

Abstract
A mechanistic understanding of the intermolecular interactions and structural changes during fibrillation is crucial for the design of safe and efficacious glucagon formulations. Amide hydrogen/deuterium exchange with mass spectrometric analysis was used to identify the interactions and amino acids involved in the initial stages of glucagon fibril formation at acidic pH. Kinetic measurements from intrinsic and thioflavin T fluorescence showed sigmoidal behavior. Secondary structural measurement of fibrillating glucagon using far-UV circular dichroism spectroscopy showed changes in structure from random coil → α-helix → β-sheet, with increase in α-helix content during the lag phase followed by increase in β-sheet content during the growth phase. Hydrogen/deuterium exchange with mass spectrometric analysis of fibrillating glucagon suggested that C-terminal residues 22–29 are involved in interactions during the lag phase, during which N-terminal residues 1–6 showed no changes. Molecular dynamics simulations of glucagon fragments showed C-terminal to C-terminal interactions with greater α-helix content for the 20–29 fragment, with hydrophobic and aromatic residues (Phe-22, Trp-25, Val-23, and Met-27) predominantly involved. Overall, the study shows that glucagon interactions during the early phase of fibrillation are mediated through C-terminal residues, which facilitate the formation of α-helix-rich oligomers, which further undergo structural rearrangement and elongation to form β-sheet-rich mature fibrils.
Introduction
Some soluble peptides and proteins can self-assemble to form ordered, insoluble, β-sheet-rich fibers called amyloid fibrils. More than 20 different human proteins are known to form amyloid fibrils and several are responsible for diseases such as Alzheimer’s disease and prion disorders (e.g., Creutzfeldt-Jakob disease) (1,2). Recent studies suggest that the toxicity of these proteins is due to the accumulation of prefibrillar intermediates, rather than the mature fibrils themselves (3–5). However, detailed structural characterization of these transient intermediates is often difficult and they typically are not amenable to conventional high-resolution methods such as x-ray crystallography. As a result, fibrillation has often been characterized by a combination of analytical methods, including thioflavin T (ThT) staining, circular dichroism (CD) spectroscopy, Fourier transform infrared spectroscopy, fiber diffraction patterns, atomic force microscopy (AFM), transmission electron microscopy, small-angle x-ray scattering, and NMR spectroscopy (6–8). These methods are limited in that they are only semiquantitative, lack structural resolution, and/or require relatively large amounts of the peptide or protein. In the studies reported here, amide hydrogen/deuterium exchange with mass spectrometric analysis (HDX-MS) has been used to identify the early stage interactions and structural changes in the fibrillation of human glucagon with peptide-level resolution.
Glucagon is a 29-amino acid peptide hormone that increases blood glucose levels and is used in the treatment of severe hypoglycemia. Though administered by injecting a solution, glucagon is unstable in the solution form (9,10) and the commercial product is marketed as a lyophilized solid. The formation of fibrillar aggregates is arguably the most serious type of degradation in glucagon, because it occurs rapidly under a wide range of conditions and increases solution viscosity, making injection difficult. For peptide and protein drugs in general, the presence of aggregates is associated with the potential for changes in potency and an increased risk for life-threatening immunogenic side effects, an even greater concern. To ensure that glucagon is administered to patients in a safe and fully potent form, the lyophilized drug product is reconstituted in dilute acidic buffer just before administration, and any surplus solution is discarded immediately.
Inhibiting glucagon fibrillation is critical to the development of a stable solution formulation and to ensuring the safety and efficacy of reconstituted solids. Various strategies have been attempted to achieve this goal. The design of aggregation-resistant glucagon analogs has been shown to be beneficial in some cases (11,12). Methionine (Met-27) oxidation has been shown to greatly delay the fibrillation of unmodified glucagon (10), suggesting that mutations near the C-terminus may be particularly effective. PEGylation has enhanced the physical stability of glucagon under various conditions (13). However, any changes to the amino acid sequence or structure of a small peptide such as glucagon may impact biological activity (14–19), and so are not without risk. Attempts to stabilize glucagon using readily available formulation additives have generally been ineffective or limited in their ability to stabilize the native structure (20–22).
The rational design of fibrillation-resistant glucagon requires a thorough understanding of the mechanisms of its self-assembly, with particular attention to the early phases of fibrillation and the regions that participate in early stage interactions. Though there have been numerous studies of glucagon fibrillation (23–29), the molecular mechanisms involved have not been well studied, due in part to the rapid fibrillation kinetics and the lack of high-resolution methods. As with other amyloidogenic peptides and proteins, glucagon fibrillation is a complex process. which includes molecular interactions, the formation of intermediate states, structural transitions, and the assembly of oligomers to form fibrils (23,24). Complexity is further increased by structural polymorphism in the fibrils themselves, which results from changes in concentration, solvent composition, and temperature (24,30). Several studies have shown that both hydrophobic and charged amino acids in the glucagon sequence are critical for fibrillation (28,29,31–34). Some suggest that the hydrophobic residues in the N- and C-terminus are involved (32), whereas others argue that only the N-terminal region participates (28). The results from these studies have been based either on analysis of mutants or characterization of the mature fibrils, rather than on studies of native glucagon itself, and may depend on solution composition and temperature.
In this study, we used HDX-MS and other biophysical measurements to monitor the sequence of structural events in glucagon during fibrillation under acidic conditions, from loss of monomer to the development of mature fibrils. Pulsed HDX with MS analysis has been used successfully to capture the molecular interactions and transient intermediates during fibrillation of other proteins (35–38). HDX-MS has also been used to characterize glucagon fibrillation at neutral pH (34), though local structural changes during the early stages of fibril formation were not investigated. We conducted HDX at pH 4.5, where the rate of exchange is ∼1000-fold slower than at pH 7.4, the pH used in the previous study (34). The lower pH allows the molecular interactions and conformational changes during the early stages of glucagon fibrillation to be monitored at a range of HDX reaction times. The results from HDX-MS, far-UV CD spectroscopy, and molecular dynamics (MD) simulations show that the unstructured monomers self-associate through the C-terminus to form α-helix-rich oligomers during the lag phase of glucagon fibrillation. The oligomers then undergo structural changes with an increase in β-sheet content during the transition to the mature fibrils. Overall, the work identifies the C-terminus as the region responsible for the early stages of glucagon fibrillation at acidic pH. Thus, inhibiting C-terminal self-association is expected to be an effective approach to stabilizing solution formulations of glucagon under these conditions.
Materials and Methods
Materials
Research grade human glucagon was purchased from ProSpec (East Brunswick, NJ). 96-well black and crystal-clear microplates were purchased from Greiner Bio-One (Frickenhausen, Germany). ThT was purchased from Abcam (Cambridge, MA). MS spectrometry grade water, acetonitrile (ACN) and formic acid (FA) were from ThermoFisher Scientific (Waltham, MA). Deuterium oxide (D2O) (99.9%) was purchased from Cambridge Isotope Laboratories (Andover, MA). Poroszyme Immobilized Pepsin, Bulk Media was purchased from Applied Biosystems (Foster City, CA) and packed into a high-performance liquid chromatography column (50 × 2.1 mm, Grace Davison Discovery Sciences, Deerfield, IL). All other chemicals used were at least reagent grade and used as received.
Intrinsic fluorescence measurement
The fluorescence emission from Trp-25 in glucagon was measured over time during fibrillation. A 0.6 mg/mL glucagon solution was prepared in 3.2 mM HCl, 0.9% NaCl (w/v) (pH 2.5), and centrifuged at 14,000 rpm for 3 min to remove any insoluble material. Samples in triplicate were quickly transferred to a 96-well black microtiter plate and placed in a BioTek Synergy 4 Multi-Detection microplate reader (BioTek Instruments, Winooski, VT). The excitation and emission wavelengths were set to 295 nm and 355 nm, respectively, and measurement was carried out for 8 h at 23°C at 15-min intervals preceded by 5 s automixing before each reading. The data were fitted to the Boltzmann equation (a sigmoidal function) as given by
| (1) |
where A1 is the minimum signal, A2 is the maximum signal, x0 is the time at which the change in signal is 50% (t50), dx is the time constant, and the lag time is estimated by x0 – 2 dx.
ThT fluorescence measurement
Glucagon at 0.6 mg/mL in 3.2 mM HCl, 0.9% NaCl (w/v) (pH 2.5) was incubated with 50 μM ThT. Samples in triplicate were quickly transferred to a 96-well black flat bottom microtiter plate and placed in the BioTek Synergy 4 Multi-Detection microplate reader (BioTek Instruments, Winooski, VT). Fibrillation was followed by measuring the fluorescence intensity of ThT with the excitation and emission wavelengths set to 440 nm and 482 nm, respectively. Measurements were carried out at 15-min intervals for 8 h at 23°C with 5 s automixing before each reading. The data were fitted to the Boltzmann equation (Eq. 1) as described previously.
Turbidity measurement
Samples containing 0.6 mg/mL of glucagon in 3.2 mM HCl, 0.9% NaCl (w/v) (pH 2.5) were prepared in triplicate and incubated in 96-well crystal-clear microtiter plates. The turbidity of the solutions was measured by UV absorbance at 405 nm at 15-min intervals using a BioTek Synergy 4 Multi-Detection microplate reader (BioTek Instruments). Measurements were carried out for 8 h at 23°C with 5 s automixing before each reading. The data were fitted to the Boltzmann equation (Eq. 1).
CD spectroscopy
To monitor structural changes in glucagon during fibrillation, glucagon at 0.6 mg/mL in 3.2 mM HCl, 0.9% NaCl (pH 2.5) was allowed to fibrillate for 24 h. Samples were withdrawn at regular intervals, diluted to 0.3 mg/mL in 3.2 mM HCl, 0.9% NaCl (pH 2.5), and subjected to CD spectroscopy using a JASCO J-815 spectrometer (JASCO Analytical Instruments, Easton, MD). Spectra were collected in the far-UV range between 190 and 250 nm using a 1 mm path length cell with 0.2 nm bandwidth at 20°C. Each spectrum was an average of three scans with a scanning speed of 50 nm/min. CD spectra were analyzed using the online K2D3 program to estimate the secondary structure at each time point (39).
Amide HDX-MS
To determine the extent of deuterium uptake by glucagon, samples were prepared containing 0.6 mg/mL of glucagon in 3.2 mM HCl, 0.9% NaCl (w/v), pH 2.5, and centrifuged at 14,000 rpm for 3 min to remove any insoluble material. In-exchange was initiated by diluting the freshly prepared sample 10-fold in deuterated buffer containing 10 mM sodium citrate and 0.9% NaCl (w/v) (pHread 4.5), resulting in a final concentration of 90% D2O and 17 μM glucagon. Exchange was allowed to proceed at 23°C and samples withdrawn at various times (0.5–120 min). Each 20 μL sample was quenched by rapidly mixing with 40 μL of ice-cold solution containing 5 M urea, 5% methanol, and 0.2% FA (pH 2.5) and then flash frozen in liquid nitrogen and stored at −80°C until analysis.
To monitor glucagon fibrillation kinetics using HDX-MS, glucagon samples (0.6 mg/mL in 3.2 mM HCl, 0.9% NaCl, pH 2.5) were prepared and allowed to fibrillate at 23°C for 24 h. At various times during fibrillation, 5 min pulse labeling with deuterium was carried out. Pulse labeling was initiated by diluting a 2 μL sample of fibrillating glucagon in 18 μL of deuterated buffer containing 10 mM sodium citrate and 0.9% NaCl (w/v) (pHread 4.5), resulting in a final concentration of 90% D2O and 17 μM glucagon. The pulse-labeled samples were quenched and stored at −80°C as described previously until analysis.
To determine the extent of deuterium uptake by the mature fibrils, glucagon at 0.6 mg/mL was allowed to fibrillate at 23°C for 48 h. Triplicate 50 μL samples were centrifuged at 4000 rpm for 5 min to separate the fibril from supernatant. After discarding the supernatant, the fibrils were washed thrice with 500 μL of blank buffer (3.2 mM HCl, 0.9% NaCl (w/v), pH 2.5) to remove any nonfibrillar glucagon present in the samples. HDX was initiated by incubating fibrils in 50 μL of deuterated buffer (99% D2O) containing 10 mM sodium citrate and 0.9% NaCl (w/v) (pHread 4.5) at 23°C for 48 h. Reactions were quenched by quickly adding 100 μL of ice-cold solution containing 5 M urea, 5% methanol, and 0.2% FA (pH 2.5). Samples were flash frozen in liquid nitrogen and stored at −80°C as described previously.
Deuterium uptake was assessed for both intact glucagon and for proteolytic digests. For intact glucagon, deuterium uptake was measured using a high-performance liquid chromatography mass spectrometry (LC/MS) system (1200 series LC, 6520 Q-TOF; Agilent Technologies, Santa Clara, CA) equipped with a custom-built refrigeration unit that maintained the column at ∼0°C. The samples were quickly thawed and ∼28 pmole of glucagon was injected into a peptide microtrap (Michrom Bioresources, Auburn, CA). Samples were desalted for 2 min with 15% ACN, 85% water, and 0.1% FA and eluted in 2.3 min using a gradient to 90% ACN, 10% water, and 0.1% FA. Mass spectra were obtained over the m/z range 100–1700. For samples subjected to proteolytic digestion, ∼55 pmole of glucagon was injected into an immobilized pepsin column. Online digestion was carried out in water containing 0.1% FA at a flow rate of 0.2 mL/min. The digested sample was desalted in a peptide microtrap for 1.7 min and eluted using a gradient of ACN (10–60%) in 0.1% FA onto a reverse phase analytical column (Zorbax 300SB-C18; Agilent Technologies, Santa Clara, CA) at 50 μL/min. Undeuterated glucagon samples were also analyzed and their mass measured by tandem mass spectrometry (MS/MS) analysis (collision-induced dissociation fragmentation; MassHunter Software; Agilent Technologies). Peptides identified using undeuterated glucagon were mapped onto subsequent deuteration experiments using the semiautomated HX-Express software to obtain the average number of deuterons exchanged (40). For peptides showing a bimodal distribution of deuterium uptake, the percent deuteration was calculated from the weighted average of deuterium uptake in the accessible and protected populations. The bimodal spectral envelopes were fitted to the sum of two Gaussian distributions using OriginPro 8.6, and the relative area under each peak used to obtain the percentage of each population at different times during fibrillation. As the time between thawing and mass spectrometric analyses for all the HDX samples were very similar, the values obtained were not subjected to back exchange correction. All values are the mean of at least three independent HDX experiments.
The HDX kinetics of freshly prepared glucagon were fitted to a monoexponential association model (Eq. 2) for deuterium uptake:
| (2) |
where Dmax is the maximum deuteration at equilibrium, k is the rate constant, and t is deuteration time (min).
Deuterium uptake data from the peptides of fibrillating glucagon were fitted to a monoexponential decay model (Eq. 3):
| (3) |
where D0 is the deuterium uptake at t = 0 of fibrillation, D∞ is the deuterium uptake at t = ∞ of fibrillation; and k and t are the rate constant and the fibrillation time, respectively.
MD simulation
Initial structures of the N-terminal (residues 1–8) and C-terminal (residues 22–29) fragments were generated from reported NMR structures of glucagon (Protein Data Bank (PDB) ID: 1KX6) (41). Three different models were selected as starting configurations for MD simulations, and are referred to as models 1, 5, and 10 in keeping with the numbering in the ensemble of NMR models in the original PDB file. In simulations of the interactions of two molecules of either the 1–8 fragment or the 22–29 fragment, the molecules were initially placed close to one another with arbitrary relative initial orientation, maintaining at least a 4 Å distance between any two atoms in the two fragments. Combining the conformations of the three NMR models for each fragment, three starting configurations were generated for each of the N-terminal and C-terminal fragment simulations. Specifically, starting configurations for both the N-terminal fragment (1–8) and the C-terminal fragment (22–29) simulations were: model 1 with model 5, model 1 with model 10, and model 5 with model 10. All simulations were performed on capped peptides (i.e., N-terminus acetylated and C-terminus amidated) and the side chains of His residues in the N-terminal fragments were doubly protonated to represent the most likely state in solution at pH 2.5.
To simulate the interactions of peptide fragments, the molecules were solvated in a preequilibrated octahedron of TIP3P water molecules, with a minimum distance of 10 Å between the octahedron boundary and solute atoms (42). Production simulations were performed in an NPT ensemble using the AMBER-99SB force field with periodic boundary conditions and an integration time step of 2 fs, applying the particle mesh Ewald method to treat electrostatic interactions (43). All bonds involving hydrogen atoms were constrained using the SHAKE algorithm (44) and van der Waals interactions were truncated at a distance of 10 Å. A Langevin thermostat (45) with collision frequency of 1 ps−1 was used to maintain the temperature at 298 K, and pressure was maintained at 1 atm using isotropic position scaling with a pressure relaxation time of 2 ps. The N-terminal 1–8 fragment simulation was neutralized by the addition of one Cl− ion per fragment. In a simulation, the water molecules with constrained peptide(s) first were energy minimized. The system was then gradually heated from 0 K to 298 K over a 20 ps MD simulation period. The system was then equilibrated at constant temperature and pressure for 200 ps and final production runs performed for 100 ns. Snapshots were saved every 0.05 ns, resulting in 2000 snapshots for each production simulation.
A contact between residues from two molecules was identified if a distance <5 Å was observed between any pair of atoms. Only contacts formed between two different peptide molecules were analyzed, and not those within a single strand. All MD snapshots of the simulations were considered for contact analysis. The frequencies of observing contacts were first analyzed for the three separate simulations of two molecules, and then averaged over all three simulations to obtain a single mean contact frequency. The α-helix content of each snapshot was analyzed using the DSSP software (46,47). In simulations of two interacting peptides, snapshots were analyzed separately for each peptide. For each amino acid, the percentage of snapshots in which it was part of an α-helix substructure was computed for all simulations, and the mean structural content of the various single and two peptide molecule(s) simulations was computed. Molecules were visualized in PyMOL (48–50) and the graphs were generated using Python and matplotlib (51).
Results
Fibrillation kinetics using fluorescence and turbidity measurements
During fibrillation, a decrease in intrinsic fluorescence and an increase in ThT fluorescence with time were observed (Fig. 1 and Table 1). The decrease in intrinsic fluorescence is consistent with a change in the environment of the single fluorescent amino acid (Trp-25), which is solvent exposed in monomeric glucagon (24) and may become buried in the relatively hydrophobic environment produced by the fibrils. The increase in ThT fluorescence is consistent with the formation of β-sheet structures in the developing fibrils. An increase in turbidity was observed (Fig. 1 and Table 1) and is consistent with increasing particle number and/or size of particles as fibrillation proceeds. Fibrillation kinetics were characterized by lag, growth, and equilibrium phases, as reported previously for glucagon (24). Both fluorescence and turbidity signals follow a sigmoidal curve, which is characteristic of nucleation-dependent fibrillation (21,24). The lag times for fibrillation as measured by turbidity, intrinsic fluorescence, and ThT fluorescence were 148 ± 3 min, 164 ± 2 min, and 204 ± 2 min, respectively. Similarly, the t50 values for turbidity, intrinsic fluorescence, and ThT fluorescence measurements were found to be 230 ± 1 min, 212 ± 1 min, and 263 ± 2 min, respectively (Table 1). Because the lag time and t50 value were greater when measured by the ThT assay than when measured by intrinsic fluorescence, suggests that the formation of β-sheet-rich amyloid fibrils is preceded by the formation of oligomeric intermediates. The shortest lag time and t50 values were observed with turbidity measurements, implying that particles are detected before the detection of changes in structure by the other two methods. Due to differences in the methods and their limits of detection, however, these quantitative comparisons should be regarded as approximate.
Figure 1.
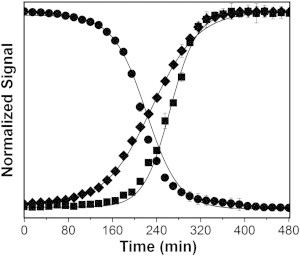
Kinetics of glucagon fibrillation by intrinsic fluorescence (solid circle), ThT fluorescence (solid square), and turbidity (solid diamond) measurements. Glucagon at 0.6 mg/mL was allowed to fibrillate and the change in signals were measured. Lines are fits to the Boltzmann equation (n = 3, mean ± SD). The regression parameters are given in Table 1.
Table 1.
Kinetic parameters for glucagon fibrillation as determined by intrinsic (Trp) fluorescence, ThT fluorescence, and turbidity measurements
| Measurement | Lag timea (min) | t50a (min) |
|---|---|---|
| Trp fluorescence | 164 ± 2 | 212 ± 1 |
| ThT fluorescence | 204 ± 2 | 263 ± 2 |
| Turbidity | 148 ± 3 | 230 ± 1 |
n = 3, mean ± SD.
Secondary structure changes during fibrillation by CD spectroscopy
CD spectroscopy has been used to study structural changes during amyloid fibrillation of proteins and peptides (52–57). Here, a total of 17 far-UV CD spectra acquired at different times were overlaid to show secondary structural changes during fibrillation (Fig. 2 A). Changes in the CD spectra are consistent with changes in secondary structure from random coil → α-helix → β-sheet during fibrillation. The solution structure of freshly prepared (t = 0 min) glucagon was primarily random coil with a negative band around 195 nm. An increase in α-helix content was observed for fibrillating glucagon, with the emergence of negative bands around 220 and 208 nm. The predicted structural composition shows maximal α-helix content at 150 min (Fig. 2). CD spectra at later times (180–1440 min) are consistent with increased β-sheet content, with negative and positive bands at 218 nm and 195 nm, respectively (Fig. 2).
Figure 2.
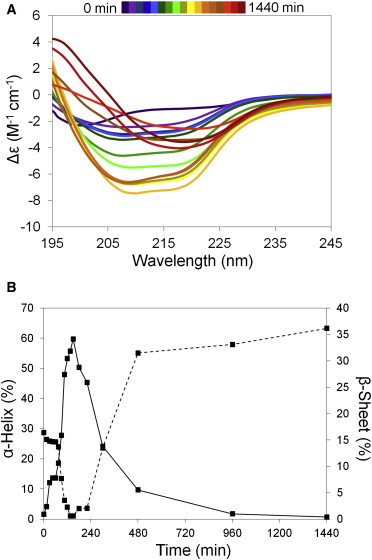
Structural changes during glucagon fibrillation. CD measurements were carried out at regular intervals for the fibrillating glucagon. (A) Overlay of CD spectra from fibrillating glucagon (0–1440 min). (B) Percent α-helix (solid line) and β-sheet (dashed line) content determined using the K2D3 program. To see this figure in color, go online.
Amide HDX-MS for monomeric glucagon
HDX of freshly prepared glucagon was carried out by diluting the samples 10-fold with D2O to give a final glucagon concentration of 60 μg/mL. Dilution is assumed to effectively quench fibrillation, although having little to no effect on the extent of deuterium uptake by monomeric glucagon. An increase in deuterium uptake was observed over the HDX time course, together with a single Gaussian-shaped isotope distribution, with no new peaks or significant peak broadening at any time point (Fig. S1 in the Supporting Material). The relationship between the number of deuterons incorporated and time was fitted to a monoexponential association model, with a resulting half-life (t1/2) of 5.3 min (Fig. 3 A). The half-life suggests that 5-min pulse labeling is adequate to monitor the structural changes during glucagon fibrillation, providing sufficient deuterium uptake (Fig. 3 A) without requiring too large a fraction of the fibrillation time course (Fig. 1).
Figure 3.
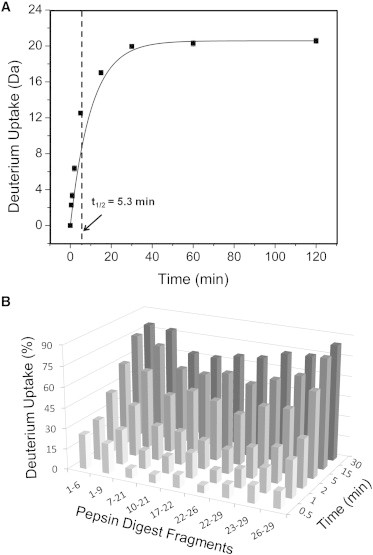
HDX kinetics for monomeric glucagon. HDX reactions were carried out for freshly prepared glucagon. Samples were withdrawn at various times (0.5–120 min) and analyzed using MS. Deuterium uptake was assessed for both intact glucagon and for proteolytic digests. (A) Deuterium uptake for intact glucagon as a function of time (0.5–120 min). Lines are fits to a monoexponential association function (Eq. 2) (n = 3). Standard deviations are less than the size of the symbols. The vertical dotted line indicates the half-life (t1/2 = 5.3 min). (B) Percent deuterium uptake for nine pepsin digest fragments from glucagon as a function of time (0.5–30 min) (n = 3).
Deuterium uptake in monomeric glucagon was also monitored at the peptide level following pepsin digestion. A total of 18 peptic fragments were detected by LC/MS. Of these, nine were selected (1–6, 1–9, 7–21, 10–21, 17–22, 22–26, 22–29, 23–29, 26–29) having reproducible strong MS signals and providing 100% sequence coverage with some overlap (Fig. S2). After 30 min of deuterium exposure, peptides 1–6, 1–9, and 26–29 showed 80% deuterium uptake, whereas fragments 7–21, 10–21, 17–22, and 22–26 showed ∼65% deuterium uptake (Fig. 3 B). Although this suggests some protection in the central portion of the glucagon molecule, the overall pattern and kinetics of exchange suggest that the molecule is unstructured, with uniform increase in deuterium incorporation along the sequence.
Amide HDX-MS for fibrillating glucagon
During fibrillation, the initial (t = 0) deuterated mass envelope is unimodal. After 15 min of fibrillation, a new peak envelope was observed (Peak I) with lower m/z and greater protection from exchange (Fig. 4 A). Generally, reduced deuterium uptake in HDX is consistent with greater conformational stability and/or greater protection from solvent (36). Here, we infer that Peak I corresponds to α-helical glucagon, or to glucagon oligomers, or early fibrils, which are protected from deuterium incorporation by intermolecular association and dissociate into monomers before LC/MS analysis. The intensity of Peak I increases with time, and the overall deuterium uptake (i.e., the weighted average of deuterium incorporation into the two peaks) decreases over time, providing additional support for this inference (Fig. 4 B). By 480 min of fibrillation, Peak II was no longer detected, indicating a complete shift in the population from monomeric to oligomeric glucagon. The amount of deuterium uptake as a function of fibrillation time (15–480 min) was fitted to a monoexponential decay model. A significantly shorter half-life (t50 = 114 ± 2 min) was observed for HDX than for Trp fluorescence (t50 = 212 ± 1 min), turbidity (t50 = 230 ± 1 min), or ThT fluorescence (t50 = 263 ± 2 min) measurements. This suggests that HDX probes structural changes and interactions that precede the changes measured by the other methods. A small decrease in the level of deuterium uptake was also observed for Peak I and Peak II with increase in fibrillation time (Fig. S3), suggesting the existence of intermediates within each population.
Figure 4.
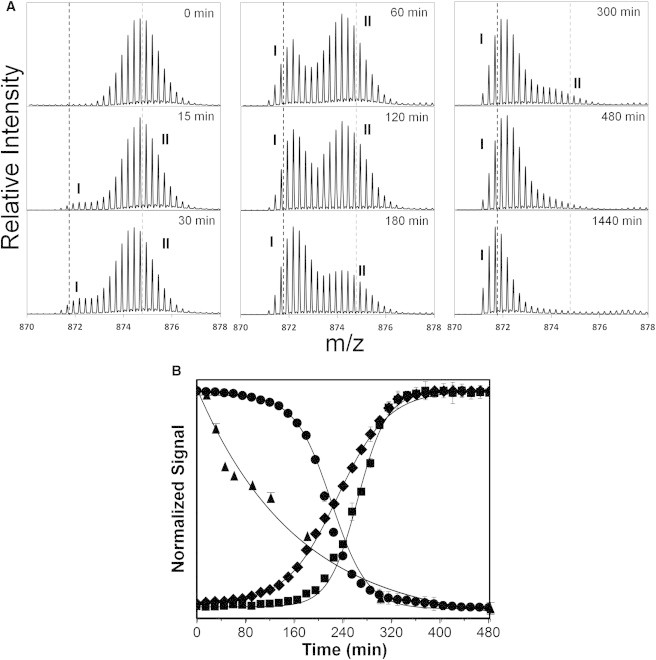
HDX-MS of fibrillating glucagon at the intact level. A 5 min pulse labeling with deuterium was carried out for the fibrillating glucagon at regular intervals (0–480 min) and deuterium uptake measured by MS analysis. (A) Enhanced deuterated mass envelope of intact glucagon (m/z = 871.1729, z = +4) showing bimodal peak distribution (Peak I and II) during fibrillation. Gray and black broken lines represent the centroid of the mass envelope after 0 min and 1440 min of fibrillation, respectively. (B) Glucagon fibrillation as monitored by tryptophan (Trp-25) emission at 355 nm (solid circle), ThT florescence at 482 nm (solid square), turbidity at 405 nm (solid diamond), and deuterium uptake by amide HDX-MS (solid triangle) (n = 3, mean ± SD). See text for details of methods and calculations.
Amide HDX-MS for fibrillating glucagon at the peptide level
To identify the particular regions of the glucagon sequence that are protected from HDX in the early stages of fibril formation, samples were subjected to proteolytic digestion before MS analysis. A total of nine overlapping peptides covering 100% of the glucagon sequence were identified and analyzed (Fig. S2). A decrease in deuteration with time was observed for all peptides (Figs. S4, 5 A, and S5), with the rate of decrease varying among the fragments. During fibrillation, overlapping fragments, 1–6 and 1–9 from the N-terminal region showed a decrease in deuterium uptake at a much slower rate than fragments from other regions, with t50 values ∼5× and ∼3× greater, respectively, than for the other peptides (Fig. 5 B). Peptic fragments 1–6, 1–9, and 10–21 still showed ∼25%, ∼14%, and ∼10% deuterium uptake, respectively, after 1440 min, whereas deuterium incorporation for the C-terminus (residues 22–29) was <1% after only 480 min of fibrillation (Figs. 5 A and S4). For the C-terminus, both the relatively rapid decrease in deuterium incorporation and the near-zero incorporation after 480 min suggest that this region is involved in the early stages of glucagon fibrillation. This result is consistent with the prediction of aggregation prone regions using AGGRESCAN and TANGO software (Fig. S6). Conversely, peptide 1–6 showed ∼60% deuterium uptake from t = 0 to 120 min with decreased deuteration only after 180 min, suggesting that this region does not participate in early fibrillation (Figs. 5 A and S4). Peptides 1–9, 10–21, and 22–29 showed intermediate behavior.
Figure 5.
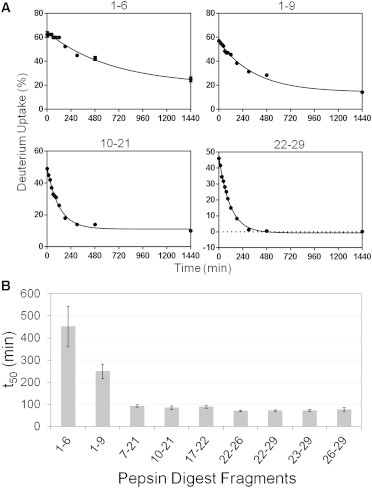
Interactions during glucagon fibrillation monitored by amide HDX-MS. A 5 min pulse labeling with deuterium was carried out for fibrillating glucagon at regular intervals (0–480 min). The deuterium uptake was measured by MS analysis following online pepsin digestion (A) Percent deuterium uptake for peptides 1–6, 1–9, 10–21, and 22–29 as a function of time of fibrillation. Lines represent fits to monoexponential decay; see text for details (n = 3, mean ± SD). (B) t50 values for the decrease in deuterium incorporation in the nine glucagon fragments (n = 3 ± SE).
Amide HDX-MS of mature glucagon fibrils
To identify the solvent accessible regions in mature fibrils, glucagon was allowed to fibrillate at pH 2.5 for 48 h and then exposed to D2O for up to an additional 48 h. The deuterated fibrils showed a bimodal mass envelope on MS analysis, with a second peak (Peak II′) showing increased deuterium incorporation appearing after 48 h of HDX (Fig. S7). Deuterium uptake values for the Peak I′ populations of peptic fragments 1–9, 10–21, and 22–29 after 48 h were ∼2%, ∼6%, and ∼1%, respectively (Fig. 6), indicating high protection from exchange consistent with interactions within the fibril. In contrast, peptic fragments 1–9, 10–21, and 22–29 in Peak II′ populations showed ∼64%, ∼80%, and ∼77% deuterium uptake, respectively, with region 10–29 more exposed than the N-terminal fragment (Fig. 6). The relative intensities of Peak II′ relative to Peak I′ (i.e., [Peak II′ intensity/Peak I′ intensity] × 100) for fragments 1–9, 10–21, and 22–29 are ∼36%, ∼28%, and ∼8%, respectively (Figs. S7 and 6).
Figure 6.
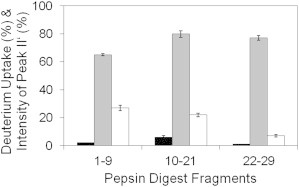
HDX-MS of mature glucagon fibrils. Glucagon at 0.6 mg/mL was allowed to fibrillate and subjected to HDX. Deuterium uptake was measured by MS analysis following online pepsin digestion. Percent deuterium uptake for the three glucagon peptides in Peaks I′’ (black bars) and II′ (gray bars) following 48 h of HDX (n = 3, mean ± SD). Intensity of Peak II′ relative to Peak I′ (white bars) (i.e., [Peak II′ intensity/Peak I′ intensity] × 100) for the three peptic fragments following 48 h of HDX (n = 3, mean ± SD).
Glucagon interactions by MD simulation
MD simulations were performed to provide insight into structural changes and early interactions involved in glucagon fibrillation. The α-helix content of fragment 1–8 was negligible in simulations of either one or two molecules, the latter allowing for effects of interaction on secondary structure (Fig. S8, A and B). In contrast, the C-terminal fragment 22–29 formed α-helices in both one- and two-molecule simulations (Fig. S8, C and D), with greater α-helix content in simulations of two molecules. To mimic the experimental conditions, MD simulations were repeated in the presence of 0.9% NaCl for a system containing two N-terminal fragments (model 1 with model 10) and two C-terminal fragments (model 1 with model 10). The simulations were performed for 15 ns and compared to the first 15 ns of the salt-free simulations. Though a slight increase in secondary structure was observed in the presence of salt, the difference in the α-helix content was minimal (data not shown).
In light of the experimental evidence that C-terminal interactions are involved in the early stages of fibrillation, we aimed to identify the critical contacts for the C-C-terminal interactions. When analyzing the contacts between amino acids, the C-terminal fragment 22–29 showed at least one contact in >94% of snapshots for all models tested. To highlight the preferred side-chain interactions, the 10 most frequently observed contacts averaged from three independent simulations of two molecules of the C-terminal fragment 22–29 were identified (Fig. 7). Hydrophobic interactions between amino acids are most frequently observed and account for eight of the 10 most frequent interactions. In particular, Trp-25 participates in four of the 10 most frequent interactions, i.e., with Phe-22, Val-23, Leu-26, and Met-27. Amino acids adjacent to Trp-25 also participated in hydrophobic contacts. Phe-22, for example, is engaged in five out of the top 10 most frequent interactions, four with hydrophobic or aromatic residues. An aromatic T-shaped interaction between Phe-22 and Trp-25 is also among the most frequent contacts.
Figure 7.
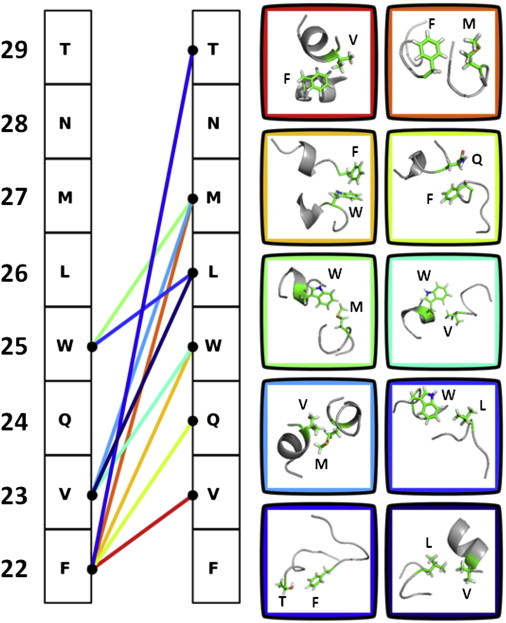
Identification of critical contacts for the C-terminal interactions in glucagon fibrillation under acidic conditions. The 10 most frequent contacts observed in simulations of two molecules of glucagon fragment 22–29 are shown. Each line represents one of the 10 interactions, which are ordered from red to blue based on frequency. The amino acid residues are indicated by their single letter code with residue numbers on the left. To see this figure in color, go online.
Discussion
Various mechanisms have been proposed for the molecular assembly of glucagon fibrils (23,27,28,32,58–60). One such mechanism is based on solution-state NMR studies, in which a glucagon trimer was shown to interact with the existing fibrils (28). HDX-NMR of mature glucagon fibrils in the same study suggested a mechanism in which the N-terminal residues are protected from deuterium exchange and form the core of the fibrils (28), though the molecular interactions at the early stage of glucagon fibrillation were not reported. AFM studies have shown that hollow disc-shaped oligomers are formed during the early stages of fibrillation, perhaps consistent with the presence of glucagon trimers. The oligomers observed by AFM were proposed to act as precursors for the formation of protofibrils, which in turn assemble to form mature fibrils (23,27,58,60). A study of alanine mutants of residues Phe-6, Tyr-10, Val-23, and Met-27 showed that the mutations alter glucagon hydrophobicity and impair its ability to form β-sheets, suggesting that these residues may be important in fibril formation (32). However, substitution of even a single residue can change the physicochemical properties of the peptide, so that inferences regarding the behavior of native glucagon based on these mutants may not be valid.
In the studies reported here, the fibrillation of native, monomeric glucagon was monitored using HDX-MS, intrinsic and ThT fluorescence, turbidity, CD-spectroscopy, and MD simulation, with the goal of identifying both the structural changes and the residues involved in glucagon fibrillation. HDX-MS is particularly well suited to these goals, providing high-resolution structural information with minimal perturbation of the native structure or the developing oligomers and fibrils.
The fibrillation kinetics of glucagon under acidic conditions were found to be sigmoidal, with a lag phase followed by rapid fibril growth, as reported previously (24). Kinetic analysis by turbidity and fluorescence measurements were also consistent with previous reports and followed the profile expected for nucleation-dependent fibrillation (Fig. 1). In a nucleation-dependent process, oligomeric species are formed during the lag phase until a sufficient number of nuclei have been produced to initiate fibril growth. Our HDX-MS results for fibrillating glucagon suggest the presence of two conformationally distinct populations. Unlike the fluorescence and turbidity measurements, the kinetic events observed with HDX-MS do not follow a sigmoidal curve. Rather, HDX-MS data show changes in deuterium exposure as early as 15 min from the initiation of the fibrillation experiment, whereas the fluorescence and turbidity measurements show significant changes only after ∼150 min. This suggests that HDX-MS monitors conformational changes involved in the lag phase of glucagon fibrillation, preceding fibrillation as detected by other methods. With proteolytic digestion, HDX-MS can also be used to identify the regions responsible for these early interactions, as discussed below.
At the intact level, HDX-MS of fibrillating glucagon suggests conformational changes between and within each population during the transition from monomer to fibril (Figs. 4 and S3). Results from both HDX-MS and far-UV CD spectroscopy suggest that glucagon fibril formation is a multistep process during which intermediates are formed at various stages of the reaction. In solution at concentrations below 1 mg/mL, glucagon has been shown to exist as a monomer with flexible random coil structure (25,61–63). At higher concentrations, glucagon self-associates and undergoes a structural transition from random coil to α-helix (61,64). CD spectral analysis of the freshly prepared sample (0.6 mg/mL) shows that glucagon is primarily a random coil (Fig. 2), and the single Gaussian peak distribution in the HDX-MS data (Fig. S1) confirms that glucagon is mostly monomeric at t = 0. An increase in α-helix content by CD was observed during the lag phase, reaching a maximum during the late-lag phase (∼150 min) (Fig. 2). This observation is consistent with a decrease in deuterium uptake with increasing fibrillation time (Fig. 4), and suggests that the α-helix formation during the early phase is mediated by intermolecular interactions. CD spectra at later times (220–1440 min) and ThT fluorescence measurements suggest that fibril growth during the exponential phase is accompanied by a structural transition from α-helix to β-sheet.
Following pepsin digestion, HDX-MS data at the peptide level suggest that the C-terminal region is involved in the interactions and structural changes during the early stages of glucagon fibrillation (Fig. 5). Identification of the regions involved is based on quantitative measurement of deuterium incorporation and protection from exchange. Quantitative determination of sequence-dependent protection from HDX shows that the N-terminal segment (residues 1–9) does not participate in the interactions involved in the early stages of fibrillation, because deuterium incorporation is relatively unchanged for this segment. In contrast, residues (22–29) spanning the C-terminal segment show decreases in deuterium incorporation consistent with intermolecular association (Fig. 5). Deuterium uptake for fragments 22–29 and 10–21 decreases from 46% to 15% and 49% to 26%, respectively, during the lag phase (0–120 min), whereas fragments 1–6 and 1–9 did not show significant changes during this period (Figs. 5 A and S4). These results support the idea that the intermolecular interactions involving the C-terminus occur during the lag phase.
HDX-MS of mature glucagon fibrils allowed the conformations of glucagon within the fibrils to be captured (Figs. 6 and S7). The high deuterium uptake (∼60% for 1–9, ∼80% for 10–21, and 77% for 22–29) for Peak II′ and low deuterium uptake (∼2% for 1–9, ∼6% for 10–21, and ∼1% for 22–29) for Peak I′ population suggest that the former corresponds to residues on the fibril surface, whereas the later corresponds to those in the core of fibril. The intensity of Peak II′ relative to Peak I′ for 22–29 (∼8%) is less than for other regions (∼36% for 1–9 and ∼28% for 10–21) (Fig. 6), again consistent with involvement of the C-terminus in fibril formation.
MD simulation of glucagon-derived peptides allowed the interactions and structural changes to be mapped at the residue level. MD simulation of two molecules of fragment 22–29 showed a greater increase in α-helix content than was observed in simulations of a single 22–29 fragment, suggesting that intermolecular interactions induce the mutual folding of C-terminal fragments. Simulations also showed that the side chains from residues Phe-22, Trp-25, Val-23, and Met-27 are predominantly involved in these interactions (Fig. 7), in agreement with previous claims that hydrophobic intermolecular interactions stabilize the α-helical conformation of glucagon (65). The high contact frequency observed in simulations of two molecules of fragment 22–29 indicates that C-terminal fragments tend to be engaged in intermolecular interactions, providing additional support for the involvement of the C-terminus in the early stages of fibrillation.
The results presented here are not in agreement with previous HDX-NMR studies at pH 2.0, in which the HDX reaction was carried out for mature glucagon fibrils that were then solubilized and analyzed by NMR spectroscopy (28). In that study, amide hydrogen atoms in the N-terminal region were found to be 20% solvent protected, whereas other regions showed 10% protection, suggesting the involvement of the N-terminal region in fibrillation. However, this inference is based on characterization of the end product (mature fibril) of fibrillation and not earlier monomeric and oligomeric species. Here, we have used HDX-MS to probe molecular interactions continuously from the early stages of fibrillation and provide evidence for the initial involvement of the C-terminus.
Overall, our results support a model for glucagon fibrillation in which interactions of hydrophobic residues in the C-terminal region, especially residues Phe-22, Trp-25, Val-23, and Met-27, initiate glucagon oligomerization at acidic pH. The C-terminal interactions lead to the formation of α-helix-rich oligomeric intermediates during the lag phase. The accumulation of oligomers is followed by structural rearrangements and elongation to form β-sheet-rich mature fibrils in the growth phase (Fig. 8). A similar mechanism has been proposed for insulin fibrillation, in which α-helical oligomers are thought to be the structural nucleus that elongates to form β-sheet-rich fibrils (66). However, the mechanisms by which α-helix-rich glucagon oligomers undergo a transition to β-sheet-rich amyloid fibrils were not elucidated by this study, and glugacon trimers were neither detected experimentally nor predicted by MD simulation. The progression from early oligomers to mature fibrils thus is not defined by the results reported here (Fig. 8).
Figure 8.
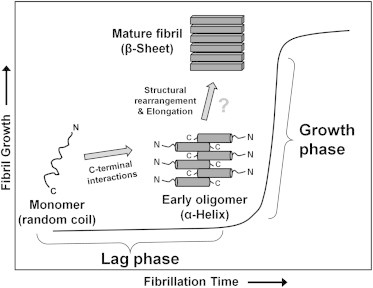
Proposed model for the aggregation of glucagon at acidic pH. In solution, glucagon exists as unstructured monomers. Early stage interactions in glucagon fibrillation are mediated through the C-terminal region, which results in the formation of α-helix-rich oligomers. Oligomers formed during the lag phase undergo structural rearrangements and elongation to form β-sheet-rich amyloid fibrils by mechanisms not defined by the results reported here.
Finally, the results support the use of amide HDX-MS to probe early structural changes in protein/peptide fibrillation. Due to its rapid analysis time, HDX-MS can provide high resolution information on conformational changes during fibrillation that are not easily monitored by methods such as NMR and x-ray crystallography. The results from HDX-MS and MD simulations further suggest that residues 22–29 may be a suitable target for inhibitors designed to inhibit glucagon fibrillation.
Conclusions
HDX-MS, far-UV CD, fluorescence spectroscopy, and MD simulation were used to map the structural transitions of glucagon during fibrillation. At acidic pH, the early stage interactions are mediated through the C-terminal region, which results in the formation of species with stable α-helical structure. These species precede the formation of fibrils as observed by fluorescence spectroscopy.
Acknowledgments
The authors thank Lavanya K. Iyer and Jainik P. Panchal from the Topp laboratory for their helpful suggestions and scientific discussions.
The authors gratefully acknowledge financial support from National Institutes of Health (NIH) R01 GM085293 (P.I. E. Topp) and from the College of Pharmacy at Purdue University.
Supporting Material
References
- 1.Chiti F., Dobson C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 2.Knowles T.P., Vendruscolo M., Dobson C.M. The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 2014;15:384–396. doi: 10.1038/nrm3810. [DOI] [PubMed] [Google Scholar]
- 3.Kayed R., Head E., Glabe C.G. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 4.Chimon S., Shaibat M.A., Ishii Y. Evidence of fibril-like β-sheet structures in a neurotoxic amyloid intermediate of Alzheimer’s β-amyloid. Nat. Struct. Mol. Biol. 2007;14:1157–1164. doi: 10.1038/nsmb1345. [DOI] [PubMed] [Google Scholar]
- 5.Flach K., Hilbrich I., Holzer M. Tau oligomers impair artificial membrane integrity and cellular viability. J. Biol. Chem. 2012;287:43223–43233. doi: 10.1074/jbc.M112.396176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Langkilde A.E., Vestergaard B. Methods for structural characterization of prefibrillar intermediates and amyloid fibrils. FEBS Lett. 2009;583:2600–2609. doi: 10.1016/j.febslet.2009.05.040. [DOI] [PubMed] [Google Scholar]
- 7.Lindgren M., Hammarström P. Amyloid oligomers: spectroscopic characterization of amyloidogenic protein states. FEBS J. 2010;277:1380–1388. doi: 10.1111/j.1742-4658.2010.07571.x. [DOI] [PubMed] [Google Scholar]
- 8.Myers J.K. Spectroscopic characterization of amyloid fibril formation by lysozyme. J. Chem. Educ. 2014;91:730–733. [Google Scholar]
- 9.Beaven G.H., Gratzer W.B., Davies H.G. Formation and structure of gels and fibrils from glucagon. Eur. J. Biochem. 1969;11:37–42. doi: 10.1111/j.1432-1033.1969.tb00735.x. [DOI] [PubMed] [Google Scholar]
- 10.Pedersen J.S. The nature of amyloid-like glucagon fibrils. J. Diabetes Sci. Tech. 2010;4:1357–1367. doi: 10.1177/193229681000400609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li P., Rogers T., Zhang F. Design, synthesis and crystallization of a novel glucagon analog as a therapeutic agent. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2007;63:599–601. doi: 10.1107/S1744309107028655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chabenne J., Chabenne M.D., Dimarchi R. A glucagon analog chemically stabilized for immediate treatment of life-threatening hypoglycemia. Mol. Metab. 2014;3:293–300. doi: 10.1016/j.molmet.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stigsnaes P., Frokjaer S., Moeller E.H. Characterisation and physical stability of PEGylated glucagon. Int. J. Pharm. 2007;330:89–98. doi: 10.1016/j.ijpharm.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 14.Nooijen W.J., Kempen H.J. Immunogenicity and bioactivity of glucagon, modified at methionine-27. Horm. Metab. Res. 1979;11:459–463. doi: 10.1055/s-0028-1092761. [DOI] [PubMed] [Google Scholar]
- 15.Unson C.G., Macdonald D., Merrifield R.B. Position 9 replacement analogs of glucagon uncouple biological activity and receptor binding. J. Biol. Chem. 1991;266:2763–2766. [PubMed] [Google Scholar]
- 16.Xiao Q., Giguere J., Wheeler M.B. Biological activities of glucagon-like peptide-1 analogues in vitro and in vivo. Biochemistry. 2001;40:2860–2869. doi: 10.1021/bi0014498. [DOI] [PubMed] [Google Scholar]
- 17.Joshi A.B., Kirsch L.E. The relative rates of glutamine and asparagine deamidation in glucagon fragment 22–29 under acidic conditions. J. Pharm. Sci. 2002;91:2331–2345. doi: 10.1002/jps.10213. [DOI] [PubMed] [Google Scholar]
- 18.Rodbell M., Birnbaumer L., Sundby F. The reaction of glucagon with its receptor: evidence for discrete regions of activity and binding in the glucagon molecule. Proc. Natl. Acad. Sci. USA. 1971;68:909–913. doi: 10.1073/pnas.68.5.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Unson C.G., Merrifield R.B. Identification of an essential serine residue in glucagon: implication for an active site triad. Proc. Natl. Acad. Sci. USA. 1994;91:454–458. doi: 10.1073/pnas.91.2.454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fang W.J., Qi W., Carpenter J.F. Effects of excipients on the chemical and physical stability of glucagon during freeze-drying and storage in dried formulations. Pharm. Res. 2012;29:3278–3291. doi: 10.1007/s11095-012-0820-7. [DOI] [PubMed] [Google Scholar]
- 21.Matilainen L., Larsen K.L., Jarho P. The effect of cyclodextrins on chemical and physical stability of glucagon and characterization of glucagon/gamma-CD inclusion complexes. J. Pharm. Sci. 2008;97:2720–2729. doi: 10.1002/jps.21209. [DOI] [PubMed] [Google Scholar]
- 22.Macchi F., Eisenkolb M., Otzen D.E. The effect of osmolytes on protein fibrillation. Int. J. Mol. Sci. 2012;13:3801–3819. doi: 10.3390/ijms13033801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dong M., Hovgaard M.B., Besenbacher F. AFM study of glucagon fibrillation via oligomeric structures resulting in interwoven fibrils. Nanotechnology. 2006;17:4003–4009. doi: 10.1088/0957-4484/17/16/001. [DOI] [PubMed] [Google Scholar]
- 24.Pedersen J.S., Dikov D., Otzen D.E. The changing face of glucagon fibrillation: structural polymorphism and conformational imprinting. J. Mol. Biol. 2006;355:501–523. doi: 10.1016/j.jmb.2005.09.100. [DOI] [PubMed] [Google Scholar]
- 25.Oliveira C.L., Behrens M.A., Pedersen J.S. A SAXS study of glucagon fibrillation. J. Mol. Biol. 2009;387:147–161. doi: 10.1016/j.jmb.2009.01.020. [DOI] [PubMed] [Google Scholar]
- 26.Andersen C.B., Hicks M.R., Otzen D.E. Glucagon fibril polymorphism reflects differences in protofilament backbone structure. J. Mol. Biol. 2010;397:932–946. doi: 10.1016/j.jmb.2010.02.012. [DOI] [PubMed] [Google Scholar]
- 27.De Jong K.L., Incledon B., DeFelippis M.R. Amyloid fibrils of glucagon characterized by high-resolution atomic force microscopy. Biophys. J. 2006;91:1905–1914. doi: 10.1529/biophysj.105.077438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Svane A.S., Jahn K., Nielsen N.C. Early stages of amyloid fibril formation studied by liquid-state NMR: the peptide hormone glucagon. Biophys. J. 2008;95:366–377. doi: 10.1529/biophysj.107.122895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Onoue S., Iwasa S., Yajima T. Structural transition of glucagon in the concentrated solution observed by electrophoretic and spectroscopic techniques. J. Chromatogr. A. 2006;1109:167–173. doi: 10.1016/j.chroma.2005.11.130. [DOI] [PubMed] [Google Scholar]
- 30.Pedersen J.S., Flink J.M., Otzen D.E. Sulfates dramatically stabilize a salt-dependent type of glucagon fibrils. Biophys. J. 2006;90:4181–4194. doi: 10.1529/biophysj.105.070912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jeppesen M.D., Westh P., Otzen D.E. The role of protonation in protein fibrillation. FEBS Lett. 2010;584:780–784. doi: 10.1016/j.febslet.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 32.Pedersen J.S., Dikov D., Otzen D.E. N- and C-terminal hydrophobic patches are involved in fibrillation of glucagon. Biochemistry. 2006;45:14503–14512. doi: 10.1021/bi061228n. [DOI] [PubMed] [Google Scholar]
- 33.Tartaglia G.G., Pawar A.P., Vendruscolo M. Prediction of aggregation-prone regions in structured proteins. J. Mol. Biol. 2008;380:425–436. doi: 10.1016/j.jmb.2008.05.013. [DOI] [PubMed] [Google Scholar]
- 34.Scavenius C., Ghodke S., Enghild J.J. Hydrogen exchange mass spectrometry as an analytical tool for the analysis of amyloid fibrillogenesis. Int. J. Mass Spectrom. 2011;302:167–173. doi: 10.1016/j.ijms.2010.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Y., Rempel D.L., Gross M.L. Pulsed hydrogen-deuterium exchange mass spectrometry probes conformational changes in amyloid beta (Aβ) peptide aggregation. Proc. Natl. Acad. Sci. USA. 2013;110:14604–14609. doi: 10.1073/pnas.1309175110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kheterpal I., Wetzel R. Hydrogen/deuterium exchange mass spectrometry—a window into amyloid structure. Acc. Chem. Res. 2006;39:584–593. doi: 10.1021/ar050057w. [DOI] [PubMed] [Google Scholar]
- 37.Kheterpal I., Chen M., Wetzel R. Structural differences in Abeta amyloid protofibrils and fibrils mapped by hydrogen exchange—mass spectrometry with on-line proteolytic fragmentation. J. Mol. Biol. 2006;361:785–795. doi: 10.1016/j.jmb.2006.06.066. [DOI] [PubMed] [Google Scholar]
- 38.Singh J., Udgaonkar J.B. Dissection of conformational conversion events during prion amyloid fibril formation using hydrogen exchange and mass spectrometry. J. Mol. Biol. 2013;425:3510–3521. doi: 10.1016/j.jmb.2013.06.009. [DOI] [PubMed] [Google Scholar]
- 39.Louis-Jeune C., Andrade-Navarro M.A., Perez-Iratxeta C. Prediction of protein secondary structure from circular dichroism using theoretically derived spectra. Proteins. 2012;80:374–381. doi: 10.1002/prot.23188. [DOI] [PubMed] [Google Scholar]
- 40.Weis D.D., Engen J.R., Kass I.J. Semi-automated data processing of hydrogen exchange mass spectra using HX-Express. J. Am. Soc. Mass Spectrom. 2006;17:1700–1703. doi: 10.1016/j.jasms.2006.07.025. [DOI] [PubMed] [Google Scholar]
- 41.Braun W., Wider G., Wüthrich K. Conformation of glucagon in a lipid-water interphase by 1H nuclear magnetic resonance. J. Mol. Biol. 1983;169:921–948. doi: 10.1016/s0022-2836(83)80143-0. [DOI] [PubMed] [Google Scholar]
- 42.Jorgensen W.L., Chandrasekhar J., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 43.Darden T., York D., Pedersen L. Particle mesh Ewald: an N-log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- 44.Ryckaert J.P., Ciccotti G., Berendsen H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 1977;23:327–341. [Google Scholar]
- 45.Loncharich R.J., Brooks B.R., Pastor R.W. Langevin dynamics of peptides: the frictional dependence of isomerization rates of N-acetylalanyl-N′-methylamide. Biopolymers. 1992;32:523–535. doi: 10.1002/bip.360320508. [DOI] [PubMed] [Google Scholar]
- 46.Joosten R.P., te Beek T.A., Vriend G. A series of PDB related databases for everyday needs. Nucleic Acids Res. 2011;39:D411–D419. doi: 10.1093/nar/gkq1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kabsch W., Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 48.Schrödinger, L. 2010. The AxPyMOL Molecular Graphics Plugin for Microsoft PowerPoint, Version 1.0.
- 49.Schrödinger, L. 2010. The JyMOL Molecular Graphics Development Component, Version 1.0.
- 50.Schrödinger, L. 2010. The PyMOL Molecular Graphics System, Version 1.3r1.
- 51.Hunter J.D. Matplotlib: a 2D graphics environment. Comput. Sci. Eng. 2007;9:90–95. [Google Scholar]
- 52.Goldsbury C., Goldie K., Aebi U. Amyloid fibril formation from full-length and fragments of amylin. J. Struct. Biol. 2000;130:352–362. doi: 10.1006/jsbi.2000.4268. [DOI] [PubMed] [Google Scholar]
- 53.Wilkins D.K., Dobson C.M., Gross M. Biophysical studies of the development of amyloid fibrils from a peptide fragment of cold shock protein B. Eur. J. Biochem. 2000;267:2609–2616. doi: 10.1046/j.1432-1327.2000.01270.x. [DOI] [PubMed] [Google Scholar]
- 54.Jha N.N., Anoop A., Maji S.K. Characterization of amyloid formation by glucagon-like peptides: role of basic residues in heparin-mediated aggregation. Biochemistry. 2013;52:8800–8810. doi: 10.1021/bi401398k. [DOI] [PubMed] [Google Scholar]
- 55.Arora A., Ha C., Park C.B. Insulin amyloid fibrillation at above 100 degrees C: new insights into protein folding under extreme temperatures. Protein Sci. 2004;13:2429–2436. doi: 10.1110/ps.04823504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Otzen D.E., Kristensen O., Oliveberg M. Designed protein tetramer zipped together with a hydrophobic Alzheimer homology: a structural clue to amyloid assembly. Proc. Natl. Acad. Sci. USA. 2000;97:9907–9912. doi: 10.1073/pnas.160086297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Giehm L., Svergun D.I., Vestergaard B. Low-resolution structure of a vesicle disrupting α-synuclein oligomer that accumulates during fibrillation. Proc. Natl. Acad. Sci. USA. 2011;108:3246–3251. doi: 10.1073/pnas.1013225108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhou X., Liu J., Zhang Y. Assembly of glucagon (proto)fibrils by longitudinal addition of oligomers. Nanoscale. 2011;3:3049–3051. doi: 10.1039/c1nr10332f. [DOI] [PubMed] [Google Scholar]
- 59.Ghodke S., Nielsen S.B., Otzen D. Mapping out the multistage fibrillation of glucagon. FEBS J. 2012;279:752–765. doi: 10.1111/j.1742-4658.2011.08465.x. [DOI] [PubMed] [Google Scholar]
- 60.Christensen P.A., Pedersen J.S., Otzen D.E. Spectroscopic evidence for the existence of an obligate pre-fibrillar oligomer during glucagon fibrillation. FEBS Lett. 2008;582:1341–1345. doi: 10.1016/j.febslet.2008.03.017. [DOI] [PubMed] [Google Scholar]
- 61.Andersen C.B., Otzen D., Rischel C. Glucagon amyloid-like fibril morphology is selected via morphology-dependent growth inhibition. Biochemistry. 2007;46:7314–7324. doi: 10.1021/bi6025374. [DOI] [PubMed] [Google Scholar]
- 62.Panijpan B., Gratzer W.B. Conformational nature of monomeric glucagon. Eur. J. Biochem. 1974;45:547–553. doi: 10.1111/j.1432-1033.1974.tb03580.x. [DOI] [PubMed] [Google Scholar]
- 63.Gratzer W.B., Beaven G.H., Bradbury E.M. A conformational study of glucagon. Eur. J. Biochem. 1968;3:276–283. doi: 10.1111/j.1432-1033.1968.tb19527.x. [DOI] [PubMed] [Google Scholar]
- 64.Srere P.A., Brooks G.C. The circular dichroism of glucagon solutions. Arch. Biochem. Biophys. 1969;129:708–710. doi: 10.1016/0003-9861(69)90231-8. [DOI] [PubMed] [Google Scholar]
- 65.Sasaki K., Dockerill S., Blundell T. X-ray analysis of glucagon and its relationship to receptor binding. Nature. 1975;257:751–757. doi: 10.1038/257751a0. [DOI] [PubMed] [Google Scholar]
- 66.Vestergaard B., Groenning M., Svergun D.I. A helical structural nucleus is the primary elongating unit of insulin amyloid fibrils. PLoS Biol. 2007;5:e134. doi: 10.1371/journal.pbio.0050134. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.