

Highlights
-
•
The clinical presentation of mixed gonadal dysgenesia is known as criptorcidism and undesending testis.
-
•
Mixed Gonadal Dysgenesia with mosaism 45, X/46, X, +mark karyotype are rare case.
-
•
We present a rare patient case with mixed gonadal dysgenesis as a disorder of sex development (DSD) and a new pattern of chromosome in the karyotype, undergoing laparoscopic procedure for sex correction.
Abbreviations: MGD, mixed gonadal dysgenesis; mark, mar; TH, true hermaphroditism; AMH, anti-Müllerian hormone; UDT, undescended testis; SRY, sex determined region of Y chromosome; kg, kilogram; cm, centimeter; EMS, external masculinization scores; HRT, hormone replacement therapy; HCG, human chorionic gonadotropin; IGF-I, insulin like grows factor 1; PCR, polymerase chain reaction; GTG, generate test generator; MPH, mid-parental height; GH, growth hormone
Keywords: Mixed gonadal dysgenesis, Karyotype, Laparoscopic surgery
Abstract
Introduction
We present a rare patient case with mixed gonadal dysgenesis as a disorder of sex development (DSD) and a new pattern of chromosome in the karyotype, 45, X/46, X, +mar(Y).
Presentation of case
A ten-year-old boy, raised in a nursery center, presented with ambiguous genitalia. Two cell lines, (45, X) and [46,X, +mar(Y)] were observed utilizing cytogenetic investigation including fluorescence in situ hybridization (FISH) which were carried out on his peripheral lymphocytes. A significantly higher percentage (75%) of Y-containing cells was observed in the blood, which could be considered the major reason why the case did not have distinct ambiguous genitalia. A further explorative laparoscopic procedure was performed, during which orchiectomy was performed, and remnants of Müllerian duct were excised.
Discussion
A complete and sufficiently careful medical evaluation and genetics counseling of neonates is highly recommended in order to avoid any delayed insufficient diagnostic, conservative, and therapeutic care in children living with guardians rather than their biological parents. Both molecular and cytogenetic studies are recommended in some DSDs to help early diagnosis of the disease, which is important for further essential surgical approaches.
Conclusion
Cytogenetic studies followed by a laparoscopic exploratory and surgical survey are helpful tools for unraveling the mosaicism involving sex chromosomes and the complicated process in mixed gonadal dysgenesis patients.
1. Introduction
Gonadal dysgenesis refers to a variety of clinical conditions in which abnormal development of the fetal gonad is present. It consists of 46, XY gonadal dysgenesis, mixed gonadal dysgenesis (MGD), and 45, X Turner syndrome [1]. MGD, a disorder of sex development (DSD), is defined in individuals who typically have a differentiated gonad on one side and a streak gonad or streak testis (usually intra-abdominal) on the other side. Persistent Müllerian structures such as the ovary or uterus may also be present [2]. However, true hermaphroditism (TH) cases have both unequivocal ovarian tissue and testicular elements independent to their karyotypes. TH and MGD should be differentiated, and a counseling physician should be consulted to decide for early gonadectomy in MGD to prevent the high risk of malignancy [3,4].
Sex chromosome mosaicism (45, XO/46, XY) is the most common karyotype expressed in MGD and may cause formation of a dysgenetic or malformed gonad. This is referred to as a streak gonad that does not produce enough testosterone [2,5]. Ambiguous genitalia is the most common presentation for individuals with a 45X/46XY karyotype accounting for about 60% of involved individuals [6]. In clinical examination, undescended testis (cryptorchidism) and/or hypospadias may be found. For this reason, a chromosomal study via karyotyping is necessary to be done for any patient with penoscrotal hypospadias and undescended testis [7]. Measurement of the level of anti-Müllerian hormone (AMH), which is usually insufficient in dysgenetic gonad, can also help us to differentiate any type of intersexual condition [8]. In these conditions, the patients have female reproductive organs such as a hemi-uterus and fallopian tube in testicular dysgenesis [9]. Meanwhile, patients with impaired testosterone secretion show the level of AMH either within normal limit or slightly elevated [8].
Y chromosome behavior is a strong risk factor for germ cell tumors [10]; however, hormone production via saving testis is very varied and depends on each case individually [11–13].
The correlation of clinical phenotype in MGD with cancer risk has been shown to be as high as 52% [12]. In mild MGD, spermatogenesis may be impaired in adulthood, resulting in infertility or subfertility. However in more severe MGD, testicular cancer may be observed at a younger age [6,14].
We present a rare case with MGD and a new pattern of chromosome in the karyotype, 45, X/46, X, +mar(Y).
2. Case presentation
A ten-year-old boy was referred to the pediatrics clinic of Hazrat Rasul Akram Hospital with left-sided cryptorchidism, penoscrotal hypospadias, and growth retardation. His height was 118.7 cm, and his body weight was 22 kg (25th Percentile). He was raised in a nursery center as a result of his father’s incompetence and some family problems, which might be the reason why he had not sought any medical advice earlier. His physical examination consisted of a significant short stature, low-set hair line, a gonadal phenotype similar to male genitalia with hypospadias, chordee penis, and a left-sided undescended testis (UDT). In karyotype, 20 metaphases were assessed on the basis of GTG technique at 450–500 band resolution including five plates (25%) of 45X, and the remaining plates (75%) revealed 46 chromosomes with a small marker chromosome (45, X [5]/46, X, +mar [15]. Thus, a molecular study was planned to determine the origin of the marker chromosome. In the molecular study, DNA was extracted from peripheral blood leukocytes to seek the sex-determined region of the Y chromosome (SRY) gene by PCR technique. The specific probes for X and Y chromosomes were also applied to approach the origin of the marker in a FISH study. These results revealed presence of both X and Y chromosomes and SRY genes which were compatible with a male phenotype. The laboratory tests are included in Table 1.
Table 1.
The results of the patient’s serum tests.
| No | Lab test: | |
|---|---|---|
| 1 | FSH-ECL | 1.76 mlU/ml |
| 2 | FSH/LH Ratio | 14.67 mIU |
| 3 | LH-ECL | 0.12 mIU LOW |
| 4 | Testosterone | <.025 ng/ml LOW |
| 5 | 17OH-Prog | 0.25 ng/mlu |
| 6 | DHEA-SO4, ECL | 26.7 Micg/dl |
| 7 | Androstendione-RIA | 0.4 ng/ml |
| 8 | A.C.T.H | 8.5 pg/ml |
| 9 | CORTISOL, ECL (AM) | 14.5 |
| 10 | WBC | 4500 |
| 11 | Hb | 11.5 |
| 12 | MCV | 75.3 |
| 13 | Blood sugar | 79 |
| 14 | Sodium | 140 |
| 15 | Potassium | 4.4 |
Note: FSH: follicle-stimulating hormone, ECL: electrochemiluminescency, LH: luteinizing hormone, 17OH-Prog: 17-hydroxy-progesterone, DHEA-SO4: dehydroepiandrosterone, RIA: radioimmunoassay, A.C.T.H: adrenocorticotropic hormone, WBC: white blood cells, Hb: hemoglobin, MCV: mean corpuscular volume.
Ultrasound examination revealed no left testis in the scrotum or inguinal canal, and no sign of remnants of Müllerian structures, such as the uterus, was appreciated. After counseling with the patient, his family, and pediatric psychologist, the decision was made to pronounce the patient male gendered. Subsequently, explorative laparoscopy was offered, and left testis was seen on the left internal ring. In addition, the fallopian tube and a single ovary were seen on the same side, but no uterus was observed. (Figs. 1 and 2) In the next step, laparoscopy orchiectomy was performed, and remnants of Müllerian duct were excised. In a histopathologic review of specimen, streak ovary, fallopian tube, and testis were confirmed. The patient was referred for urology and endocrinology evaluation, and a trial of GH therapy for treatment of short stature was considered.
Fig. 1.
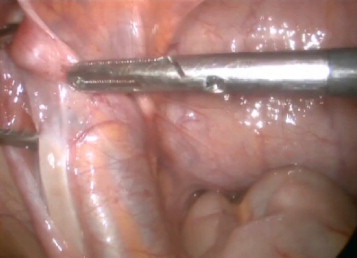
Intra-abdominal testis.
Fig. 2.
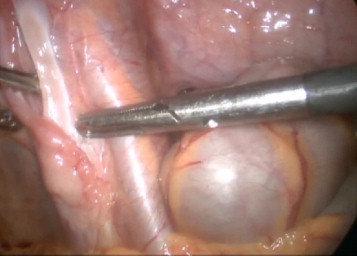
Fallopian tube.
3. Discussion
Sex chromosomal mosaicism is the common karyotype in MGD. Johansen and coworkers reported structural rearrangement of the Y chromosome in 63% of MGD patients [15,16]. However, normal XY karyotype was reported by Anand et al. [17]. In our case, mosaic chromosomal pattern with a chromosome marker was found. Therefore, PCR was performed for the sex-determining region of the Y chromosome (SRY) upon proposal of MGD as the primary diagnosis. Most MGD cases require corrective surgery because of the two mismatched gonads. In case of the decision to convert to male gender, female ductal structures should be removed. If the child’s phallus is small, she would function better as a female; therefore, the testis should be removed [2,5,15]. Normal right side testis in our patient produced adequate testosterone to enhance puberty and body shape formation. In addition, the patient was raised as a male, and he preferred to stay as such. Although there is no exact prediction of tumor risk in normal appearing males with mosaic (45X, 46XY) karyotype, tumor risk was anticipated low in our patient due to normal testicular differentiation and maturation process present in the clinical manifestation, including a normal external masculinization score (EMS) and bilaterally descended testes [18]. Therefore, according to this observation, we decided to preserve our patient’s right testis with close follow-up in order to prevent gonadoblastoma formation.
However, early diagnosis and evaluation is very important for preventing psychological sequels as well as ruling out other abnormalities. Meanwhile, a complete and sufficiently careful medical evaluation and genetics counseling of neonates referred to nurseries is highly recommended to avoid any delayed and insufficient diagnostic, conservative, therapeutic, and psychological care in children with guardians rather than their biological parents. These also include exclusion other congenital and/or genetic abnormalities such as congenital adrenal hyperplasia.
Johansen et al. reported an adequate gonadal function in male gender to produce pubertal phase. They showed that EMS, defined by Ahmed et al. [19], acts as a sign of clinically malfunctioning gonads in patients with a very low EMS [15]. Phonotypical males with a 45, X/46, XY karyotype and its variants assume to have normal testicular function and spontaneous puberty even in patients with ambiguous genitalia [18,20]. However, some patients might need hormone replacement therapy (HRT) after spontaneous pubertal onset [20]. In male patients with MGD, the cause of infertility is UDT and dysgenetic testis.
The prevalence of growth retardation is high although it seems not to be due to growth hormone (GH) deficiency. In our case, trial of GH therapy for was considered treatment of short stature.
Providing a multidisciplinary approach to managing these children is crucial, like our medical team, which included a pediatric endocrinologist, genetics counselor, laparoscopic surgeon, and psychologist.
4. Conclusion
Patients with mixed gonadal dysgenesis have a different presentation and variant of chromosomal abnormalities. A multidisciplinary approach is necessary and crucial for early diagnosis and proper management of these patients in order to prevent mental and social sequels.
Conflict of interest
All authors declare that they have no financial conflict of interest, and the supporting organization has supported the scientific aspect of the research.
Funding
All authors declare that they have received no funding for this case report.
Author contribution
| Author name | Study concept and design | Acquisition of data | Statistical analysis and interpretation of data | Drafting of the manuscript | Critical revision of the manuscript for important intellectual content | Administrative, technical, and material support |
|---|---|---|---|---|---|---|
| Fahimeh Soheilipour | ✓ | ✓ | ✓ | ✓ | ||
| Ommolbanin Abed | ✓ | ✓ | ||||
| Babak Behnam | ✓ | ✓ | ✓ | |||
| Mohammadreza Abdolhosseini | ✓ | ✓ | ✓ | |||
| Peyman Alibeigi | ✓ | ✓ | ||||
| Abdolreza Pazouki (corresponding author) |
✓ | ✓ | ✓ | ✓ |
Consent
Written informed consent was obtained from the patient for publication of this case report and accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal on request, however it is in Persian.
Acknowledgement
A special note of thanks is forwarded to Ms. Pontia Fallahi for her valuable English edition.
Contributor Information
Fahimeh Soheilipour, Email: fsoheilipour@yahoo.com.
Ommolbanin Abed, Email: abed.surgery@gmail.com.
Babak Behnam, Email: b_behnam@yahoo.com.
Mohammadreza Abdolhosseini, Email: mra_75@yahoo.com.
Peyman Alibeigi, Email: dralibeigi@yahoo.com.
Abdolreza Pazouki, Email: apazouki@yahoo.com.
References
- 1.Berkovitz G.D., Fechner P.Y., Zacur H.W., Rock J.A., Snyder H.M., III, Migeon C.J. Clinical and pathologic spectrum of 46, XY gonadal dysgenesis: its relevance to the understanding of sex differentiation. Medicine. 1991;70(6):375–383. doi: 10.1097/00005792-199111000-00003. [DOI] [PubMed] [Google Scholar]
- 2.Sagodi L., Solyom E., Toth A., Kekesi A., Borbas E.P.T. [Mixed gonadal dysgenesis associated with an isodicentric Y chromosome] Orv. Hetil. 2007;148(33):1567–1571. doi: 10.1556/OH.2007.28108. [DOI] [PubMed] [Google Scholar]
- 3.Robboy S., Bernhardt P., Parmley T. Embryology of the female genital tract and disorders of abnormal sexual development. In: Kurman R., editor. Blaustein’s Pathology of the Female Genital Tract. Springer; New York: 1994. pp. 3–29. [Google Scholar]
- 4.Kim K.-R., Kwon Y., Joung J.Y., Kim K.S., Ayala A.G., Ro J.Y. True hermaphroditism and mixed gonadal dysgenesis in young children: a clinicopathologic study of 10 cases. Mod. Pathol. 2002;15(10):1013–1019. doi: 10.1097/01.MP.0000027623.23885.0D. [DOI] [PubMed] [Google Scholar]
- 5.Crone J., Amann G., Gheradini R., Kirchlechner V., Fékété C.-N. Management of 46, XY partial gonadal dysgenesis–revisited. Wien. Klin. Wochenschr. 2002;114(12):462. [PubMed] [Google Scholar]
- 6.Telvi L., Lebbar A., Del Pino O., Barbet J.P., Chaussain J.L. 45, X/46, XY mosaicism: report of 27 cases. Pediatrics. 1999;104(2):304–308. doi: 10.1542/peds.104.2.304. [DOI] [PubMed] [Google Scholar]
- 7.Jawaheer D., Juo S.H., Le Caignec C., David A., Petit C., Gregersen P. Mapping a gene for 46, XY gonadal dysgenesis by linkage analysis. Clin. Genet. 2003;63(6):530–535. doi: 10.1034/j.1399-0004.2003.00082.x. [DOI] [PubMed] [Google Scholar]
- 8.Rey R.A., Belville C., Nihoul-Fékété C., Michel-Calemard L., Forest M.G., Lahlou N. Evaluation of gonadal function in 107 intersex patients by means of serum antimüllerian hormone measurement. J. Clin. Endocrinol. Metab. 1999;84(2):627–631. doi: 10.1210/jcem.84.2.5507. [DOI] [PubMed] [Google Scholar]
- 9.Donahoe P.K., Crawford J.D., Hendren W.H. Mixed gonadal dysgenesis, pathogensis, and management. J. Pediatr. Surg. 1979;14(3):287–300. doi: 10.1016/s0022-3468(79)80486-8. [DOI] [PubMed] [Google Scholar]
- 10.Verp M.S., Simpson J.L. Abnormal sexual differentiation and neoplasia. Cancer Genet. Cytogenet. 1987;25(2):191–218. doi: 10.1016/0165-4608(87)90180-4. [DOI] [PubMed] [Google Scholar]
- 11.Kersemaekers A.-M.F., Honecker F., Stoop H., Cools M., Molier M., Wolffenbuttel K. Identification of germ cells at risk for neoplastic transformation in gonadoblastoma: an immunohistochemical study for OCT3/4 and TSPY. Hum. Pathol. 2005;36(5):512–521. doi: 10.1016/j.humpath.2005.02.016. [DOI] [PubMed] [Google Scholar]
- 12.Cools M., Stoop H., Kersemaekers A.-M.F., Drop S.L., Wolffenbuttel K.P., Bourguignon J.-P. Gonadoblastoma arising in undifferentiated gonadal tissue within dysgenetic gonads. J. Clin. Endocrinol. Metab. 2006;91(6):2404–2413. doi: 10.1210/jc.2005-2554. [DOI] [PubMed] [Google Scholar]
- 13.Cools M., Drop S.L., Wolffenbuttel K.P., Oosterhuis J.W., Looijenga L.H. Germ cell tumors in the intersex gonad: old paths, new directions, moving frontiers. Endocr. Rev. 2006;27(5):468–484. doi: 10.1210/er.2006-0005. [DOI] [PubMed] [Google Scholar]
- 14.Chang H.J., Clark R.D., Bachman H. The phenotype of 45, X/46, XY mosaicism: an analysis of 92 prenatally diagnosed cases. Am. J. Hum. Genet. 1990;46(1):156. [PMC free article] [PubMed] [Google Scholar]
- 15.Johansen M.L., Hagen C.P., Rajpert-De Meyts E., Kjærgaard S., Petersen B.L., Skakkebæk N.E. 45, X/46, XY mosaicism: phenotypic characteristics, growth, and reproductive function—a retrospective longitudinal study. J. Clin. Endocrinol. Metab. 2012;97(8):E1540–E1549. doi: 10.1210/jc.2012-1388. [DOI] [PubMed] [Google Scholar]
- 16.Yamakita N., Yasuda K., Mori H., Kuriyama M., Kumamoto Y., Miura K. A case of mixed gonadal dysgenesis (MGD)–with a review of MGD patients reported in Japan. Jpn. J. Med. 1989;28(6):744. doi: 10.2169/internalmedicine1962.28.744. [DOI] [PubMed] [Google Scholar]
- 17.Anand A., Gupta N.P., Singh M., Mathur S.R., Nayyar R. Mixed gonadal dysgenesis with normal karyotype: a rare case report. Indian J. Pathol. Microbiol. 2010;53(2):313. doi: 10.4103/0377-4929.64297. [DOI] [PubMed] [Google Scholar]
- 18.Cools M., Pleskacova J., Stoop H., Hoebeke P., Van Laecke E., Drop S. Gonadal pathology and tumor risk in relation to clinical characteristics in patients with 45, X/46, XY mosaicism. J. Clin. Endocrinol. Metab. 2011;96(7):E1171–E1180. doi: 10.1210/jc.2011-0232. [DOI] [PubMed] [Google Scholar]
- 19.Ahmed S.F., Khwaja O., Hughes I.A. The role of a clinical score in the assessment of ambiguous genitalia. BJU Int. 2000;85(1):120–124. doi: 10.1046/j.1464-410x.2000.00354.x. [DOI] [PubMed] [Google Scholar]
- 20.Martinerie L., Morel Y., Gay C.-L., Pienkowski C., De Kerdanet M., Cabrol S. Impaired puberty, fertility, and final stature in 45, X/46, XY mixed gonadal dysgenetic patients raised as boys. Eur. J. Endocrinol. 2012;166(4):687–694. doi: 10.1530/EJE-11-0756. [DOI] [PubMed] [Google Scholar]