

Highlights
-
•
We reported the 80th case of chordoid glioma and reviewed the literature.
-
•
Clinical outcomes reported have been poor.
-
•
If possible, efficient treatment depends upon radical surgical resection, however partial resection with adjuvant radiosurgery can be the most recommend due to local tumor and morbid-mortality relation.
-
•
No chemotherapeutic regimen has been shown to be effective for CG.
Keywords: Chordoid glioma, Glioma/surgery, Third ventricle/pathology, Treatment outcome
Abstract
Introduction
Chordoid glioma is a rare low-grade brain tumor originating from the anterior wall of the third ventricle.
Case presentation
A 13-year-old female with progressive intermittent holocranial headaches and a diagnosis of chordoid glioma underwent tumor resection in our neuro-oncology unit.
Discussion
We review all 79 cases of chordoid glioma reported in the literature so far, focusing on the diagnostic criteria, treatment options and prognosis.
Conclusion
Efficient treatment of chordoid glioma depends upon radical surgical resection. Based on the reviewed data, which showed high morbi-mortality rates for this kind of tumor, we recommend a more conservative treatment approach.
1. Introduction
Chordoid glioma (CG) is a rare low-grade tumor that arises from the anterior wall or roof of the third ventricle. It was first described in 1998 by Brat et al. [1]. In 2000, it was incorporated into the World Health Organization (WHO) classification as grade II [2]. Chordoid glioma affects women at a rate of 2:1, and most patients are between 30 and 60 years of age [3].
This tumor was named chordoid glioma because of its distinctive histologic appearance, reminiscent of chordoma, and its avid staining with glial fibrillary acidic protein (GFAP) in immunohistochemical analyses [4]. After a cross-referenced PubMed search that yielded 79 published cases, we present one illustrative case and review the diagnostic criteria, treatment options and prognosis.
2. Case presentation
A 13-year-old girl presented with 2 weeks of progressive intermittent holocranial headaches, with no specific behavioral alterations. Magnetic resonance imaging (MRI) showed a well-defined lesion adjacent to the floor of the third ventricle, with little surrounding edema. The initial diagnosis was intraventricular meningioma, which was resected in 2003 at a regional hospital in Sao Paulo, Brazil.
In 2011, a new resection was performed due to lesion regrowth. The new histological diagnosis was gliosarcoma. An oncologist conducted 28 sessions of adjuvant radiotherapy (50, 4 Gy). Post-operatively, the patient evolved with diabetes insipidus (DI) and hypothyroidism. A follow-up with serial MRI was conducted and three months after this surgery, the patient began experiencing convulsive seizures.
In 2012, tumor regrowth in the hypothalamic region was detected via MRI. At this time, the patient presented with behavioral alterations characterized by psychomotor impairment, despite a Karnofsky score of 90 and an Eastern Cooperative Oncology Group (ECOG) score of 1 (Fig. 1).
Fig. 1.
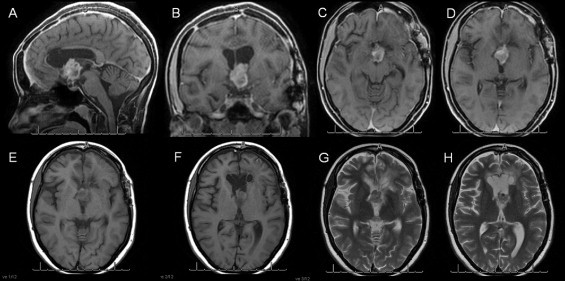
Coronal, sagittal and axial T1 gadolinium-contrast and T2 axial images showing a tumor in the 3rd ventricle.
When the patient was then referred to our oncology unit, we opted for a total transcortical resection guided by neuronavigation (Fig. 2). The material from the second surgery was sent to a different laboratory for a second histological diagnosis, along with material from the third surgery (Fig. 3). The anatomo-pathological report revealed a CG of the third ventricle. Postoperatively, the patient had improved psychomotor function and mild mental confusion, and maintained her previous Karnofsky and ECOG scores (90 and 1, respectively).
Fig. 2.
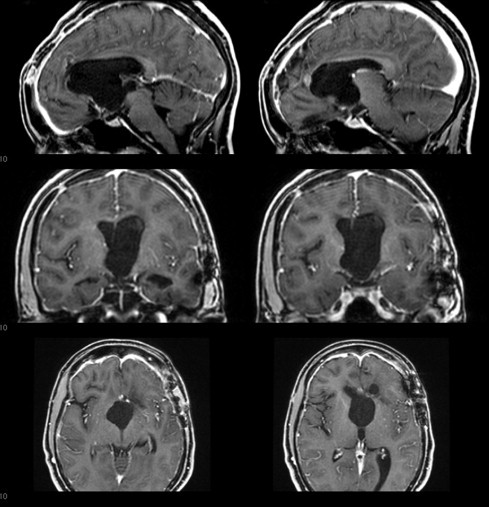
Immediate Postoperative coronal, sagittal and axial T1 gadolinium-contrast images.
Fig. 3.
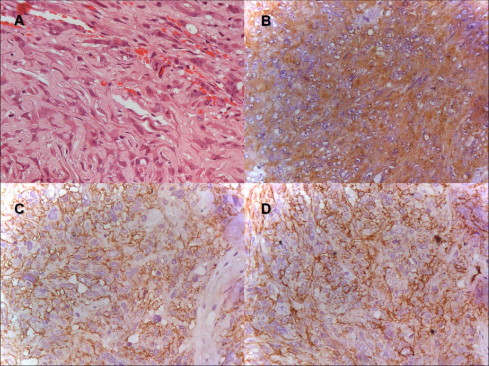
Photomicrographs of the tumor (original magnification x400). (A) Hematoxylin and Eosin stained slides show strands and ribbons of eosinophilic abundant cytoplasm and epitheliod neoplastic cells on myxoid basophilic matrix. (B) Immunostained slide for glial-fibrillary acid protein (GFAP) shows diffuse and strong expression in neoplastic cells. (C) Immunostained slide for epithelial membrane antibody (EMA) shows diffuse and strong expression in neoplastic cells. (D) Immunostained slide for CD 34 shows focal and strong expression in neoplastic cells.
3. Discussion
3.1. Clinical presentation
Initial presentations of CG vary from asymptomatic to aggressive, and most cases show slow and mild progression. Specific symptoms are attributable to the specific tumor location and to the involvement of adjacent structures responsible for possible endocrine alterations, visual deficits and behavioral alterations, as well as symptoms secondary to an obstructive hydrocephalus [4–6].
In the case reported herein, the patient initially presented with a headache and as the tumor progressed, developed behavioral and endocrine complications.
3.2. Differential diagnoses
Suprasellar masses (including pituitary macroadenomas, craniopharyngiomas, meningiomas, metastases, optic and hypothalamic pilocytic astrocytomas) account for more than 75% of these lesions. Tumors of the third ventricle constitute an uncommon subset of suprasellar masses, and offer differential diagnostic value, excluding meningiomas, craniopharyngioma, hypothalamic gliomas, hypothalamic hamartoma, germ cell tumors, and others [7,8].
Meningioma is among these differential diagnoses, which was our patient’s initial diagnosis. Kobayashi et al [2] also reported three cases with initial diagnoses of ventricular meningioma. These neoplasms had histologic appearance of cords and clusters of epithelioid cells within a mucinous background, along with a low-grade lymphoplasmacytic infiltrate reminiscent of a chordoma or chordoid meningioma. Unlike meningiomas, which are not glial in origin, all CG stained avidly for the glial cell marker GFAP [2,9].
3.3. Diagnostic imaging
CG appears hyperdense on computed tomography (CT) in 65% of cases, and enhances homogeneously following contrast administration in 68%. CG may be confused with other hyperdense lesions, such as lymphoma, meningioma and aneurysms.
MRI is preferred for CG because it clearly depicts the hypothalamic involvement through multiplanar imaging. The MRI findings characteristic of CG are an isointense ovoid mass on T1-weighted MR (63% of cases) and slightly hyperintense T2-weighted images, with homogeneous enhancement by gadolinium (70%). Most tumors are solid, but 25% of them display some cystic area within their periphery. Sagittal images clearly depict the infundibulum displaced posteriorly, and the largest portion of these ovoid masses generally occurs in the superoinferior orientation [10,11].
3.4. Pathological diagnosis
Histopathologically, the tumor consists of clusters and cords of oval to polygonal epithelioid-shaped cells with eosinophilic cytoplasms, which are embedded in a mucinous matrix. Mitoses are rare and necrosis is absent. Lymphoplasmacytic infiltrates and Russell bodies are often found in the tumor [9,12].
Immunohistochemically, CG is strongly and diffusely positive for GFAP and vimentin, and shows focal reactivity to EMA and cytokeratin in some cases, but little or no reactivity to S-100 protein.
Some authors reported immunoreactivity for CD34, which is useful for making differential diagnoses between chordoid meningioma, pilocytic astrocytoma, and/or ependymoma [2,9,12].
The diagnosis of CG was supported in our case by both the well-characterized morphology and immunohistochemistry findings. The pathologic examination disclosed cords and clusters of epithelioid cells within a mucinous matrix (Fig. 3A). No anaplastic features were identified. Immunohistochemically, tumor cells were positive for GFAP (Fig. 3B), EMA (Fig. 3C) and focal for CD 34 (Fig. 3D), as well as negative for pan-cytokeratin, protein S-100 and synaptophysin. The Ki-67 labeling index was 2%. No accumulation of p53 was observed.
The diagnosis in the current report was challenged by the location of the tumor and by secondary alterations related to previous treatment, which could also be associated with chordoid meningioma, chordoma and metastatic epithelial neoplasms. Given that necrosis, vascular alterations and hemorrhage are morphologic features of primary high-grade neoplasms or metastasis, these diagnoses are ruled out by the fact that we did not observe these characteristics and that surgery and previous radiotherapy had therapeutic effects. Moreover, the neoplastic cells showed glial differentiation and a low proliferative index, as well as a lack of epithelial or chondro-osseous markers. These findings ruled out the diagnoses of meningioma, chordoma and carcinoma.
The histogenesis of CG remains uncertain. Two recent reports suggested a subcommissural origin based on ultrastructural findings. Electron microscopy findings are also suggestive of glial origin, especially of ependymal cells known as tanycytes, which are located in the anterior third ventricle and cover the circumventricular organs in the anatomic vicinity of the third ventricle, the organum vasculosum of the lamina terminalis and the subfornical organ [6,10,13].
In all cases of CG reported to date, the anatomic site of origin can be traced to the lamina terminalis. This origin accounts for a number of the tumor’s observed properties, including the circumscribed gross morphology, the fusiform shape, and certain specific endocrine disturbances that are regulated by the organum vasculosum [8,12].
The vasculature of these circumventricular organs lacks a blood-brain barrier, which is associated with the typical uniform contrast-enhancement observed in neuroimaging studies of CG [10].
3.5. Treatment
There is controversy over the most appropriate treatment for CG. Most reported cases were surgically resected (either with complete or subtotal resection) while others were submitted to stereotactic biopsy.
The current treatment of choice for CG is surgical resection. Gross total resection (GTR) is the ideal treatment, although tumor size and location often impede radical resection. When this is the case, the risk of surgical morbidity (often associated with optical and endocrinological symptoms) cannot be ignored. Low grade, incompletely resected tumors will continue to grow slowly and may require later resection.
The few cases that reported using chemotherapy and/or radiotherapy showed these therapies to be ineffective [2] Gamma knife radiosurgery (GKRS), indicated 2–4 months after a partial resection, has also shown acceptable effectiveness [2,13].
There is enough evidence to support indication of stereotactic radiosurgery after partial resection or biopsy, as patients who received this treatment had better overall survival rates without major morbidity (they mainly experienced optic neuropathy). Nevertheless, total radiation dose and treatment volume remain to be determined [13].
3.6. Complications
Mortality rates and complications related to the surgical procedures vary according to surgical technique. GTR has led to postoperative death in 29% of cases, and a 67% chance of complications among survivors. After partial resection, mortality and morbidity decreased to 14% and 50%, respectively. In cases where only biopsy was performed, no deaths or complications were reported. GTR carries significant risk because of the proximity between the tumor and critical neurovascular structures [13,14]. In 2010, DeSouza et al. [15] reported that death is most likely to occur in the first month postoperatively due mostly to pulmonary embolism (42%) or cardiovascular causes.
The most commonly-reported complications are: cognitive disorders, pulmonary embolism and hypothalamic dysfunction. There is also a high incidence of postoperative thromboembolic events, but the causes behind these events have yet to be identified [2,5,13].
3.7. Outcome
Clinical outcomes for CG cases reported so far have been poor, which is partially due to the deep location of the tumor, and also due to typical tumor features such as solid consistency and adherence to critical neurovascular structures, which often render radical resection difficult or impossible [13–15].
4. Conclusion
CG has distinct features that allow for a definitive diagnosis. Efficient treatment depends upon radical surgical resection. Based on the reviewed data, which showed high morbi-mortality rates for this kind of tumor, we recommend a more conservative treatment approach.
In cases where local circumstances preclude radical resection, partial resection with adjuvant radiosurgery should be performed. To date, no chemotherapeutic regimen has been shown to be effective for CG.
Conflict of interest
There is no conflict of interests involving the authors and the contents in this manuscript.
Funding
None of the authors received grant money or other financial support for this study. This project was not funded.
Ethical approval
This manuscript was approved by the Ethics Committee of our neuro-oncology unit of the Hospital das Clínicas, Universidade de Sao Paulo. Written informed consent will be sent in the attached submission.
Author contribution
Barbara A. Morais and Djalma F.S. Menendez were responsible for the conception and design of the study, acquisition of data and analysis and interpretation of data.
Guilherme A. Lepski, Raphael S.S. Medeiros and Manoel J. Teixeira were responsible for drafting the article and revising it critically for important intellectual content, furthermore they issued the final approval of the version to be submitted.
Consent
There are no characteristics altered which distort scientific meaning. We have obtained written consent from the patient’s guardian (father) which is attached on submission online.
Guarantor
Guilherme A. Lepski.
References
- 1.Brat D.J., Scheithauer B.W., Staugaitis S.M., Cortez S.C., Brecher K., Burger P.C. Third ventricular chordoid glioma: a distinct clinicopathologic entity. J. Neuropathol. Exp. Neurol. 1998;57:283–290. doi: 10.1097/00005072-199803000-00009. [DOI] [PubMed] [Google Scholar]
- 2.Kobayashi T., Tsugawa T., Hashizume C., Arita N., Hatano H., Iwami K. Therapeutic approach to chordoid glioma of the third ventricle–three case reports and review of the literature. Neurol. Med. Chir. 2013;53:249–255. doi: 10.2176/nmc.53.249. [DOI] [PubMed] [Google Scholar]
- 3.Zarghouni M., Vandergriff C., Layton K.F., McGowan J.B., Coimbra C., Bhakti A. Chordoid glioma of the third ventricle. Proceedings (Bayl. Univ. Med. Center) 2012;25(3):285–286. doi: 10.1080/08998280.2012.11928853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pomper M.G., Passe T.J., Burger P.C., Scheithauer B.W., Brat D.J. Chordoid glioma: a neoplasm unique to the hypothalamus and anterior third ventricle. AJNR Am. J. Neuroradiol. 2001;22:464–469. [PMC free article] [PubMed] [Google Scholar]
- 5.Dziurzynski K., Delashaw J.B., Gultekin S.H., Yedinak C.G., Fleseriu M. Diabetes insipidus, panhypopituitarism, and severe mental status deterioration in a patient with chordoid glioma: case report and literature review. Endocr. Pract. 2009;15(3):240–245. doi: 10.4158/EP.15.3.240. [DOI] [PubMed] [Google Scholar]
- 6.Baehring J.M., Bannykh S. Chordoid glioma of the third ventricle. J. Neurooncol. 2006;76(3):269. doi: 10.1007/s11060-006-6054-y. [DOI] [PubMed] [Google Scholar]
- 7.Hinai Q.S.A., Petrecca K. Rarest of the rare: chordoid glioma infiltrating the optic chiasm. Surg. Neurol. Int. 2011;2:53. doi: 10.4103/2152-7806.80118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Scheurkogel M.M., van Duinen S.G., Verstegen M.J., Lycklama à Nijeholt G.J. Chordoid glioma: a rare suprasellar mass. Acta Neurol. Belg. 2012;112(3):311–314. doi: 10.1007/s13760-012-0084-3. [DOI] [PubMed] [Google Scholar]
- 9.Ni H.C., Piao Y.S., Lu D.H., Fu Y.J., Ma X.L., Zhang X.J. Chordoid glioma of the third ventricle: four cases including one case with papillary features. Neuropathology. 2013;33(2):134–139. doi: 10.1111/j.1440-1789.2012.01333.x. [DOI] [PubMed] [Google Scholar]
- 10.Sanches P., Yamashita S., Freitas C.C.M., Resende L.A.L. Chordoid glioma of the third ventricle: a new case report. Radiol. Bras. 2012;45(5):288–290. [Google Scholar]
- 11.Tu A., Yeo T., Steinke D., Resch L., Mehta V. Chordoid glioma: imaging pearls of a unique third ventricular tumor. Can. J. Neurol. Sci. 2010;37(5):677–680. [PubMed] [Google Scholar]
- 12.Brat D.J. The elusive origin of chordoid glioma. Arch. Pathol. Lab. Med. 2006;130(4):437–438. doi: 10.5858/2006-130-437-TEOOCG. [DOI] [PubMed] [Google Scholar]
- 13.Vij M., Jaiswal S., Jaiswal A.K., Jain M., Behari S. Chordoid glioma of the third ventricle: a case report with review of literature. Neurol. India. 2011;59:469–471. doi: 10.4103/0028-3886.82740. [DOI] [PubMed] [Google Scholar]
- 14.Jung T.Y., Jung S. Third ventricular chordoid glioma with unusual aggressive behavior. Neurol. Med. Chir. 2006;46(12):605–608. doi: 10.2176/nmc.46.605. [DOI] [PubMed] [Google Scholar]
- 15.DeSouza R.M., Bodi I., Thomas N., Marsh H., Crocker M. Chordoid glioma: ten years of a low-grade tumor with high morbidity. Skull Base. 2010;20:125–138. doi: 10.1055/s-0029-1246223. [DOI] [PMC free article] [PubMed] [Google Scholar]