

Abstract
Background
RBM10 is an RNA binding protein involved in the regulation of transcription, alternative splicing and message stabilization. Mutations in RBM10, which maps to the X chromosome, are associated with TARP syndrome, lung and pancreatic cancers. Two predominant isoforms of RBM10 exist, RBM10v1 and RBM10v2. Both variants have alternate isoforms that differ by one valine residue, at amino acid 354 (RBM10v1) or 277 (RBM10v2). It was recently observed that a novel point mutation at amino acid 354 of RBM10v1, replacing valine with glutamic acid, correlated with preferential expression of an exon 11 inclusion variant of the proliferation regulatory protein NUMB, which is upregulated in lung cancer.
Findings
We demonstrate, using the GLC20 male-derived small cell lung cancer cell line - confirmed to have only one X chromosome - that the two (+/−) valine isoforms of RBM10v1 and RBM10v2 result from alternative splicing. Protein modeling of the RNA Recognition Motif (RRM) within which the alteration occurs, shows that the presence of valine inhibits the formation of one of the two α-helices associated with RRM tertiary structure, whereas the absence of valine supports the α-helical configuration. We then show 2-fold elevated expression of the transcripts encoding the minus valine RBM10v1 isoform in GLC20 cells, compared to those encoding the plus valine isoform. This expression correlates with preferential expression of the lung cancer-associated NUMB exon 11 inclusion variant.
Conclusions
Our observations suggest that the ability of RBM10v1 to regulate alternative splicing depends, at least in part, on a structural alteration within the second RRM domain, which influences whether RBM10v1 functions to support or repress splicing. A model is presented.
Keywords: RBM10, RNA binding protein, RRM, Alternative splicing, Regulation, NUMB
Findings
Background
The RNA binding protein RBM10 is capable of regulating the expression of a number of genes including some involved in apoptosis (e.g., FASR) [1] and cell proliferation (e.g., NUMB) [2]. In all of the reported cases but one, this expression regulation takes the form of alternative splicing regulation [1-4]. The one gene whose expression is not regulated by RBM10-mediated alternative splicing is the angiotensin receptor 1 (AT1), where RBM10 influences AT1 expression by binding within the transcript’s 3′-untranslated region (UTR), stabilising the message and subsequently contributing to a decreased rate of transcription [4]. How RBM10 accomplishes these disparate functions remains to be determined.
The region(s) within the RBM10 protein that is involved in any of the RNA-protein interactions identified to date has not been defined although it likely involves at least one of two RNA Recognition Motifs (RRM) and two zinc fingers (ZnF) [5]. On the other hand, two consensus motifs within the target RNA with which the RBM10 protein binds have been defined as CUCUGAACUC and CGAUCCCU [2]. Using HeLa cells, Bechara et al. [2] reported that RBM10 interactions occurred predominantly within the upstream intron of an excluded alternate exon but predominantly downstream of an included alternate exon. On the other hand, using HEK293 cells, Wang et al. [3] reported that RBM10 interactions occurred in the vicinity of the 5′- and 3′-splice sites within both the upstream and downstream introns of alternate exons, though predominant binding in the vicinity of the upstream 3′-splice site was associated with less exon skipping. Obviously, much remains to be elucidated concerning the regulation of these interactions.
Mutations in RBM10 have been noted in cells associated with lung and pancreatic cancers and the neuromuscular disorder TARP syndrome [3,6-10] (summarized in Table 1). In one lung study, 12/183 (7%) adenocarcinoma specimens had RBM10 mutations, each patient having a different mutation (5/12 being missense, 5/12 truncating and 2/12 splice-site) [10]. In another lung study, a mutation - caused by a T to A substitution and consequent valine (V) to glutamic acid (E) substitution within the second RRM (RRM2) of RBM10 - was reported in A549 lung adenocarcinoma cells, with consequences for NUMB alternative splicing [2]. In the blood cells of patients with TARP syndrome, six different mutations were identified [6-8], and a six exon-spanning deletion (aa 651–889) that codes for a truncated RBM10 isoform with an inability to regulate alternative splicing [3].
Table 1.
RBM10 mutations
| Phenotype | Mutation effect | Mutation | Exon | Protein | Reference |
|---|---|---|---|---|---|
| NSCLC | Missense | 2 | E4K | Imielinski et al. [10] | |
| NSCLC | Missense | 2 | R6H | Imielinski et al. [10] | |
| TARP syndrome | Frameshift | c.159delC | 3 | p.Lys54SerfsX80 | Gripp et al. [7] |
| NSCLC | Nonsense | 3 | E67 | Imielinski et al. [10] | |
| TARP syndrome | c.448C>T | 4 | p.Gln150X | Johnston et al. [8] | |
| NSCLC | Nonsense | 5 | R157fs | Imielinski et al. [10] | |
| NSCLC | Nonsense | 7 | Y206 | Imielinski et al. [10] | |
| NSCLC | Nonsense | 8 | R230 | Imielinski et al. [10] | |
| TARP syndrome | c.724+2T>C | 8 | Johnston et al. [8] | ||
| NSCLC | Missense | 10 | 1316F | Imielinski et al. [10] | |
| TARP syndrome | Nonsense | c.1235G>A | 12 | p.Trp412X | Johnston et al. [6] |
| NSCLC | Missense | 16 | Y580F | Imielinski et al. [10] | |
| NSCLC | Splice site | 17 | Y596 | Imielinski et al. [10] | |
| Pancreatic neoplasm | Frameshift | c.1817-1818insA | 17 | p.E606EfsX37 | Furukawa et al. [9] |
| TARP syndrome | Frameshift | c.1893-1894insA | 17 | p.Pro632ThrfsX41 | Johnston et al. [6] |
| TARP syndrome | Deletion | aa651-889 | 18-23 | Wang et al. [3] | |
| NSCLC | Missense | 18 | R685L | Imielnski et al. [10] | |
| TARP syndrome | c.2176C>T | 20 | p.Arg726X | Johnston et al. [8] | |
| NSCLC | Nonsense | 21 | E810 | Imielinski et al. [10] | |
| NSCLC | Splice site | 22 | V846 | Imielinski et al. [10] |
The pre-mRNA for RBM10 is alternatively spliced to yield two predominant protein isoforms: a 930 amino acid (aa) (~103.5 kDa) isoform that includes exon 4, referred to as RBM10 variant 1 (RBM10v1), and an 853 aa (~94.5 kDa) isoform that lacks exon 4, referred to as RBM10v2. In addition to the two major RBM10 variants, the Ensembl Database lists a variant of RBM10v2 that is one amino acid shorter (852 aa). The 7 April 2003 GenBank deposition version 1 for this RBM10v2(V277del) isoform (NM_152856.1) describes it as having an “alternate donor splice site” compared to RBM10v1. Indeed, a tandem donor splice site of the configuration GYNGYN was identified in RBM10 exon 10 (as GTGGTG) from a screen of expressed sequence tags (ESTs) for variants generated from upstream and downstream tandem repeat triplets [11]. Utilization of the downstream triplate (the “e” site referred to in [11]) would result in the inclusion of a valine at amino acid 277 for RBM10v2 and 354 for RBM10v1. For clarity throughout this manuscript, we will refer to the longer RBM10v2 isoform as RBM10v2(V277), the shorter RBM10v2 isoform as RBM10v2(V277del), the longer RBM10v1 isoform as RBM10v1(V354) and the shorter RBM10v1 isoform as RBM10v1(V354del).
Our comprehensive review of the literature and various protein databases suggested that the presence of RBM10v1(V354del) is by no means generally recognized and that there appears to be some confusion as to its legitimacy and the mechanism by which it is generated. For instance, RBM10v1(V354del) has been identified as RBM10 “missing” valine 354 (UniProt – P98175-2), and as a mismatch of I353 with valine (on the Protein Data Bank website). For an overview of RBM10v1 isoforms described in various databases and references, see Table 2.
Table 2.
RBM10v1 isoforms reported in various database and references
| Origin | Sequence | Reference |
|---|---|---|
| Databases | ||
| Ensembl | STIVEAA | ENST00000377604 |
| NIH GenBank | STIVEAA | NM_005676 |
| STIEAA | NM_001204467 | |
| NIH GenBank | STIVEAA | NP_005667 |
| STIEAA | NP_001191396 | |
| EMBL-EBI InterPro | STIVEAA | P98175 |
| UniProtKB | STIVEAA | P98175-1 |
| STI-EAA | P98175-2 | |
| neXtprot beta | STIVEAA | iso1 |
| STI-EAA | iso2 | |
| USCSC | STIVEAA | uc004dhf.3 |
| STI-EAA | uc004dhh.3 | |
| HUGE | STI-EAA | KIAA0122/GenBank D50912 |
| References | ||
| Bechara et al. [2] | Minus valine isoform; STI(E)EAA | |
| Inoue et al. [1] | STI-EAA | KIAA0122 |
In the study presented herein using a small cell lung cancer cell line, we confirm the existence of two full-length RBM10v2 transcripts, RBM10v2(V277) and RBM10v2(V277del), and prove the existence of two full-length RBM10v1 transcripts, RBM10v1(V354) and RBM10v1(V354del). We then demonstrate that the presence or absence of valine alters the tertiary structure of the second RNA Recognition Motif (RRM2) within the RBM10 protein. Finally, we show that 2-fold higher levels of the transcripts encoding the minus - compared to the plus - valine isoforms of both RBM10v1 and RBM10v2 correlates with higher levels of the lung cancer associated NUMB exon 11 inclusion variant, compared to the exon 11 exclusion variant.
Results
Evidence that alternative splicing occurs within exon 10
The GLC20 cell line was established from a small cell lung cancer (SCLC) of male origin [12]. It is RBM5-null and was determined, by FISH, to contain only one X chromosome (Figure 1A). Alternative splicing of the X-linked RBM10 gene [13], to generate both RBM10v1 and RBM10v2, was confirmed in the GLC20 cells, using RT-PCR (Figure 1B). Most of the cancer, or transformed, cell lines that we have tested express more RBM10v1 than RBM10v2, at both the mRNA (Figure 1B) and protein (Figure 1C and data not shown) levels. This observation is particularly true for GLC20 cells, which express such a small amount of RBM10v2 protein that it is technically challenging to detect.
Figure 1.
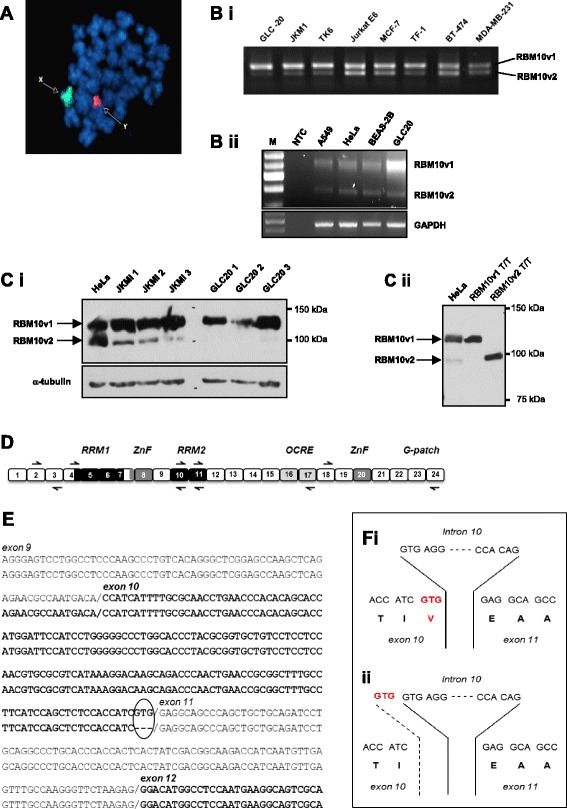
Alternative splicing of RBM10. (A) FISH analysis of GLC20 cells, with painted X and Y chromosomes, demonstrating the presence of only one X chromosome. (B) RBM10v1 and RBM10v2 RNA expression in various cell lines, including GLC20. Representative raw RT-PCR data using RBM10 exon 4 spanning primers. Bi: RBM10F with RBM10RS primers. Bii: RBM10F with RBM10v1/v2R primers. M: 100 bp DNA ladder (FroggaBio Inc., Toronto, Canada). NTC: no template control. (C) Protein expression by Western blot. Ci shows RBM10 expression in whole cell lysates from three cell lines, including GLC20. The numbers 1, 2 and 3 after JKM1 and GLC20 delineate cells from three biological replicates. Cii includes control HeLa protein and in vitro translated RBM10v1 and RBM10v2 protein, to confirm the location of RBM10v2 is the cell line extracts. (D) Cartoon of full-length RBM10v1 mRNA, not drawn to scale. Boxes represent exons. Left and right black arrows represent primer placement for sequencing. Approximate positioning of consensus functional motifs is indicated by text and differential shading. (E) Alignment of the two GLC20 RBM10v1 isoform sequences. Circled area indicates the region that differs between the two RBM10v1 isoforms. (F) Nucleotide and amino acid sequences of the RBM10v1 exon10/intron 10/exon 11 donor and acceptor sites for (i) RBM10v1(V354), and (ii) RBM10v1(V354del).
To estimate functionality of RBM10 in the GLC20 cells, sequencing of cDNA was carried out. Only full-length RBM10v1 cDNA was sequenced because full-length RBM10v2 cDNA could not be amplified from the GLC20 cells. Mixed sequence was observed beginning at the 3′-end of exon 10, with either the forward or reverse primer. At that point in time (June 2014), the Ensembl Database listed two different RBM10v2 isoforms, we are herein designating RBM10v2(V277) and RBM10v2(V277del) (corresponding to GenBank Accession Numbers NM_001204466.1 and NM_152856.2). To determine if the mixed sequencing read in the GLC20 RBM10v1 sample was the result of a mixture of transcripts with different exon 10 3′-end sequences, we designed primers immediately 3′- to the anticipated modified triplet site in the cDNA (refer to Figure 1D for primer locations), thereafter expecting an unambiguous sequence read. Indeed, clear sequence was obtained, thereby delineating the altered region as the last three nucleotides of exon 10 (Figure 1E).
The presence of a mixture of two different isoforms of RBM10v1 suggested either an alternative splicing event or two RBM10 alleles, the later possibility being unlikely since we had confirmed only one X chromosome by FISH analysis (Figure 1A). Since there remained the possibility of an RBM10 gene duplication with allelic variation, we decided to sequence RBM10v1 cDNA from additional cell lines, theorizing that if the same mixed read was observed in other cells it would suggest alternative splicing. We sequenced transcripts from both male and female-derived cell lines, since one RBM10 allele is silenced as the result of X chromosome inactivation in female somatic cells [14,15], therefore whether male or female-derived, cell lines would theoretically have only one RBM10 gene that is transcribed. RBM10v1 and RBM10v2 cDNA was sequenced in A549 (a male-derived lung adenocarcinoma cell line), HeLa (a female-derived cervical adenocarcinoma cell line) and BEAS-2B (a male-derived non-cancerous SV40/adenovirus-transformed lung cell line). Sequencing data revealed the same nucleotide variation in exon 10 of RBM10v1 and exon 9 of RBM10v2 in all three cell lines, therefore suggesting that the two isoforms of RBM10v1 are, as originally surmised within GenBank deposition NM_152856.1 and by Hiller et al. [11], the result of an alternative splicing event. An example of how this might occur is presented diagrammatically in Figure 1F.
Sequence variation that occurs as a result of alternative splicing at the 3′-ends of exons with tandem repeats such as GYNGYN, is regulated partly by U1snRNA binding and partly by the presence of CGGG and GGGT sequons in the downstream intron, particularly if the downstream intron is shorter than 200 nucleotides (nts) [11]. Intron 10 of RBM10 is 171 nts and contains three CGGG sequons, two GGGT sequons and one CGGGGT sequon, suggesting that co-expression of the two RBM10v1 isoforms is indeed a regulated alternative splicing event. The stimuli that control this regulation remain to be determined, but are unlikely limited to male, lung or cancer cells, since both RBM10v1(V354) and RBM10v1(V354del) were present in A549, HeLa, BEAS-2B and GLC20 cells.
Structural consequences of alternative splicing of RBM10v1
RBM10v1 and RBM10v2 both contain two RRMs (refer to Figure 1D) defined as ~75-85 amino acids that three dimensionally form four β-sheets flanked by two α-helices. The most conserved sequences within any RRM comprise the RNP2 and RNP1 domains (reviewed in [5]), RNP1 having the most highly conserved sequence of the two. Notably, amino acid 354 in RBM10v1 is located in approximately the middle of RRM2, at the beginning of the second α-helix and near to the end of β-sheet three, which is encoded by the highly conserved RNP1 domain [16].
Valines have a high free energy that is associated with α-helix disruption and since α-helices are known to contribute to the stabilization of RNA/protein interactions, disruption of the α-helix by a valine insertion would be predicted to destabilize any RNA/protein interaction. In RBM10, the placement of the altered amino acid adjacent to the highly conserved β-sheet three structure suggested that any potential alteration to the second α-helical structure within RRM2 might have a significant effect on overall protein conformation, and consequently, the ability to interact with RNA. Indeed, Bechara et al. [2] recently described a G to T mutation in the terminal exon 10 codon of RBM10, which changed the amino acid coded from valine (V) to a glutamic acid (E). This single mutation had dramatic functional consequences, manifesting as an altered ability to splice specific downstream targets, such as pre-mRNA encoding the proliferation regulatory protein NUMB [2].
To better understand how a change to this residue might contribute to RBM10 functional alterations we compared the structures of a valine-retaining, a glutamic acid-substituting and a valine-lacking amino acid within RRM2. We uploaded these altered RBM10v1 RRM2 sequences into SwissProtKB/Swiss-Prot (www.expasy.org), a program that predicts a two-dimensional configuration and ranks it against similar configurations of previously crystalized structures. A crystal structure for a minus-valine RBM10 RRM2 (designated 2m2d) [17,18] was the reference structure for the V354, V354E and V354del RBM10v1 RRM2 predictions. We also uploaded the RRM2 sequences into Phyre2 (the Protein Homology/analogY Recognition Engine v2.0, www.sbg.bio.ic.ac.uk). To visualize a rotatable three-dimensional structure, the structure predictions for V354, V354E and V354del from both SwissProtKB and Phyre2 were uploaded into the Yasara modeling program (Yet Another Scientific Artificial Reality Application, www.yasara.org). A comparison of all the predictions (Figure 2) revealed that the addition of valine did, as anticipated, disrupt the α-helical structure and thus the classic configuration of an RRM domain (shown as a colour change from dark blue to cyan by the Yasara software). Exclusion of the valine was associated with an α-helix. Substitution of the V for an E resulted in two slightly different configurations, depending on the prediction program used: both programs, however, predicted a change to, but a retention of, an α-helical structure compared to either the V354del or the V354. These modelling results suggest that conformational changes to the RBM10v1 protein could be responsible for altering the protein’s ability to interact with RNA.
Figure 2.
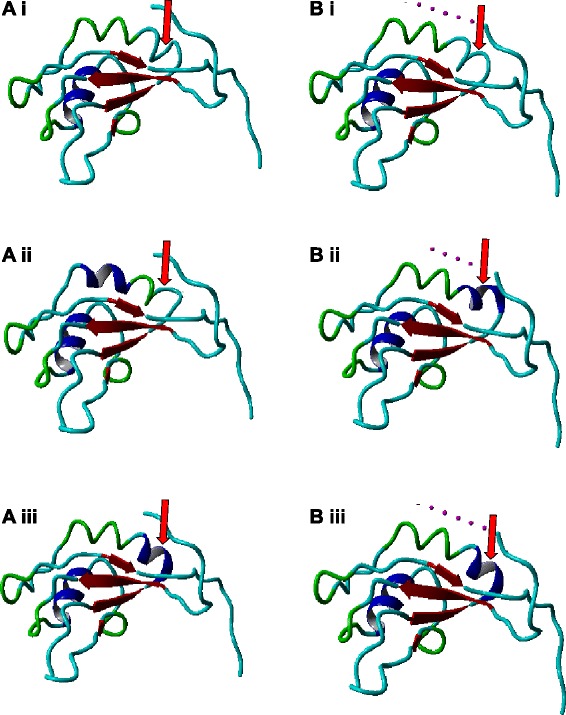
Conformation of RBM10v1 RRM2. The RBM10 RRM2 conformation was modeled using SwissProtKB (A) or Phyre2 (B) . (i) Isoforms V354. (ii) Isoforms V354E. (iii) Isoforms V354del. Arrow indicates the position of the +/− valine. Yasara structure colors indicate beta-sheets (red), alpha-helixes (dark blue), turns (green) and random coils (cyan).
Functional consequences of alternative splicing of RBM10v1
Using PAR-CLIP technology Bechara et al. [2] identified an RBM10 cluster in the 3′-splice site region preceding NUMB exon 11 (the current Ensembl exonic designation for the previously referred to NUMB exon 9) [2]. They then went on to demonstrate, using an A549 non-small cell lung cancer (NSCLC) cell line with an RBM10v1(V354E) mutation (herein referred to as A549-JV), that expression of recombinant RBM10v1 protein, with either a valine or a glutamic acid at amino acid 354, altered NUMB splicing. With valine present (RBM10v1(V354)), there was preferential NUMB exon 11 exclusion - associated with NOTCH repression and decreased proliferation. On the other hand, the glutamic acid-containing isoform (RBM10v1(V354E)) demonstrated preferential NUMB exon 11 inclusion – associated with NOTCH activation and increased proliferation. Recognising that a V354E substitution would not necessarily have a similar effect as a V354del, but taking into consideration our structural predictions, the fact that the valine to glutamic acid mutation occurred at exactly the same site as the RBM10v1 alternative splicing of RBM10v1(V354) to RBM10v1(V354del) suggested to us that regulated alternative splicing of RBM10v1 has functional significance and is important to lung cancer.
Considering that RBM10v1(V354) expression correlated with preferential NUMB exon 11 exclusion and RBM10v1(V354E) expression correlated with preferential NUMB exon 11 inclusion [2] and that expression of the exon 11 retaining NUMB transcript is frequently increased in lung adenocarcinomas [19], one might predict that (a) downregulation of RBM10v1(V354) is one means by which lung cancer cells circumvent proliferation controls, and (b) more RBM10v1(V354del) than RBM10v1(V354) is expressed in lung cancers. Interestingly, we have whole transcriptome-sequencing data (manuscript in preparation) demonstrating that in the GLC20 small cell lung cancer cells and three stable GLC20 sublines, transcripts encoding RBM10v1(V354del) and RBM10v2(V277del) have ~2-fold higher expression than transcripts encoding RBM10v1(V354) and RBM10v2(V277) (Figure 3A). When NUMB exon 11 alternative splicing was examined in the GLC20 cells, as predicted, preferential expression of the NUMB exon 11 inclusion variant was observed (Figure 3B).
Figure 3.
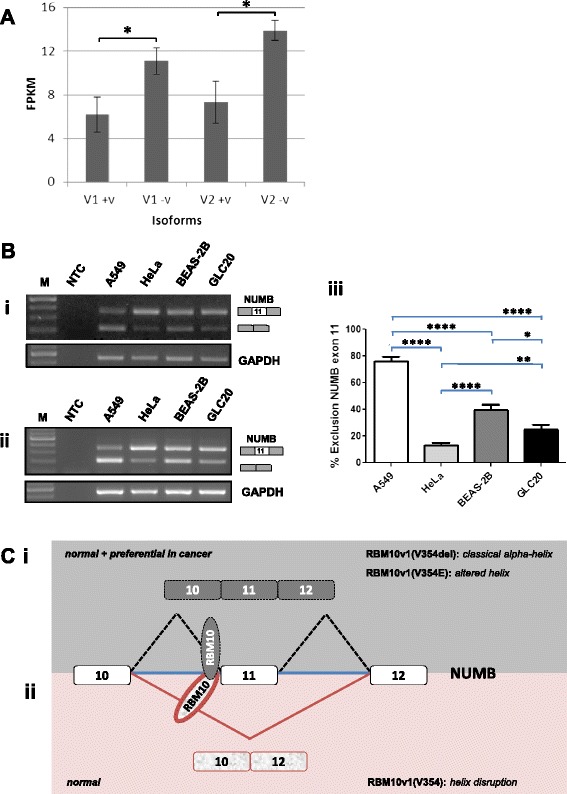
Functional affects associated with RBM10 variant expression. (A) In GLC20 cells, comparative expression levels of RBM10v1 and RBM10v2 transcripts encoding the valine-retaining and valine-lacking isoforms, as determined by RNA-seq. *p < 0.05. (B) NUMB alternative splicing in A549, HeLa, BEAS-2B and GLC20 cells. (i) and (ii) are 2% agarose gels with SYBR®safe, showing representative amplicon expression levels following end-point PCR with (i) 28 cycles, or (ii) 40 cycles, using NUMB exon 11-spanning primers and (i) 19 cycles or (ii) 25 cycles for GAPDH. (iii) Following densitometry of amplicons from six end-point PCR reactions (three at 28 cycles and three at 40 cycles) of the two different cDNA preparations from one RNA extraction for each cell line, the percentage of the NUMB exon 11 exclusion variant was calculated and plotted. Error bars represent the standard error of the mean. Significances were calculated using an unpaired Student’s t-test, with *p < 0.05, **p < 0.01 and ****p < 0.0001. (C) Model depicting how the +/− valine isoforms of RBM10v1 might influence NUMB exon 11 alternative splicing. The plus and minus valine isoforms are present, at low levels, in normal cells, and contribute to the production of both the exon 11 inclusion and exclusion variants of NUMB. Both RBM10 isoforms are able to bind NUMB pre-mRNA, but the minus valine isoform of RBM10v1 does so with higher affinity, having the classical α-helix structure. The plus valine isoform does not bind as efficiently, thereby interfering with recognition of the intron 10 3′splice site, resulting in NUMB exon 11 exclusion.
NUMB splicing was also examined in HeLa and BEAS-2B cells as well as our A549 cells (herein referred to as A549-LS) that, unlike those used by Valcárcel and colleagues [2], express transcripts encoding both RBM10v1(V354) and RBM10v1(V354del), as opposed to only RBM10v1(V354E). If RBM10v1(V354del) functions in a similar manner to RBM10v1(V354E) - to generate the NUMB exon 11 inclusion transcript - then we anticipated A549-LS cells would express both the NUMB exon 11 inclusion and exclusion transcripts. We also anticipated more of the NUMB exon 11 exclusion transcript in the A549-LS cells (resulting from RBM10v1(V354) expression), compared to the A549-JV subline (which lacks RBM10v1(V354)). As shown in Figure 3B, and confirming the observations of Valcárcel and colleagues [2], the A549-LS subline expressed both NUMB variants, but significantly more of the NUMB exclusion variant than the inclusion variant (as opposed to the A549-JV subline that expressed predominantly the NUMB exon 11-inclusion transcript: see [2], Figure Six C, upper panel, lane 14). In the transformed but non-tumourigenic BEAS-2B bronchial epithelial cells, we predicted a lower NUMB exon 11 inclusion to exclusion ratio, based on the previously reported observations that the ratio of NUMB exon 11 inclusion to exon 11 exclusion is higher in lung cancer than in normal cells [19] and our hypothesis that the RBM10v1(V354del) isoform is more prevalent in cancer cells. As shown in Figure 3B, the BEAS-2B cells did indeed have the lowest NUMB exon 11 inclusion to exclusion ratio of the four cell lines examined. Finally, as expected, and previously shown by Bechara et al. [2], HeLa cells had a clearly higher inclusion to exclusion expression ratio. The only unexpected observation from this cell line RNA analysis was the high level of the NUMB exon 11 exclusion variant in the A549-LS cells, considering they are a lung cancer cell line. Determination of the relative expression levels of the transcripts encoding RBM10v1(V354) and RBM10v1(V354del) isoforms in this subline would help to resolve this conundrum.
Discussion
RBM10 is an apoptosis regulatory protein [20] and its paralogue, RBM5 functions in the same capacity, but has the added ability to regulate the cell cycle [21,22]. Based on the findings of Bechara et al. [2], it appears that RBM10 is also able to regulate the cell cycle, via the NOTCH signaling pathway [2]. Is the regulation of RBM10v1(V354) versus RBM10v1(V354del) alternative splicing a means by which normal cells temporarily modulate proliferation prior to a repair versus apoptosis event? Have cancer cells hijacked this alternative splicing mechanism as one means of circumventing proliferation controls? Of the mutations identified so far in pancreatic cancer and NSCLCs, none occurs at this site to permanently generate the minus valine isoform, except the mutation described by Bechara et al. [2] in a lung adenocarcinoma cell line (see Table 1). It is therefore difficult to predict how important the regulation of this alternative splicing event is to cancer initiation and/or progression. We have initiated studies to more thoroughly characterise the expression of transcripts encoding the two RBM10v1 and RBM10v2 isoforms in primary lung cancer specimens. Additional studies relating to protein expression are also warranted, as are studies concerning the regulation of this expression.
In an attempt to align our observations with those of Bechara et al. [2] and Wang et al. [3] we propose a model to explain how changes in RBM10v1 splicing might be predicted to regulate NUMB splicing. The model takes into consideration the following observations. Firstly, overexpression of RBM10 correlates with NUMB exon 11 exclusion [2,3]. Secondly, in lung cancer there is preferential expression of the NUMB exon 11 inclusion variant. Thirdly, in lung cancer RBM10 is highly expressed [10], but while in our A549, HeLa, BEAS-2B and GLC20 cells transcripts encoding both RBM10v1 isoforms were observed, there was two-fold more RBM10v1(V354del) than RBM10v1(V354) in the GLC20 SCLC cells. Our model does not attempt to reconcile the somewhat conflicting binding data previously reported [2,3].
Our model, depicted in Figure 3C, posits that RBM10v1(V354) and RBM10v1(V354del) are both expressed in normal cells, but that in cancers RBM10v1(V354del) is preferentially expressed. RBM10v1(V354) and RBM10v1(V354del) are both capable of interacting with pre-mRNA, and do so in the vicinity of the upstream intronic 3′-splice site of the alternate exon. RBM10v1(V354del), however, has a higher affinity interaction with pre-mRNA than RBM10v1(V354) (resulting from its ability to form the second α-helix within RRM2). In its capacity as a higher affinity binder, RBM10v1(V354del) is able to function as an auxiliary splicing factor. As for RBM10v1(V354), disruption of the second α-helical structure within RRM2 generates an RBM10v1 isoform that interacts with the same pre-mRNA as RBM10v1(V354del) but in a manner that impedes splicing. As a result of either less efficient or lower affinity binding or an inability to optimally interact with other auxiliary/splicing factors, expression of RBM10v1(V354) functions to increase the splicing of the alternative transcript. In our model, the increase in the NUMB exon 11 inclusion transcript that is associated with downregulation of RBM10 results from the presence of reduced levels of the competing, less efficient binding, RBM10v1(V354) isoform and consequently more binding of the higher affinity RBM10v1(V354del) isoform, resulting in more of the NUMB exon 11 inclusion product. According to our model, therefore, alternative splicing regulation by RBM10 depends not only on the ratio of RBM10v1(V354) to RBM10v1(V354del), but on the total levels of RBM10v1 protein as well.
Exactly how the structural change associated with a valine insertion at amino acid 354 of RBM10v1 results in splicing changes is unknown. Multiple binding motifs have been identified for RBM10 [2] as have multiple potential binding regions within pre-mRNA [2,3], suggesting that interaction of RBM10 with pre-mRNA targets may be a dynamic and flexible phenomenon. And while expression of RBM10 is more frequently associated with alternate exon exclusion [1-3,23], it is also associated with alternate exon inclusion [2,3]. Perhaps binding and function of RBM10 is more influenced by tertiary structure than primary sequence of the protein, and the multiple sequence motifs and binding regions identified within pre-mRNAs reflect interactions with different RBM10 isoforms that each assume a slightly different conformation.
To note just prior to submission of this manuscript, Ensembl (which had not, at least during the course of this investigation, listed RBM10v1(V354del) in its database) removed the RBM10v2(V277del) isoform, thereby eliminating all reference to the minus-valine isoforms. The minus valine isoforms are those which form the classic α-helical structure associated with the RRM domains (as shown herein), RNA transcripts encoding the minus valine isoform of RBM10v1 exist in multiple cell types (as shown herein), and functional studies demonstrate the apoptotic regulatory ability [20] and alternative splicing ability [2] of RBM10v1(V354del). We therefore suggest that the alternative splicing of RBM10v1, and likely RBM10v2, is a regulated event worthy of consideration in functional studies.
Materials and methods
Cell culture and differentiation
GLC20 cells were kindly provided by Charles Buys (University of Groningen, The Netherlands). Cells were cultured in RPMI supplemented with 10% fetal bovine serum (FBS). JKM1 [24], Jurkat Clone E6 [25], MCF-7 [26], MDA-MB-231 [27] and TF-1 cells [25] were grown as previously described. A549 (from ATCC) and HeLa cells (provided by Hoyun Lee, AMRIC) were grown in DMEM/F-12 medium supplemented with 10% FBS. TK6 cells (provided by Elliot Drobetsky, University of Montreal) were grown in DMEM/F-12 medium supplemented with 10% fetal horse serum. BEAS-2B cells (purchased from ATCC) were grown in LHC-9 medium, on plates pre-coated with 0.01 mg/ml fibronectin, 0.03 mg/ml collagen and 0.01 mg/ml bovine serum albumin. Media and sera were purchased from Life Technologies (Burlington, Canada).
Fluorescent in situ hybridization (FISH)
Slides were prepared from a cell suspension using standard cytogenetic techniques. Slides were denatured at 75°C for 2 mins and hybridized overnight at 37°C with Spectrum Green (X chromosome) and Spectrum Red (Y chromosome) (Vysis, Abbott Molecular, Mississauga, Canada). Following hybridization the slides were washed in 0.4×SSC/0.3% NP40 at 73°C for 2 mins, then 2.0×SSC/0.1% NP40 at 23°C for 1 min. Slides were dried in the dark then stained with Vysis DAPI II and the coverslips applied. Images were captured using an Olympus BX60 microscope equipped with a mercury bulb and camera. Images were processed with Cytovision software from Genetix.
RNA extraction, reverse-transcription and PCR
RNA was isolated from cell pellets using Tri-Reagent (Molecular Research Center, Inc., Cedarlane, Burlington, Canada). Reverse transcription was carried out using 1 μg of RNA, and MMLV (for expression level reactions) or SuperScript II (for sequencing) reverse transcriptase (Life Technologies). Amplification of cDNA by polymerase chain reaction (PCR) was performed using gene-specific primers RBM10F and RBM10RS (exon 4 spanning) [27] (Figure 1Bi) or RBM10v1/v2R (also exon 4 spanning) [20] (Figure 1Bii), NUMBF(exon10): 5′-TAGAAGGGGAGGCAGAGAGC-3′ and NUMBR(exon12): 5′-CTCAGAGGGAGTACGTCTAT-3′ and GAPDH [28] (all primers purchased from AlphaDNA, Montreal, Canada). End-point PCR reaction conditions: (1) 95°C for 5 minutes, (2) gene-specific cycle number (40 cycles for RBM10, 28 and 40 cycles for NUMB, and 19 (Figure 3Bi) or 25 (Figures 1Bii and 3Bii) cycles for GAPDH) of 95°C for 30 seconds, 62°C (RBM10F + RS), 55°C (RBM10F + v1/v2R), 61°C (NUMB), 59°C (GAPDH) for 30 seconds, 72°C for 45 seconds, and (3) 72°C for 10 minutes. The samples were visualized following electrophoresis through a 2% (40 mM Tris-acetate, 10 mM EDTA, pH 8.0) (TAE) agarose gel containing SYBR®safe DNA gel stain (Life Technologies).
For sequencing of full-length RBM10, nested PCR was carried out, using RBM10FNhe1: 5′-CTA GCT AGC TAG TGG CTG GGA AGT GAA ACG GAG CCA GCG-3′ and RBM10RL: 5′-TGG CTG GGG AGT GGG CTG G-3′ primers for Reaction 1 and RBM10FNhe1 and RBM10RHindIII: 5′-CCC AAG CTT GGC TGG GCC TCG TTG AAG CG-3′ primers for Reaction 2, Platinum Pfx Polymerase (Life Technologies) and 5 μl of Reaction 1 as template for Reaction 2. PCR reaction conditions: Reaction 1: (1) 95°C for 2 minutes, (2) 18 cycles of 94°C for 10 seconds, 66°C for 1 minute, 68°C for 3 minutes; Reaction 2: (3) 22 cycles of 94°C for 10 seconds, 62°C for 1 minute, 68°C for 3 minutes, (4) 72°C for 5 minutes.
Immunoblotting
Western blotting was carried out as previously described [20]. RBM10 antibody was used at a dilution of 1:500. The in vitro transcription/translation reactions were carried out using a TNT® T7 Quick Coupled Transcription/Translation Kit (Promega, through Fisher Scientific, Nepean, Canada), and plasmid constructs pcDNA3.RBM10v1 or pcDNA3.RBM10v2 [20].
Sequencing
Full-length RBM10v1 and RBM10v2 amplicons were separated by agarose gel electrophoresis (note, no full-length RBM10v2 amplicon was detectable in the GLC20 cells) and cDNA was excised using a QIAquick gel extraction kit (Qiagen, Toronto, Canada). DNA quantity (absorbance at 260 nanometers (nm)) and purity (ratio of the absorbance at 260 and 280 nm) were determined using a NanoDrop 2000C spectrophotometer (Fisher Scientific, Ottawa, Canada), and the samples were sent for sequencing. Samples were sequenced, using the Sanger technique, by the MOBIX Lab - DNA Sequencing and Oligo Synthesis Facility (McMaster University, Hamilton, Canada). Internal primers (sequences available upon request) were generated by MOBIX. Bi-directional, overlapping sequence reads of ~600 bp were generated, as detailed in Figure 1D.
Availability of supporting data
The data set supporting the results of this article is included within the article.
Acknowledgements
The authors would like to dedicate this manuscript to Charles Buys, Professor Emeritus, University of Groningen, who passed away earlier this year. Professor Buys provided the GLC20 cell line for this study. Work was funded by NSERC Grant # 9043429 to L.C.S., the Northern Cancer Foundation and the Northeastern Ontario Cancer Therapeutics Research Initiative (CTRI).
Abbreviations
- RRM
RNA recognition motif
- FISH
Fluorescent in situ hybridization
- V
Valine
- E
Glutamic acid
- PCR
Polymerase chain reaction
- RT-PCR
Reverse-transcription PCR
- nt
Nucleotide
- aa
Amino acid
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
ST carried out all the initial molecular biology experiments and prepared the first draft of the manuscript. ST is responsible for Figure 1D,E,F and Figure 2. JL and ST performed the protein modeling. CP performed the final RT-PCR experiment presented in Figure 1B. BK performed the final Western blot presented in Figure 1Ci. JR performed the final NUMB RNA expression experiment, and produced the graph. AM performed the FISH analysis. LS conceived the project and developed the model. LS wrote the final draft of the manuscript. All authors have reviewed and approve of the final version of the manuscript.
Contributor Information
Sarah J Tessier, Email: sarah.tessier@live.ca.
Julie J Loiselle, Email: loiselle.julie@gmail.com.
Anne McBain, Email: amcbain@hsnsudbury.ca.
Celine Pullen, Email: butlerc@timiskaminghu.com.
Benjamin W Koenderink, Email: bkoenderink@amric.ca.
Justin G Roy, Email: jroy@amric.ca.
Leslie C Sutherland, Email: lsutherland@amric.ca.
References
- 1.Inoue A, Yamamoto N, Kimura M, Nishio K, Yamane H, Nakajima K. RBM10 regulates alternative splicing. FEBS Lett. 2014;588:942–7. doi: 10.1016/j.febslet.2014.01.052. [DOI] [PubMed] [Google Scholar]
- 2.Bechara EG, Sebestyen E, Bernardis I, Eyras E, Valcarcel J. RBM5, 6, and 10 Differentially Regulate NUMB Alternative Splicing to Control Cancer Cell Proliferation. Mol Cell. 2013;52:720–33. doi: 10.1016/j.molcel.2013.11.010. [DOI] [PubMed] [Google Scholar]
- 3.Wang Y, Gogol-Doring A, Hu H, Frohler S, Ma Y, Jens M, et al. Integrative analysis revealed the molecular mechanism underlying RBM10-mediated splicing regulation. EMBO Mol Med. 2013;5:1431–42. doi: 10.1002/emmm.201302663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mueller CF, Berger A, Zimmer S, Tiyerili V, Nickenig G. The heterogenous nuclear riboprotein S1-1 regulates AT1 receptor gene expression via transcriptional and posttranscriptional mechanisms. Arch Biochem Biophys. 2009;488:76–82. doi: 10.1016/j.abb.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 5.Sutherland LC, Rintala-Maki ND, White RD, Morin CD. RNA binding motif (RBM) proteins: a novel family of apoptosis modulators? J Cell Biochem. 2005;94:5–24. doi: 10.1002/jcb.20204. [DOI] [PubMed] [Google Scholar]
- 6.Johnston JJ, Teer JK, Cherukuri PF, Hansen NF, Loftus SK, Chong K, et al. Massively parallel sequencing of exons on the X chromosome identifies RBM10 as the gene that causes a syndromic form of cleft palate. Am J Hum Genet. 2010;86:743–8. doi: 10.1016/j.ajhg.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gripp KW, Hopkins E, Johnston JJ, Krause C, Dobyns WB, Biesecker LG. Long-term survival in TARP syndrome and confirmation of RBM10 as the disease-causing gene. Am J Med Genet A. 2011;155A:2516–20. doi: 10.1002/ajmg.a.34190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnston JJ, Sapp JC, Curry C, Horton M, Leon E, Cusmano-Ozog K, et al. Expansion of the TARP syndrome phenotype associated with de novo mutations and mosaicism. Am J Med Genet A. 2014;164A:120–8. doi: 10.1002/ajmg.a.36212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Furukawa T, Kuboki Y, Tanji E, Yoshida S, Hatori T, Yamamoto M, et al. Whole-exome sequencing uncovers frequent GNAS mutations in intraductal papillary mucinous neoplasms of the pancreas. Sci Rep. 2011;1:161. doi: 10.1038/srep00161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012;150:1107–20. doi: 10.1016/j.cell.2012.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hiller M, Huse K, Szafranski K, Rosenstiel P, Schreiber S, Backofen R, et al. Phylogenetically widespread alternative splicing at unusual GYNGYN donors. Genome Biol. 2006;7:R65. doi: 10.1186/gb-2006-7-7-r65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smit EF, de Vries EG, Timmer-Bosscha H, de Leij LF, Oosterhuis JW, Scheper RJ, et al. In vitro response of human small-cell lung-cancer cell lines to chemotherapeutic drugs; no correlation with clinical data. Int J Cancer. 1992;51:72–8. doi: 10.1002/ijc.2910510115. [DOI] [PubMed] [Google Scholar]
- 13.Nagase T, Seki N, Tanaka A, Ishikawa K, Nomura N. Prediction of the coding sequences of unidentified human genes. IV. The coding sequences of 40 new genes (KIAA0121-KIAA0160) deduced by analysis of cDNA clones from human cell line KG-1. DNA Res. 1995;2:167–210. doi: 10.1093/dnares/2.4.167. [DOI] [PubMed] [Google Scholar]
- 14.Coleman MP, Ambrose HJ, Carrel L, Nemeth AH, Willard HF, Davies KE. A novel gene, DXS8237E, lies within 20 kb upstream of UBE1 in Xp11.23 and has a different X inactivation status. Genomics. 1996;31:135–8. doi: 10.1006/geno.1996.0022. [DOI] [PubMed] [Google Scholar]
- 15.Thiselton DL, McDowall J, Brandau O, Ramser J, d’Esposito F, Bhattacharya SS, et al. An integrated, functionally annotated gene map of the DXS8026-ELK1 interval on human Xp11.3-Xp11.23: potential hotspot for neurogenetic disorders. Genomics. 2002;79:560–72. doi: 10.1006/geno.2002.6733. [DOI] [PubMed] [Google Scholar]
- 16.Song Z, Wu P, Ji P, Zhang J, Gong Q, Wu J, et al. Solution structure of the second RRM domain of RBM5 and its unusual binding characters for different RNA targets. Biochemistry. 2012;51:6667–78. doi: 10.1021/bi300539t. [DOI] [PubMed] [Google Scholar]
- 17.Kopp J, Schwede T. The SWISS-MODEL Repository of annotated three-dimensional protein structure homology models. Nucleic Acids Res. 2004;32:D230–4. doi: 10.1093/nar/gkh008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kiefer F, Arnold K, Kunzli M, Bordoli L, Schwede T. The SWISS-MODEL Repository and associated resources. Nucleic Acids Res. 2009;37:D387–92. doi: 10.1093/nar/gkn750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Misquitta-Ali CM, Cheng E, O’Hanlon D, Liu N, McGlade CJ, Tsao MS, et al. Global profiling and molecular characterization of alternative splicing events misregulated in lung cancer. Mol Cell Biol. 2011;31:138–50. doi: 10.1128/MCB.00709-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang K, Bacon ML, Tessier JJ, Rintala-Maki ND, Tang V, Sutherland LC. RBM10 Modulates Apoptosis and Influences TNA-a Gene Expression. Journal of Cell Death. 2012;5:1–19. doi: 10.4137/JCD.S9073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mourtada-Maarabouni M, Keen J, Clark J, Cooper CS, Williams GT. Candidate tumor suppressor LUCA-15/RBM5/H37 modulates expression of apoptosis and cell cycle genes. Exp Cell Res. 2006;312:1745–52. doi: 10.1016/j.yexcr.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 22.Oh JJ, Razfar A, Delgado I, Reed RA, Malkina A, Boctor B, et al. 3p21.3 tumor suppressor gene H37/Luca15/RBM5 inhibits growth of human lung cancer cells through cell cycle arrest and apoptosis. Cancer Res. 2006;66:3419–27. doi: 10.1158/0008-5472.CAN-05-1667. [DOI] [PubMed] [Google Scholar]
- 23.Zheng S, Damoiseaux R, Chen L, Black DL. A broadly applicable high-throughput screening strategy identifies new regulators of Dlg4 (Psd-95) alternative splicing. Genome Res. 2013;23:998–1007. doi: 10.1101/gr.147546.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sutherland LC, Edwards SE, Cable HC, Poirier GG, Miller BA, Cooper CS, et al. LUCA-15-encoded sequence variants regulate CD95-mediated apoptosis. Oncogene. 2000;19:3774–81. doi: 10.1038/sj.onc.1203720. [DOI] [PubMed] [Google Scholar]
- 25.Shu Y, Rintala-Maki ND, Wall VE, Wang K, Goard CA, Langdon CE, et al. The apoptosis modulator and tumour suppressor protein RBM5 is a phosphoprotein. Cell Biochem Funct. 2007;25:643–53. doi: 10.1002/cbf.1366. [DOI] [PubMed] [Google Scholar]
- 26.Rintala-Maki ND, Abrasonis V, Burd M, Sutherland LC. Genetic instability of RBM5/LUCA-15/H37 in MCF-7 breast carcinoma sublines may affect susceptibility to apoptosis. Cell Biochem Funct. 2004;22:307–13. doi: 10.1002/cbf.1106. [DOI] [PubMed] [Google Scholar]
- 27.Rintala-Maki ND, Goard CA, Langdon CE, Wall VE, Traulsen KE, Morin CD, et al. Expression of RBM5-related factors in primary breast tissue. J Cell Biochem. 2007;100:1440–58. doi: 10.1002/jcb.21134. [DOI] [PubMed] [Google Scholar]
- 28.Rintala-Maki ND, Sutherland LC. LUCA-15/RBM5, a putative tumour suppressor, enhances multiple receptor-initiated death signals. Apoptosis. 2004;9:475–84. doi: 10.1023/B:APPT.0000031455.79352.57. [DOI] [PubMed] [Google Scholar]