

Summary
Despite the large evolutionary distances, metazoan species show remarkable commonalities, which has helped establish fly and worm as model organisms for human biology1,2. Although studies of individual elements and factors have explored similarities in gene regulation, a large-scale comparative analysis of basic principles of transcriptional regulatory features is lacking. We mapped the genome-wide binding locations of 165 human, 93 worm, and 52 fly transcription-regulatory factors (RFs) generating a total of 1,019 data sets from diverse cell-types, developmental stages, or conditions in the three species, of which 498 (48.9%) are presented here for the first time. We find that structural properties of regulatory networks are remarkably conserved and that orthologous RF families recognize similar binding motifs in vivo and show some similar co-associations. Our results suggest that gene-regulatory properties previously observed for individual factors are general principles of metazoan regulation that are remarkably well-preserved despite extensive functional divergence of individual network connections. The comparative maps of regulatory circuitry provided here will drive an improved understanding in the regulatory underpinnings of model organism biology and how these relate to human biology, development, and disease.
Keywords: Transcription Factor, Regulatory Information, Gene Regulation, Single Nucleotide Polymorphisms, ChIP-seq
Transcription-regulatory factors (RFs) guide the development and cellular activities of all organisms through highly cooperative and dynamic control of gene expression programs. RF-coding genes are often conserved across deep phylogenies, their DNA-binding protein domains are preferentially conserved at the amino-acid level, and their in vitro binding specificities are also frequently conserved across large distances3,4. However, the specific DNA targets and binding partners of regulators can evolve much more rapidly than DNA-binding domains, making it unclear whether the in vivo binding properties of RFs are conserved across large evolutionary distances.
Comparisons of the locations of regulatory binding across species has been controversial, with some studies suggesting extensive conservation1,2,5–10 while others suggest extensive turnover11–14. While it is generally assumed that across very large evolutionary distances regulatory circuitry is largely diverged, there exist highly-conserved sub-networks15–18. Thus, confusion exists in the level of regulatory turnover between related species, possibly due to the small number of factors studied. Moreover, despite recent observations of the architecture of metazoan regulatory networks a direct comparison of their topology and structure –such as clustered binding and regulatory network motifs– has not been possible owing to large differences in the procedures employed to assay RF binding in distinct species. Here we present a systematic and uniform comparison of regulation using many factors across distantly related species to help address these questions on a scale not previously possible.
To compare regulatory architecture and binding across diverse organisms, the modENCODE and ENCODE consortia mapped the binding locations of 93 C. elegans RFs, 52 D. melanogaster RFs, and 165 human RFs as a community resource (Fig. 1, Supplementary Table 1). These RF binding datasets represent a substantial increase over those previously published for worm (194 new datasets for a total of 219) and human (211 new, 707 total) and a substantial improvement in data quality in fly with a move from ChIP-chip to ChIP-seq (93 new, 93 total)2,8,19,20. The majority of RFs are site-specific transcription factors (TFs) (83 in worm, 41 in fly, and 119 in human), although general regulatory factors such as RNA Pol II were also assayed.
Figure 1. Datasets overview.
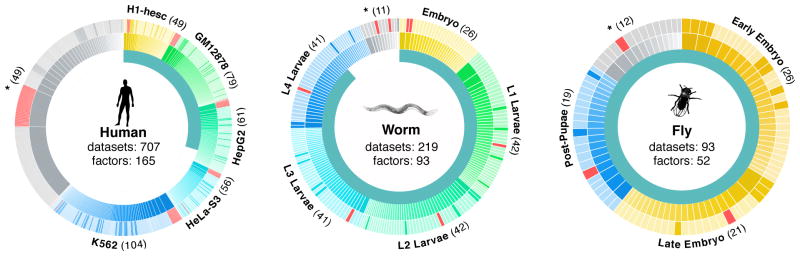
Data generated by the modENCODE and ENCODE consortium used in these analyses. The inner circle represents the fraction of datasets being presented for the first time in this paper. Each major context (cell lines in human and developmental stages in worm and fly) in each organism is colored a different hue in the outer two circles surrounding each organism and labeled on the edges of the diagrams. Datasets not in one of the main contexts are marked with asterisks. Each ChIP’d factor is depicted in the middle ring and the count is shown in parenthesis on the edges of the diagram (a given factor can be represented in multiple contexts). Every dataset is depicted in the outer ring, scaled by the number of peaks, and shaded to represent polymerase (red), transcription factor (lighter shade) and other (darker shade). In total 165, 93, and 52 unique factors were ChIP’d across all conditions and cell lines in human, worm, and fly respectively.
All RFs were analyzed by ChIP-seq according to modENCODE/ENCODE standards: antibodies were extensively characterized, and at least two independent biological replicates were analyzed21. Worm RFs were assayed in embryo (EX) and stage 1–4 larvae (L1-L4 larvae), fly RFs in early embryo (EE), late embryo (LE) and post embryo (PP), and human RFs in myelocytic leukemia K562 cells, lymphoblastoid GM12878 cells, H1 embryonic stem cells, cervical cancer HeLa cells, and liver eptihelium HepG2 cells. Binding sites were scored using a uniform pipeline that identifies reproducible targets using IDR analysis (Extended Data Figure 1)22 and quality-filtered experiments (see Supplementary Information). These rigorous quality metrics insure that the data sets used here are robust. All data presented are available at www.ENCODEProject.org/comparative/regulation/.
In order to explore motif conservation, we examined the 31 cases in which we had members of orthologous TF families profiled in at least two species (Extended Data Figure 2a; Supplementary data) we examined whether regulatory features were conserved across species. Sequence enriched motifs were found for 18 of the 31 families and for 12 orthologous families (41 RFs), the same motif is enriched in both species (Extended Data Figure 2b–c). For 18 of 31 families (64 of 93 RFs), the motif from one species is enriched in the bound regions of another species (one-sided hypergeometric, p-value=3.3×10−4). These findings indicate that many factors retain highly similar in vivo sequence specificity within orthologous families, a feature noted previously for only a limited number of factors.
Next, we used RNA-seq data3 to determine whether targets of orthologous RFs are specifically expressed at similar developmental stages between fly and worm. As a class, orthologous RFs (both assayed here and not) are significantly expressed at similar stages (Extended Data Figure 3a–c). However, expression of orthologous targets of orthologous RFs in worm and fly shows little significant target overlap (Extended Data Figure 3d) and the large majority of orthologous RFs did not show conserved target functions (Extended Data Figure 4a–c), suggesting extensive re-wiring of regulatory control across metazoans. Nevertheless, human and worm orthologous RFs were more likely to show conserved target gene functions than non-orthologous RFs (Extended Data Figure 4d, Wilcoxon test p-value < 3.9 × 10−6), highlighting RFs with conserved target functions.
RF binding is not randomly distributed throughout the genome, but rather, in all three species, approximately 50% of binding events are found in highly-occupied clusters, termed HOT regions1,2,5,8,10. HOT regions show enhancer function in integrated transcriptional reporters11 and are stabilized by cohesin15,17. HOT regions show no significant enrichment with non-specific antibodies (Extended Data Figure 5), in contrast to recent work using raw signal19 rather than IDR peaks, although the possibility that they are artifacts has been raised.
By comparing HOT regions across different developmental times and cells types, we find that 5–10% of HOT regions are constitutive, and the remaining are context-specific, indicating HOT regions are dynamically established, rather than an intrinsic property of specific regions. In humans we find that ~90% of constitutive HOT regions fall within promoter chromatin states compared to only ~10–20% of context-specific HOT regions (Fig. 2a, Extended Data Figure 6). Instead, ~80–90% of context-specific HOT regions fall within enhancer states. Moreover, these context-specific HOT regions are specifically enriched for enhancers in matching cell types or developmental stages. For example, 80% of GM12878-called HOT regions fall within GM12878-specific enhancers but only ~10% of GM12878-called HOT regions fall within enhancers called in other cell-types (Fig. 2b). These patterns remain similar for all cell types (Extended Data Figure 7), suggesting the two types of HOT regions are established concordantly and dynamically between cell types, though these patterns are weaker in the worm and fly data.
Figure 2. HOT regions.

HOT regions contain binding sites for a large number of factors. (a) A total of 2,283, 2,948, and 46,348 HOT regions exist of which 29.1%, 13.7%, and 9.7% are constitutive in worm, fly, and human respectively. A large fraction of HOT regions are shared across multiple contexts but the majority of HOT regions are specific to a single context. (b) Constitutive human HOT (cHOT) regions show strong enrichment for promoters while cell-type specific [GM12878 (GM), H1hesc (H1), HepG2 (HG), HelaS3 (HL), K562 (K5)] HOT regions show more enhancer enrichment (see also Extended Data Figure 3).
We next constructed regulatory networks in each species by predicting gene targets of each RF using TIP23 and used simulated annealing to reveal the organization of RFs in three layers of master-regulators, intermediate regulators, and low-level regulators (Fig. 3a–b). The algorithm found only 7% of RFs at the top layer of the network in fly and 13% in worm, compared to 33% in human. We also found that more edges are upward flowing in human (30%) than worm and fly (22% and 7%). This suggests differences in the global network organization with more extensive feedback and a higher number of master regulators in human.
Figure 3. Networks.

(a) Statistics of the transcription regulatory networks in human, worm, fly and their hierarchical organization. (b) An example of the hierarchical network for worm. (c) Network motif enrichment. The human, worm and fly networks are mostly consistent in terms of motif enrichment. The motif feed-forward loop is the most enriched motif in all three networks. (d) Different transcription factors have different tendencies to appear as top, middle and bottom regulators in a FFL. The lists of human, worm, fly TFs with corresponding tendencies are displayed.
We next assessed the local structure of regulatory networks, by searching for enriched sub-graphs known as network motifs (Fig. 3c). We found that the same network motifs were most and least enriched in the three species. In each case, the most abundant was the feed-forward loop (FFL), while the least abundant were cascade motifs, and both divergent and convergent regulation. Moreover, specific RFs were enriched for origin, target, or intermediate regulators in these FFLs in each species (Fig. 3d). Surprisingly, the number of FFLs varied by developmental stage in both worm and fly, with L1 stage in worm and late-embryo stage in fly showing the highest number of FFLs (Extended Data Figure 8), suggesting increased filtering fluctuations and accelerating responses in these stages24.
We next determined whether the three species showed conserved RF co-associations. We first focused on global co-associations where two factors co-associate frequently regardless of context, either by intermolecular interactions or independent recruitment (Extended Data Figure 9). With the exception of a small number of conserved global RF co-associations (e.g. SIN3A with HDAC1, HDAC2, and NR2C2 in fly and human25–27 and MXI1 with E2F1, E2F4, and E2F6 in worm and human), the majority of global co-associations were not conserved in the contexts and species pairs analyzed.
Because RF co-association at distinct binding regions is local and contextual (i.e. different combinations of factors co-associate at different genomic locations), we next used an approach to detect co-association at distinct regions of the genome based on conserved patterns of RF binding. This method uses Self Organizing Maps (SOMs) to analyze co-association patterns at specific loci by better exploring the full combinatorial space of RF binding than traditional co-association approaches (Fig. 4a–c)28. We demonstrate that co-associations at distinct genomic regions reveal a more complex view of regulatory structure and bring forth categorical enrichments that are lost in a larger, genomic context.
Figure 4. TF co-association.

Many instances of TF co-association are under very specific contexts and are likely not observed in a simple genome-wide co-association study. (a) We combined the patterns of orthologous factors and genomic regions from two organisms to train a SOM where each ‘hexagon’ contains genomic regions from either organism with the same binding pattern of orthologous factors for worm (b) and fly (g). Each hexagon is shaded by the frequency of the pattern in the pairs of organisms. We show an example of binding patterns of 4 hexagons from the human-fly (c–d) and the human-worm (e–f). Names above the heatmaps are human factor names while those below are their ortholog names. Dark shaded boxes indicate binding of that factor. (c) A binding pattern shared at equal frequency between human and fly with only CTCF and SETDB1 (CTCF and SuVar3-9 in fly) binding. (d) A binding pattern that occurs more frequently in human shows ELF1, RNA Pol II, STAT, and TBP binding. (e) A binding pattern at similar frequencies in human and worm that is an example of a HOT region. (f) A pattern more frequent in humans than worms shows RNA Pol II, E2F, FOS, MYBL2, HDAC1, MXI1, FOXA, and TBP binding. (h) Co-localization patterns that occur more frequently near promoters (<500bp) in humans are highly likely to also occur at promoters in worm (80%) and fly (100%).
We examined whether specific contextual co-associations are conserved for orthologous RFs by using binding data from each organismal pair i.e. human-worm and human-fly (Fig. 4b,g). Specific RF co-associations were observed; most are conserved to varying degrees across each organism with very few that are entirely organism-specific (Fig. 4b,g). These co-associations result in expected sets of factors such as the previously noted SIN3A+HDAC co-association. In addition, we find new co-associations such as the pattern in Fig. 4f for human-worm, which in worm is highly enriched for GO terms associated with sex determination. We further examined which co-associations are conserved at distinct gene locations (i.e. proximal and distal). We found distinct combinations of conserved co-associations in relation to TSS regions. Interestingly, virtually all TSS-proximal co-associations in human remain TSS-proximal in worm (~80%) and fly (~100%), indicating that co-associations that occur at promoters are often highly conserved (Fig. 4h). On the other hand, co-associations at distal regions are much less conserved.
Using a large resource of regulatory binding information, our results suggest that there is little conservation of individual regulatory targets and binding patterns for these highly divergent metazoans. However, we do find strong conservation of overall regulatory architecture, both in network motif usage and in concentrated regulatory binding at dynamically established HOT regions. We observe an increased conservation of in vivo sequence preferences and some target gene functions, with context-specific RF partners still be observed at specific loci in these distal comparisons. These findings are consistent with previous results indicating that the gene targets of regulation are typically quite divergent and likely account for many of the phenotypic differences among species12–14,16,29,30, despite conserved sequence preferences. We significantly extend these observations, both in the number of regulators studied and in the range of regulatory properties studied, and provide specific examples of conserved and diverged regulatory functions. Lastly, beyond its potential for comparative studies of gene regulation, the primary datasets provide invaluable new information of genome-wide TF binding information both in human, and in two of the most important metazoan models of human biology, development, and disease.
Methods
Detailed methods are in the supplement. Data sets described here can be obtained from the ENCODE project website at www.ENCODEProject.org/comparative/regulation/.
Extended Data
Extended Data Figure 1. Outline of data processing pipeline.
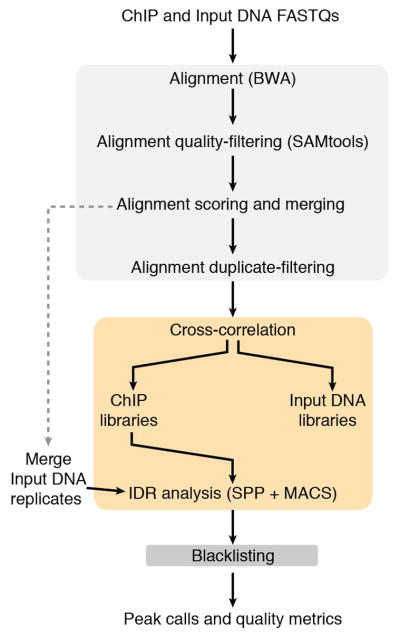
All data sets were processed using a uniform processing pipeline with identical alignment and filtering criteria and standardized IDR peak calling using SPP (Human + Worm) and MACS2 (Fly).
Extended Data Figure 2. Motifs.
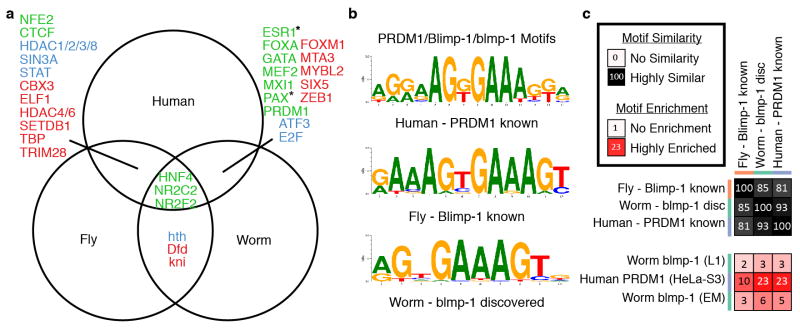
(a) 32 TF gene families with a binding dataset for at least two species (names abbreviated). Cross enrichment indicates the enrichment of motifs from one species in the datasets of another. For 13 families, we observed no cross enrichment (red). For 7 families (blue) we observed cross enrichment and for an additional 12 (green) we also had matching motifs. For two cases marked by an asterisk a known fly motif matches the human motif but no worm motif matches. (b) PRDM1/Blimp-1/blmp-1 gene family. We discovered a motif in worm datasets that match literature derived known motifs from human and fly. (c) All three motifs are highly similar and enriched in human PRDM1 and worm blmp-1 datasets. Cell-type and treatment are indicated for each dataset in parenthesis. Enrichments in each box are the fraction of motif instances that are inside the bound regions and dividing that by the fraction of shuffled motif instances. Additional motifs known and discovered for these and other datasets are included in Supplementary Information.
Extended Data Figure 3. Orthologous expression in worm/fly.

(a) Fly-worm stage alignment of expression using all fly-worm orthologs. (b) Fly-worm stage alignment by using all TF orthologs. (c) Fly-worm stage alignment by using ChIP’d TF ortholog. (d) Fly-worm stage alignment by using proximal genes to ChIP’d TF binding sites. The stage-mapped data exhibit two sets of collinear patterns between the two species (distinct diagonals). In the bottom diagonal, expression from worm embryos and larvae are matched with fly embryos and larvae, respectively; worm adults are matched with fly early embryos and fly female adults, possibly due to the orthologous gene expression in eggs of both species; worm dauers are matched with fly late embryo to L1 and L3 stages, which is similar to the position of dauer stages in the worm lifecycle (between worm L1 and L4 stages). In the upper diagonal, worm middle embryos are matched with fly L1 stage; worm late embryos are matched with fly prepupae and pupae stages; worm L4 male larvae are matched with fly male adults. This collinear pattern may be attributable to fly genes with two-mode expression profiles and many-to-one fly-worm orthologous gene pairs. For more details, please refer to the companion paper31.
Extended Data Figure 4. Comparison of GO enrichment of orthologous TF pairs.

A comparison of GO enrichment of orthologous TF pairs for all contexts in (a) Human vs Worm, (b) Human vs. Fly, and (c) Worm vs. Fly is shown. Red boxes indicate level of similar GO enrichment. ‘Plus’ signs mark orthologous TF pairs with white ‘pluses’ indicating the most significant enrichment for an ortholog pair. (d) Orthologous factors are more enriched for matching GO terms than non-orthologous factors.
Extended Data Figure 5. Human HOT enrichments are not overly enriched for control DNA.

HOT regions do not represent assembly or ChIP-ability artifacts. (a) Scatter plot of IgG IP/Input vs TF Occupancy. Scatterplot is shaded by density of points. Red dash line represents HOT threshold and black dashed line represent an enrichment of 1x. Black line represents best fitting line to the scatter plot (R2 = 0.0045) (b) A scatterplot of density (number of TF peaks per kb) rather than total number of peaks in a region shows a similar trend. (c) Barplot of fraction of regions with high IgG enrichment for HOT and non-HOT (RGB) regions using the same threshold (1.5x) as Teytelman et al. Figure 7 reveals little similarity between HOT regions and artifact ChIP regions. (d) The fraction of HOT (red) and non- HOT (blue) regions with high IgG enrichment is plotted as a function of threshold. Black line represents no enrichment (IgG/Input = 1x) and grey dashed line represents the enrichment cutoff (1.5x) used in (b) and in Teytelman et al. Figure 7. (e) Comparison of IgG (IgG/Input) and RNA Pol II enrichment (RNA PolII/Input) shows a different trend from Teytelman et al. Fig 3a. (e) Nearly all (99.967%) of our uniformly processed RNA PolII binding sites have IP/Input rations >2x, with a median enrichment of ~20x.
Extended Data Figure 6. HOT regions were identified in all organisms.

(a) To identify HOT region for each context, we first analyzed the number and size distribution of target binding regions (in which factor binding sites are concentrated). For each target case simulation, we randomly select an equivalent number of random binding regions with a matched size distribution. Next, for each factor assayed (in the target case), we evaluated the number and size of observed binding sites, and simulated an equivalent number and size distribution of target binding sites, restricting their placement to the simulated binding regions. We collapsed simulated binding sites from all factors into binding regions, verifying that these cluster into a similar number of simulated binding regions as the target binding regions. We identify regions at a 5% (HOT) and 1% (XOT) occupancy threshold based on this simulated data. (b) Binding of regulatory factors covers different fractions of the genomes of fly, human, and worm. Coverage is shown for constitutively HOT regions (cHOT – red), HOT regions (yellow), and non-HOT regions (RGB –green). Coverage for XOT regions is given in parenthesis.
Extended Data Figure 7. HOT enrichments with context-specific enhancer enrichments.

(a) Histone marks for HOT regions (represented by points and smoothed to show density) at proximal and (b) distal sites show similar trends of histone mark enrichment in their flanking regions. Enhancer calls for a specific developmental stage (c, e) or cell type (d) (labeled over each set of bar graphs) match HOT regions from that cell type and not HOT regions from another cell type. Each set of six bar graphs represents the same set of HOT regions called constitutively HOT or specific to each of the five cell types. Constitutive HOT (cHOT) regions are significantly enriched at promoters with the remaining regions overlapping enhancer regions.
Extended Data Figure 8. The number of feed forward loops in different stage-specific networks.

The number of FFLs in a stage is normalized by the number of TFs in the corresponding stage-specific network. Though the sets of TFs may differ, the number of TFs in each stage stays roughly the same.
Extended Data Figure 9. Co-associations.

Evolutionary retention and change in TF co-associations. The pairwise co-association strengths between orthologous TFs are shown for human-worm orthologs (a, b) and human-fly orthologs (c, d). For each pair of species-specific orthologs across multiple samples, the co-association strength, measured as the fraction of significant co-binding events between experiments, is shown (IntervalStats32). (a) Human co-association matrix for human-worm orthologs. (b) Worm co-association matrix for human-worm orthologs. (c) Human co-association matrix for human-fly orthologs. (d) Fly co-association matrix for human-fly orthologs. (e) Comparison of human-worm TF ortholog co-associations. The co-association strength of human-worm orthologs in human (x-axis) is plotted against the co-association strength in worm (y-axis). Lines depict 1 (solid) and 1.5 (dashed) standard deviations from the mean score. Factors in blue represent enrichments due to paralogous TFs in human that tend to be highly co-associated. (f) Comparison of human-fly TF ortholog co-associations. Co-association strength in human (x-axis) is plotted against co-association strength in fly (y-axis). For TF orthologs assayed in multiple developmental stages/cell-lines, the maximal co-association between contexts was selected for the comparative analyses (e, f).
Supplementary Material
Acknowledgments
This work is supported by the NHGRI as part of the modENCODE and ENCODE projects. This is funded by U01HG004264, RC2HG005679 and P50GM081892 to KPW, U54HG006996, U54HG004558, and U01HG004267 to MS, and F32GM101778 to KEG.
Footnotes
Author contributions
Data analysis: APB, CLA, YC, DX, PK, AK, PC, LM, KKY, JR, DW, CC, LH, PC, YCW
Data production: MS, RS, EJR, DV, RT, PW, RHW, CB, KG, JJ, LJ, DK, TK, WN, RS,
Paper writing: APB, MS, CLA, KW, KKY, RHW
NIH scientific project management: EAF, PJG, MJP. The role of the NIH Project Management Group in the preparation of this paper was limited to coordination and scientific management of the modENCODE and ENCODE consortia.
Overall project management: MS, MK, KPW, MG, RHW, VR
Completing Financial Interests
MPS is a cofounder and scientific advisory board (SAB) member of Personalis. MPS is on the SAB of Genapsys.
(see attached)
References
- 1.modENCODE Consortium et al. Identification of functional elements and regulatory circuits by Drosophila modENCODE. Science. 2010;330:1787–1797. doi: 10.1126/science.1198374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gerstein MB, et al. Integrative analysis of the Caenorhabditis elegans genome by the modENCODE project. Science. 2010;330:1775–1787. doi: 10.1126/science.1196914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gerstein M, et al. An Integrative Comparison of Metazoan Transcriptomes [Google Scholar]
- 4.Berger MF, et al. Variation in Homeodomain DNA Binding Revealed by High-Resolution Analysis of Sequence Preferences. Cell. 2008;133:1266–1276. doi: 10.1016/j.cell.2008.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moorman C, et al. Hotspots of transcription factor colocalization in the genome of Drosophila melanogaster. Proceedings of the National Academy of Sciences. 2006;103:12027–12032. doi: 10.1073/pnas.0605003103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lavoie H, et al. Evolutionary tinkering with conserved components of a transcriptional regulatory network. PLoS Biol. 2010;8:e1000329. doi: 10.1371/journal.pbio.1000329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.He Q, et al. High conservation of transcription factor binding and evidence for combinatorial regulation across six Drosophila species. Nat Genet. 2011;43:414–420. doi: 10.1038/ng.808. [DOI] [PubMed] [Google Scholar]
- 8.ENCODE Project Consortium et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mikkelsen TS, et al. Comparative epigenomic analysis of murine and human adipogenesis. Cell. 2010;143:156–169. doi: 10.1016/j.cell.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yip KY, et al. Classification of human genomic regions based on experimentally determined binding sites of more than 100 transcription-related factors. Genome Biol. 2012;13:R48. doi: 10.1186/gb-2012-13-9-r48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kvon EZ, Stampfel G, Yáñez-Cuna JO, Dickson BJ, Stark A. HOT regions function as patterned developmental enhancers and have a distinct cis-regulatory signature. Genes Dev. 2012;26:908–913. doi: 10.1101/gad.188052.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schmidt D, et al. Five-vertebrate ChIP-seq reveals the evolutionary dynamics of transcription factor binding. Science. 2010;328:1036–1040. doi: 10.1126/science.1186176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Odom DT, et al. Tissue-specific transcriptional regulation has diverged significantly between human and mouse. Nat Genet. 2007;39:730–732. doi: 10.1038/ng2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Borneman AR, et al. Divergence of transcription factor binding sites across related yeast species. Science. 2007;317:815–819. doi: 10.1126/science.1140748. [DOI] [PubMed] [Google Scholar]
- 15.Yan J, et al. Transcription Factor Binding in Human Cells Occurs in Dense Clusters Formed around Cohesin Anchor Sites. Cell. 2013;154:801–813. doi: 10.1016/j.cell.2013.07.034. [DOI] [PubMed] [Google Scholar]
- 16.Peter IS, Davidson EH. Evolution of gene regulatory networks controlling body plan development. Cell. 2011;144:970–985. doi: 10.1016/j.cell.2011.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Faure AJ, et al. Cohesin regulates tissue-specific expression by stabilizing highly occupied cis-regulatory modules. Genome Res. 2012;22:2163–2175. doi: 10.1101/gr.136507.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spitz FCO, Furlong EEM. Transcription factors: from enhancer binding to developmental control. Nat Rev Genet. 2012;13:613–626. doi: 10.1038/nrg3207. [DOI] [PubMed] [Google Scholar]
- 19.Teytelman L, Thurtle DM, Rine J, van Oudenaarden A. Highly expressed loci are vulnerable to misleading ChIP localization of multiple unrelated proteins. Proceedings of the National Academy of Sciences. 2013;110:18602–18607. doi: 10.1073/pnas.1316064110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Negre N, et al. A cis-regulatory map of the Drosophila genome. Nature. 2011;471:527–531. doi: 10.1038/nature09990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Landt SG, et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012;22:1813–1831. doi: 10.1101/gr.136184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Q, Brown JB, Huang H, Bickel PJ. Measuring reproducibility of high-throughput experiments. The Annals of Applied Statistics. 2011;5:1752–1779. [Google Scholar]
- 23.Cheng C, Min R, Gerstein M. TIP: a probabilistic method for identifying transcription factor target genes from ChIP-seq binding profiles. Bioinformatics. 2011;27:3221–3227. doi: 10.1093/bioinformatics/btr552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alon U. Network motifs: theory and experimental approaches. Nat Rev Genet. 2007;8:450–461. doi: 10.1038/nrg2102. [DOI] [PubMed] [Google Scholar]
- 25.Heinzel T, et al. A complex containing N-CoR, mSin3 and histone deacetylase mediates transcriptional repression. Nature. 1997;387:43–48. doi: 10.1038/387043a0. [DOI] [PubMed] [Google Scholar]
- 26.Nan X, et al. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393:386–389. doi: 10.1038/30764. [DOI] [PubMed] [Google Scholar]
- 27.Huang Y, Myers SJ, Dingledine R. Transcriptional repression by REST: recruitment of Sin3A and histone deacetylase to neuronal genes. Nat Neurosci. 1999;2:867–872. doi: 10.1038/13165. [DOI] [PubMed] [Google Scholar]
- 28.Xie D, et al. Dynamic trans-acting factor colocalization in human cells. Cell. 2013;155:713–724. doi: 10.1016/j.cell.2013.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carroll SB, Grenier J, Weatherbee S. From DNA to Diversity: Molecular Genetics and the Evolution of Animal Design. Wiley-Blackwell; 2004. at < http://www.wiley.com/WileyCDA/WileyTitle/productCd-1405119500.html>. [Google Scholar]
- 30.King MC, Wilson AC. Evolution at two levels in humans and chimpanzees. Science. 1975;188:107–116. doi: 10.1126/science.1090005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.