta. J Bacteriol 194:1639. doi: 10.1128/JB.06825-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Simon R, Priefer U, Pühler A. 1983. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in Gram negative bacteria. Nat Biotechnol 1:784–791. doi: 10.1038/nbt1183-784. [DOI] [Google Scholar]
- 49.Post DMB, Yu L, Krasity BC, Choudhury B, Mandel MJ, Brennan CA, Ruby EG, McFall-Ngai MJ, Gibson BW, Apicella MA. 2012. O-antigen and core carbohydrate of Vibrio fischeri lipopolysaccharide: composition and analysis of their role in Euprymna scolopes light organ colonization. J Biol Chem 287:8515–8530. doi: 10.1074/jbc.M111.324012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dunn AK, Millikan DS, Adin DM, Bose JL, Stabb EV. 2006. New rfp- and pES213-derived tools for analyzing symbiotic Vibrio fischeri reveal patterns of infection and lux expression in situ. Appl Environ Microbiol 72:802–810. doi: 10.1128/AEM.72.1.802-810.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]