

Abstract
A reported mixed opioid agonist - neurokinin 1 receptor (NK1R) antagonist 4 (Dmt-D-Arg-Aba-Gly-(3’,5’-(CF3)2)NMe-benzyl) was modified to identify important features in both pharmacophores. The new dual ligands were tested in vitro and subsequently two compounds (lead structure 4 and one of the new analogues 22, Dmt-D-Arg-Aba-β-Ala-NMe-Bn) were selected for in vivo behavioral assays, which were conducted in acute (tail-flick) and neuropathic pain models (cold plate and von Frey) in rats. Compared to the parent opioid compound 33 (without NK1R pharmacophore), hybrid 22 was more active in the neuropathic pain models. Attenuation of neuropathic pain emerged from NK1R antagonism as demonstrated by the pure NK1R antagonist 6. Surprisingly, despite a lower in vitro activity at NK1R in comparison with 4, compound 22 was more active in the neuropathic pain models. Although potent analgesic effects were observed for 4 and 22, upon chronic administration, both manifested a tolerance profile similar to that of morphine and cross tolerance with morphine in a neuropathic pain model in rat.
Keywords: Hybrid peptides, Opioid agonism, NK1R antagonism, Acute pain, Neuropathic pain, Tolerance
1. Introduction
Morphine has been the prime analgesic for the treatment of severe to moderate pain for centuries. Next to morphine, opioid ligands such as the fentanyl family of painkillers are commonly used in a clinical context. Despite the severe drawbacks that are associated with these analgesics, they remain in use to date. When opioid ligands are administered for a longer period of time, side effects such as, for example, constipation, physical dependence and analgesic tolerance emerge.1 Moreover, opioids have less analgesic efficacy in neuropathic pain, a pathology which is considered to be a major healthcare burden.2-6 Neuropathic pain can be caused by a damaged nerve in the nervous system, be the result of trauma, an infection, or diabetes,7 and it is characterized by an increased sensitivity to non-noxious and noxious stimuli.8, 9 Since neuropathic pain and opioid tolerance both share the feature of diminished morphine analgesia, a common underlying mechanism could be suggested.
One explanation for the lack of long-duration opioid efficiency is increased release and expression of certain endogenous ligands and receptors.10, 11 Being part of a larger biological network, activation of the opioid system is compensated for by the production of “anti-opioids” or pain-enhancing ligands, such as cholecystokinin and substance P. These neuroplastic changes in the CNS cannot be counteracted by the currently used treatments.
For this reason, and as an alternative to the co-administration of separate drugs, designed multiple ligands (DMLs, single chemical entities that modulate multiple targets) with both opioid and non-opioid activity were developed.12-14 Among other dual activity ligands,12 hybrid peptides bearing both an opioid and NK1R pharmacophore (e.g. compounds 1-3 in Figure 1) were designed by the teams of Lipkowski15-18 and Hruby.19, 20
Figure 1.
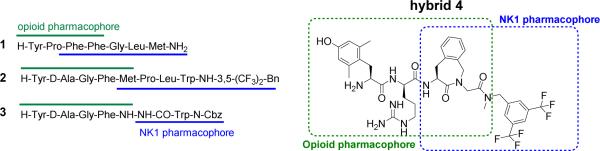
Selected examples of opioid agonist - NK1 antagonist DMLs
In these DMLs the opioid part induces the well-established analgesic effect that is characteristic of opioid ligands, whereas antagonism at the NK1 receptors blocks the signals induced by an endogenous neurotransmitter, substance P (SP), one of the pronociceptive peptides involved in pain signalling.21,22 Since prolonged pain states and sustained opioid administration lead to increased secretion of SP and enhanced expression of NK1 receptors, blockage of NK1R could potentially counteract the pain-enhancing effect induced by SP and its receptor, and eventually lead to prolonged antinociceptive efficacy. In addition, Gonzalez and coworkers showed that the selective neurokinin-1 receptor antagonist CI-1021 possessed a superior side effect profile, as compared to morphine, and may have a therapeutic use for the treatment of neuropathic pain.23 All of the above findings support the design and preparation of DMLs with a dual opioid agonists and neurokinin-1 antagonist profile for the development of more efficacious drugs for the treatment of acute and neuropathic pain.
Taking earlier work on mixed opioid-NK1R peptide DMLs into consideration,17-20 a chimeric opioid-NK1 peptidomimetic 4 (Figure 1) was previously reported by our group.24 The opioid subunit in 4 is derived from the μ opioid receptor lead peptide Dmt-D-Arg-Phe-Lys-NH2 ([Dmt1]DALDA) 5,25 and contains a constrained Aba (4-amino-2-benzazepinone)26-28 moiety in position 3. As this conformationally constrained amino acid was also the core structure of a newly developed NK1 antagonist,24 Ac-Aba-Gly-NMe-3’,5’-(CF3)2-Bn (6) (corresponding to the blue part in structure 4, Figure 1), the combination of both ligands led to the hybrid structure Dmt-D-Arg-Aba-Gly-NMe-3’,5’-(CF3)2-Bn 4, a DML with overlapping pharmacophores. The in vitro biological evaluation of 4 showed that combination of the two components gratifyingly resulted in good binding affinity for both opioid and NK1R receptors.24 Furthermore, the compound showed antinociceptive activity after intravenous administration in vivo, and hence the hybrid structure proved, similarly to the parent opioid sequence Dmt-D-Arg-Aba-Gly-NH2 7,29 capable of crossing the blood-brain barrier (BBB). Unfortunately, hybrid 4 still produced analgesic tolerance in naive animals.29 In contrast, Vanderah and coworkers reported that chronic administration of a peptidic DML with a similar dual activity profile, H-Tyr-D-Ala-Gly-Phe-Met-Pro-Leu-Trp-O-3’,5’-Bn(CF3)2 8, was able to suppress antihyperalgesic tolerance.30 We suggested that this discrepancy in results could be due to the different pain models used in both studies. Whereas Vanderah et al.30 utilized a hyperalgesia animal model, our published findings resulted from an acute pain model (tail-flick).29 Given the promising results of Vanderah et al, we pursued our efforts to develop a CNS active peptidomimetic able to attenuate opioid-related tolerance.
In this work we report structural modifications of hybrid lead structure 4. Various modifications (Figure 2) were performed to: i) increase opioid activity, ii) determine the key features for NK1R binding and antagonism, and iii) attempt to reduce the molecular weight of the lead structure.
Figure 2.
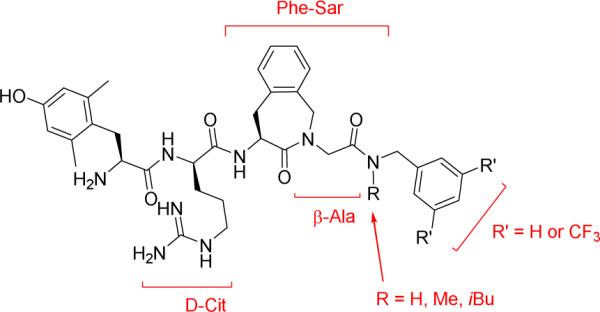
Overview of the investigated structural modifications
2. Results and discussion
2.1 Synthesis
Lead structure 4 was previously prepared in two main steps: a standard solid phase synthesis using the preassembled Fmoc-Aba-Gly-OH dipeptide,31 followed by coupling of a protected tetrapeptide precursor to NMe-3’,5’-trifluoromethyl benzylamine in solution.24
In this work, a direct solid phase assembly of the Aba building block was developed. This strategy avoids a separate solution phase synthesis of the Aba-Gly and Aba-β-Ala constrained dipeptides 13 (n=1 or 2, Scheme 1).
Scheme 1.

Solid phase assembly of the constrained opioid tetrapeptide and solution phase coupling of NK1R pharmacophoric benzyl group
Reaction conditions: a) 20% 4-methylpiperidine in DMF, 5 + 15 min: b) 2 eq. of 11 and 4 eq. NaCNBH3 in CH2Cl2/TMOF/0.5% AcOH, 30 min.; c) 3 eq. TBTU in 0.4 M NMM in DMF, 3 h; d) 6 eq. NH2NH2.H2O in DMF, 18 h; e) 3 eq. Fmoc-D-Arg(Pbf)-OH or Fmoc-D-Cit-OH, 3 eq. TBTU in 0.4 M NMM in DMF, 1.5 h; f) 20% 4-methylpiperidine in DMF, 5 + 15 min; g) 2 eq. Boc-Dmt-OH, 2 eq. DIC/HOBt in DMF, 2 h; h) 1% TFA in CH2Cl2, rt, 30 min; i) 5 eq. MeNH2, 1 eq. of 17, CH2Cl2, 50 °C, 16 h; j) 2 eq. DIPEA, 1.5 eq. DIC/HOBt, 1.5 eq. of 18, 15 h; k) TFA/TES/H2O 95/2.5/2.5, 3 h; l) preparative RP-HPLC purification.
The Aba-containing peptides were prepared using Fmoc-Gly-OH or Fmoc-β-Ala-OH linked to 2-chlorotrityl resin as the solid support, via the pathway depicted in Scheme 1. After removal of the Fmoc protective group, a reductive amination with phthaloyl-protected orthoformyl phenylalanine 11 in the presence of NaBH3CN was performed. Aldehyde 11 was obtained by reduction of the corresponding ortho-cyano analogue following a literature procedure.32 The desired secondary amine was formed to a high extent after 30 minutes, according to the Kaiser test. Longer reaction times led to overalkylation. Cyclizations with TBTU as the activating agent yielded the solid-supported aminobenzazepinones 13. To allow further elongation of the peptide, the resin-bound 13 was treated with hydrazine monohydrate for phthaloyl deprotection. The remaining amino acids were coupled by standard SPPS methods. More specifically, the coupling of Boc-Dmt-OH was performed using DIC/HOBt as the coupling mixture to avoid side reactions, caused by the unprotected phenolic group, which occur upon use of TBTU/DIPEA mixtures. The protected peptide acids 16 were obtained after mild acidic cleavage (1% TFA in CH2Cl2) from the solid support. They were of sufficient purity to allow a direct coupling with the different benzylamines in solution and thus did not require intermediate purification. The synthesis of N-methyl and N-isobutyl-3’,5’-bis(trifluoromethyl)benzylamines (18) could be achieved by reacting methyl- or isobutylamine with commercially available benzylchloride or 3’,5’-bis(trifluoromethyl)benzyl chloride following a literature procedure.33 To reduce formation of the double alkylated product, an excess of the amine was used. The crude amines were purified by flash chromatography to afford the desired benzylamines in moderate yield (43-48%). Coupling of the protected tetrapeptide analogs to the set of amines was performed using a mixture of DIC/HOBt/DIPEA. Final deprotection of the N-terminal Boc group and Pbf side chain protective group was achieved with the cleavage cocktail TFA/TES/H2O (95/2.5/2.5). Precipitation in cold ether, followed by preparative RP-HPLC purification and lyophilisation yielded pure (>95%) peptide analogues 20.
2.2 In vitro biological evaluation
In designed bifunctional ligands with high framework overlap, the risk of encountering undesired steric encumbrance increases. While highly merged structures take advantage of structural commonalities in the starting compounds - they present multiple ligands with a lower molecular weight – the close proximity of the two pharmacophores can more readily lead to steric clashes during the drug-target recognition event. This phenomenon was also apparent for hybrid 4 (Table 1). Compared to the opioid parent compound 7, creation of the opioid-NK1R hybrid ligand 4 resulted in a significantly reduced binding affinity (Kiμ (7) 0.15 nM and Kiδ (7) 0.60 nM → Kiμ (4) 0.42 nM and Kiδ (4) 10.4 nM) and activity (GPI IC50 (7) 0.32 nM and MVD IC50 (7) 0.42 nM → GPI IC50 (4) 8.51 nM and MVD IC50 (4) 43.3 nM) at both MOR and DOR. Because of the decreased binding affinity and reduced agonist potency, we decided to replace Gly4 by a β-alanine residue.34 The additional methylene, present in β-Ala, would shift the bulky C-terminal benzyl group away from the opioid pharmacophore, and potentially lower the steric hindrance upon receptor binding. Gratifyingly, the substitution improved both MOR and DOR binding 5-fold (21, Table 1). Retained DOR binding affinity in the low nanomolar range would be in favor of the proposed synergy of mixed MOR – DOR activity for enhanced in vivo antinociception.35-37
Table 1.
Opioid and NK1 Receptor Affinity and Activity of all Parent and Hybrid Compounds
| Compound Number | Compound | pA2a (NK1R) | hNK1Rb (Ki nM) | GPIc (IC50 nM) | MVDc (IC50 nM) | MORc (Ki nM) | DORc (Ki nM) | Selectivity Ki DOR/Ki MOR |
|---|---|---|---|---|---|---|---|---|
| 4 | H-Dmt-D-Arg-Aba-Gly-NMe-3’,5’-(CF3)2-Bn | 7.80 | 0.50 | 8.51 | 43.3 | 0.42 | 10.4 | 25.0 |
| 6 | Ac-Aba-Gly-NMe-3’,5’-(CF3)2-Bn | 8.40 | 27.0 | nd | nd | nd | nd | nd |
| 7 | H-Dmt-D-Arg-Aba-Gly-NH2 | nd | nd | 0.32 | 0.42 | 0.15 | 0.60 | 4.0 |
| 21 | H-Dmt-D-Arg-Aba-β-Ala-NMe-3’,5’-(CF3)2-Bn | 6.04 | 59.7 | 11.3 | 6.20 | 0.08 | 2.14 | 25.3 |
| 22 | H-Dmt-D-Arg-Aba-β-Ala-NMe-Bn | 6.44 | 13.0 | 1.86 | 2.16 | 0.08 | 0.28 | 3.5 |
| 23 | H-Dmt-D-Cit-Aba-β-Ala-NMe-3’,5’-(CF3)2-Bn | 6.27 | 34.7 | 2.03 | 1.06 | 0.37 | 0.55 | 1.5 |
| 24 | H-Dmt-D-Cit-Aba-β-Ala-NMe-Bn | nd | 3000 | 0.96 | 0.33 | 0.28 | 0.48 | 1.7 |
| 25 | H-Dmt-D-Arg-Aba-Gly-NMe-Bn | nd | 1530 | 5.97 | 8.64 | 0.09 | 2.35 | 24.8 |
| 26 | H-Dmt-D-Arg-Aba-Gly-NH-Bn | nd | 6190 | 4.44 | 3.50 | 0.14 | 0.62 | 4.4 |
| 27 | H-Dmt-D-Arg-Aba-Gly-NiBu-3’,5’-(CF3)2-Bn | nd | 35 | 19.7 d | 14.5 | nd | nd | nd |
| 28 | H-Dmt-D-Arg-Aba-Gly-NiBu-Bn | nd | 1020 | P.A e | 28.7 | nd | nd | nd |
| 29 | H-Dmt-D-Arg-Aba-β-Ala-NiBu-3’,5’-(CF3)2-Bn | nd | 743 | nd | nd | nd | nd | nd |
| 30 | H-Dmt-D-Arg-Aba-β-Ala-NiBu-Bn | nd | 308 | nd | nd | nd | nd | nd |
| 31 | H-Dmt-D-Arg-Phe-Sar-NMe-3’,5’-(CF3)2-Bn | nd | 734 | 2.95 | 2.31 | 0.62 | 1.7 | 2.7 |
| 32 | H-Dmt-D-Arg-Phe-Sar-NMe-Bn | nd | 16050 | 3.10 | 7.76 | 0.10 | 4.43 | 43.4 |
| 33 | H-Dmt-D-Arg-Aba-β-Ala-NH2 | nd | nd | 0.80 | 0.24 | 1.34 | 17.0 | 12.7 |
The pA2 values were calculated using Schild's equation.40
Inhibitory constants (Ki) of NK1 receptor ligands, measured for the receptor prototype [3H]-SP in the presence of hNK1-CHO membranes. Results are means of three independent experiments. Binding data were calculated using the nonlinear regression/one site competition fitting options of the GraphPad Prism Software.
Values represent means of 3-4 experiments. The GPI functional assay is representative of MOR activation, whereas the MVD is a DOR receptor-representative assay. Binding affinities of compounds for MOR and DOR opioid receptors were determined by displacement of [3H]DAMGO ([D-Ala2,NMePhe4,Gly-ol5]enkephalin) and [3H]DSLET ([D-Ser2,Leu5]enkephalin-Thr6) from rat brain membrane binding sites, respectively.
partial agonist, Ke = 12,7 nM (μ antagonist)
Ke = 5,91 nM (μ antagonist); nd: not determined.
Whereas the improvement in DOR binding affinity was translated into improved functional activity as determined in the MVD tissue bioassay (MVD IC50 (21) 6.20 nM), in the GPI assay, representative of MOR activation, and in comparison with 4, a slightly lower IC50 value (11.3 nM) was determined for 21. However, the drop in NK1R binding and antagonism potency was more dramatic (Table 1). Insertion of the additional methylene unit proved to be detrimental for recognition at the NK1R receptor. In small peptidomimetic NK1R antagonists the two crucial aromatic groups of the key bis-aryl motif are in close proximity of each other.38 The introduction of a methylene unit in 21 makes the linker between these groups more flexible, and hence they can move away from each other, potentially lowering binding affinity at NK1R. Here, this modification induced a 120-fold loss in NK1R binding and an almost two log unit drop in antagonism (pA2 (4) 7.80 → pA2 (21) 6.04). When the trifluoromethyl groups in 21 were removed in an attempt to lower the molecular weight of the mixed ligands resulting in compound 22, picomolar MOR binding was preserved (Kiμ 0.08 nM), DOR binding increased by a factor of 10 and in vitro agonism was improved 6- and 3-fold at MOR and DOR, respectively. Removal of the CF3 groups also improved NK1R binding affinity approximately 4 times. The increase was also apparent in the pA2 values (pA2 (21) 6.04 → pA2 (22) 6.44). In a previous study,39 we demonstrated that D-Arg2 could be replaced by D-Cit, a modification that removes one positive charge in the peptide, without loss of BBB permeability. Hence, this modification was also carried out in the present study, to give 23. Comparison with 22 showed that this change was well tolerated both at the opioid receptors and at the NK1 receptor. However, when carrying out the two modifications simultaneously (CF3 group removal and D-Arg→D-Cit), a dramatic loss in NK1R binding was observed (Ki 3000 nM for 24). Again, the opioid system was more tolerant towards this double modification. The promising results obtained with compound 22 led to the reinvestigation of the bis-trifluoromethyl removal in the Gly4-bearing tetrapeptide. In compounds 25 and 26 this removal was again well-tolerated at both MOR and DOR, but dramatically decreased NK1R binding affinity. The N-methyl amide function at the C-terminus was also confirmed to be of importance. Whereas secondary amides exist almost exclusively in the trans conformation, tertiary amides contain an appreciable amount of the cis conformer, which may be the conformer that is binding to the receptor. The cis/trans ratio of the amide bond between residue 4 and the C-terminal benzylamine moiety in lead compound 4 was investigated by NMR analysis and was estimated to be 35:65. Results obtained by molecular modeling also confirmed one of the three lowest energy conformations in the NK1R pharmacophore to adopt the cis amide geometry,24 and hence this orientation could be important for NK1R activity. In an attempt to increase the cis/trans ratio, a N-iBu group was used instead of the N-Me motif. Although the bulkyness of the iBu group was expected to promote the cis amide bond conformation, the biological data of compounds 27 to 30 clearly show that the presence of the iBu group did not enhance NK1R affinity. Only one of these ligands, compound 27, showed moderate nanomolar range NK1R binding affinity. Due to its rather limited opioid activity, this compound was not investigated any further and the cis/trans ratio was not determined experimentally.
Up to this point, the requirement of the conformational constraint, imposed by the Aba moiety, was not verified. In the ‘open ring’ compounds 31 and 32, bearing a sarcosine in position 4, the ring opening was not crucial for opioid affinity and activity, but led to a dramatic decrease in NK1R binding affinity. This could be explained by the flexibility of the Phe3 side chain in the ring opened compounds, allowing it to move away from the C-terminal benzyl amide. In contrast, in the Aba containing peptides, the methylene anchorage of the aromatic side chain to the backbone orients this side chain to the C-terminus possibly keeping the two aromatics in the bis-aryl motif in close proximity of each other.
2.3 Behavioral study in rats and mice
Among the new analogues the one with a most suited profile for in vivo evaluation was judged to be hybrid 22. This DML has a potent and quite balanced MOR and DOR binding affinity and agonist potency profile (sub- and low nanomolar range, resp.), a feature believed to be beneficial to opioid antinociception,35-37, 41, 42 while still showing low nanomolar NK1R binding affinity, and a pA2 value of 6.44 (i.e. 1.5 log unit loss in antagonism potency, compared to lead compound 4). The profile of compound 22 differs from the lead structure in terms of opioid receptor selectivity (Kiδ/Kiμ (4) 25 vs. Kiδ/Kiμ (22) 3.5), opioid potency (IC50 GPI (4) 8.51 nM and IC50 MVD (4) 43.3 nM vs. IC50 GPI (22) 1.86 nM and IC50 MVD (22) 2.16 nM), NK1R binding (K iNK1R (4) 0.5 nM vs. K iNK1R (22) 13 nM), and NK1R antagonist potency (pA2 (4) 7.8 vs. pA2 (22) 6.4). The in vivo evaluation of 4 in an acute pain model by the tail-flick assay has been published previously.29 From these results, it could be concluded that lead peptide 4 was able to cross the BBB, as it showed a potent antinociceptive effect after intravenous administration.
2.3.1 Acute pain evaluation in rats
The potencies of two new compounds (22 and 33) were compared to the effect of the initial lead peptide 4 and its parent opioid structure 7, NK-1 antagonist 6 and morphine in naïve rats (Figure 3). All tested compounds were administered i.t. at different doses in naïve rats and the effect was measured by the tail-flick test 30 min after administration. Both hybrids (4 and 22) and their parent compounds (7 and 33, resp.) increase the nociceptive threshold. The antinociceptive potencies of the parent compounds (7 and 33) are clearly much higher in comparison with those of the two hybrids and morphine. In contrast, the effect of the parent NK-1 antagonist 6 was very weak as compared to the hybrids and opioid parent compounds in naïve animals. Moreover, in the same test and after intravenous administration, structure 6 did not show any activity at all at doses up to 1.21 μmol/kg (not shown), which can be indicative of a low BBB permeation. Interestingly, lead structure 4 generates a toxic effect (observed as seizures and death of animals) at the dose of 9.5 nmol (not shown in Figure 3). Unlike hybrid 4, the modified DML hybrid 22 did not induce a toxic effect at this higher dosage. Our results also indicate that both 22 and 33 produced a long-lasting antinociceptive effect (2h) after intrathecal administration (data not shown).
Figure 3.
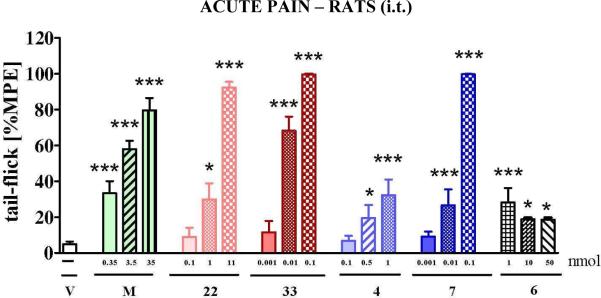
The effect of intrathecal (i.t.) administration of morphine (M, 0.35-35 nmol), hybrid 22 (0.1-11 nmol) and its parent compound 33 (opioid agonist, 0.001-0.1 nmol), hybrid 4 (0.1-1 nmol) and its parent compound 7 (opioid agonist, 0.001-0.1 nmol) and compound 6 (NK-1 antagonist 1-50 nmol) measured in tail-flick test 30 min after administration in naïve rats (7-12 animals per group). Inter-group differences were analyzed by ANOVA Bonferroni's Multiple Comparison Test; *p<0.05; ***p<0.001 vs. V – vehicle treated naïve rats. %MPE = percentage of maximal possible effect.
2.3.2 Acute pain in mice
When hybrid 22 and its parent opioid compound 33 were evaluated after i.v. administration in mice, we observed that a 10.8 μmol/kg dose of compound 22 gave way to a potent antinociceptive response that is approximatively three times higher than the one of morphine at a slightly lower dose of 12.5 μmol/kg (Figure 4). For comparison, when compound 4 was administered i.v. at a dose of 3.8 μmol/kg in a previous study, it showed an antinociceptive potency which was 3.7 times higher than i.p. administered morphine (not shown).29 From these results it appears that both compounds, hybrids 4 and 22, have a similar potency that is superior to morphine. In contrast, parent peptide 33 still showed a potent response at a dose of 1.21 μmol/kg, which largely exceeded that of morphine (Figure 4). The beneficial potency shift from 22 to 33 might be attributed to a 10-fold shift in DOR potency (see MVD assay in Table 1). Several studies have provided evidence that compounds with a dual MOR/DOR activity present beneficial pharmacological effects in comparison to highly selective MOR agonists.35
Figure 4.
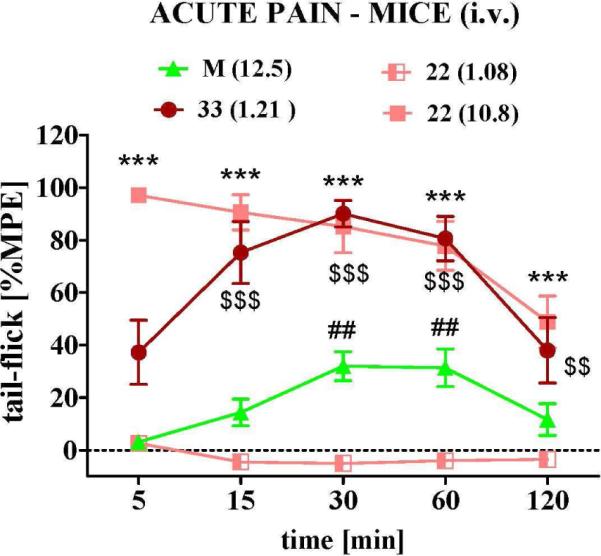
The time dependent effect of intravenous (i.v.) administration in mice of hybrid 22 (1.08; 10.8 μmol/kg), its parent compound 33 (1.21 μmol/kg), and morphine (M, 12.5 μmol/kg) in the tail-flick assay (6-8 animals per group). Statistical analysis has shown significant differences between 22 (10.8 μmol/kg) and morphine (*** p< 0.001); between 22 (1.08 μmol/kg) and morphine (## p< 0.01) as well as between 33 and morphine ($$ p< 0.01 and $$$ p < 0.001).
2.3.3 Neuropathic pain in rats
In order to verify whether compounds 4 and 22 would be capable of suppressing allodynia and hyperalgesia, induced by chronic constriction injury (CCI) to the sciatic nerve in rat, these hybrids were examined in this neuropathic pain model with i.t. administration.
Seven days after CCI the two hybrids, their parent compounds, the NK-1 antagonist and morphine were administered i.t. in rats. In the model of neuropathic pain, the antiallodynic effect of morphine and both parent compounds (33 and 7) is weaker as compared to the observed effect in the acute pain assay. This is in agreement with literature in which a lower potency of opioids for neuropathic pain treatment is described (Fig. 5A). In contrast to the pure opioids, the two hybrids (22 and 4) were significantly more active in this test. The strongest attenuation of allodynia was observed after administration of hybrid 22, reaching 80% MPE at a dose of 1 nmol (30% MPE in acute pain), and hybrid 4, reaching 60% MPE after dose of 1 nmol (30% MPE in acute pain). At a higher dose of 1 nmol, the two parent compounds 33 and 7 showed a toxic effect (marked on Fig. 5A and 5B as black bar), which was not observed after administration of hybrid compounds 22 and 4 administered at the same dose. Hybrid 4 also showed a toxic effect at the highest investigated dose (9.5 nmol, not shown). The highest decrease of allodynia and lack of toxic signs were noticed for the hybrid compound 22, although the highest investigated dose of 11 nmol was weaker in antagonizing allodynia and hyperalgesia than lower doses (not shown). This may be indicative of a weak toxic effect that was difficult to observe. At this stage the NK-1 pharmacophore appeared to be important for activity in the von Frey assay, a supposition which was supported by the antiallodynic activity of compound 6. Compound 6 was significantly more potent than the opioid parent compounds.
Figure 5.

The effect of intrathecal (i.t.) administration of following compounds in CCI neuropathic pain model in rats: morphine (M, 0.35-35 nmol), hybrid 22 (0.1-1 nmol), its parent compound 33 (0.001-1 nmol), hybrid 4 (0.1-1 nmol), its parent compound 7 (0.001-1 nmol) and NK-1 antagonist 6 (0.1 - 1 nmol) on (A) allodynia as measured by von Frey test and (B) on hyperalgesia as measured by cold plate test (6-12 animals per group). Both tests were made on day 7 after injury in CCI-subjected rats. V = vehicle-treated CCI-exposed rats, black bars = toxic effect. Inter-group differences were analyzed by ANOVA Bonferroni's Multiple Comparison Test; *p<0.05, **p<0.01, ***p<0.001 vs. V-treated CCI-exposed rats.
The ability of both parent opioid peptides, the two investigated hybrids and the NK-1R antagonist parent unit of hybrid 4 (i.e. compound 6) to lower hyperalgesia was measured by the cold plate test. As shown in Figure 5B, the hybrids, but not their parent opioid compounds, decrease hyperalgesia. In comparison to morphine and at the equal dose of 1 nmol, the effect of the hybrid compounds is significantly stronger. In contrast to parent compounds 33 and 7, compound 6 is more potent in antagonizing hyperalgesia than hybrid 4 and approximately similar to 22. Similar to the observations made in the acute pain (tail-flick) test and the allodynia test (von Frey), the only hybrid lacking toxicity at the dose of 10 nmol i.t. is the hybrid structure with reduced NK1R binding and potency, compound 22 (not shown).
The ED50 values of compounds 4, 22 and morphine are presented in Table 2. In contrast to the acute pain data, a reduction of the antinociceptive potency of morphine in the neuropathic pain model (von Frey and cold pate) is observed. In contrast, in both tests the hybrid compounds perform well in neuropathic and in acute pain attenuation. The ED50 values clearly indicate that a lower dose of 22 is needed for potent effects, as compared to 4 and morphine. Interestingly, and contrary to the classical opioids, the hybrids perform better in neuropathic pain attenuation. This observation indicates the potential value of these hybrids as drug candidates for neuropathic pain therapy.
Table 2.
Calculated ED50 values for effect of hybrids 4 and 22 compared to morphine (M) in acute pain (tail-flick test) and neuropathic pain (von Frey and cold plate tests) as measured on naïve rats (acute pain) and neuropathic pain model at day 7 after CCI.
| Response (ED50) with 95% confidence limits [nmol i.t.] | |||
|---|---|---|---|
| ACUTE PAIN | NEUROPATHIC PAIN | ||
| DRUGS | Tail-flick | von Frey | Cold plate |
| M | 0.49 (0.27-0.89) | 6.25 (2.0 - 19.44) | 377 (130 - 1093) |
| 4 | 1.66 (1.11-2.51) | 1.05 (0.65 - 1.69) | 1.14 (0.74 - 1.77) |
| 22 | 2.78 (1.80-4.30) | 0.24 (0.18 - 0.32) | 0.34 (0.25 - 0.46) |
2.3.4 Development of tolerance in neuropathic pain in rats
In this study the development of tolerance was observed after chronic administration of morphine and compounds 4 and 22, which were injected i.t. repeatedly for 6 days (once daily) starting from day 7 after CCI and lasting until day 13. The effect of the investigated compounds is similar to the long-term profile of morphine as demonstrated by the time course curves (Fig. 6A and 6C). However, in spite of the same time required for the development of tolerance with morphine, hybrid compound 22 exhibits the most potent antinociceptive effect, as demonstrated by the area under curve values in both tests (Figure 6 B and 6D).
Figure 6.

The development of tolerance to antiallodynic effect measured by (A) the von Frey test and (C) antihyperalgesic effect measured by the cold plate test after repeated once daily (6 days) intrathecal (i.t.) injections of morphine (M, 3.5 nmol), hybrid compound 4 (1 nmol) and 22 (1 nmol) from 7th day until 13th day after CCI (12-16 animals per group). The results are also presented as area under curve (AUC) in (B) von Frey test and (D) cold plate test. V = vehicle-treated CCI-exposed rats. Inter-group differences were analyzed by ANOVA Bonferroni's Multiple Comparison Test; *p<0.05 vs V-treated CCI-exposed rats.
2.3.5 Cross tolerance between morphine and hybrid compounds 22 and 4 in neuropathic pain model in rat
In order to check cross tolerance between the hybrid compounds and morphine, a cross tolerance study was conducted (Fig. 7). Starting from day 7 after CCI, animals were given daily injections of the hybrid compounds or morphine. Tolerance develops within 6 days, until day 13 after CCI (Fig. 6). The animals, that had been rendered tolerant after subsequent administrations of 4 and 22, were administered with a single dose of morphine on the 13th day of the experiment. The lack of a ‘morphine effect’ after chronic administration of the tested hybrids shows cross tolerance between these compounds. Moreover, in rats which were rendered tolerant to morphine, a single dose of the hybrid 22 was also not effective.
Figure 7.

Cross tolerance of hybrid compounds 22 and 4 with morphine measured in neuropathic pain. The influence of repeated 6 days i.t. administration of morphine (M, 3.5 nmol), hybrid 22 (1 nmol) and hybrid 4 (1 nmol) on the effect of a single dose of morphine (M, 3.5 nmol), or 22 (1 nmol) was measured on day 13 after CCI in rats (6-8 animals per group). The von Frey (A, B, C) and cold plate (D, E, F) tests were conducted 30 min after a single dose of vehicle, morphine or 22 administration. The data are presented as %MPE ± S.E.M. Inter-group differences were analyzed by ANOVA Bonferroni's Multiple Comparison Test; ***P < 0.001 indicates a significant difference compared with group which received repeated vehicle and then a single dose of vehicle (V+V-treated CCI-exposed animals); $$$P < 0.001 indicates a significant difference compared with group which received repeated vehicle and then single dose of morphine (V+M; A, B, D, E) or 22 (V+22; C, F).
3 Conclusions
Several bifunctional ligands were synthesised and tested in vitro and in vivo in acute and neuropathic pain models. The conformationally constrained peptidomimetic ligands turned out to have subnanomolar binding affinity and high agonist potency at the MOR and DOR receptors, and good to moderate binding affinity and antagonist activity at the neurokinin-1 receptor. The best in vitro activity profiles were displayed by reference compound 4 and by the newly synthesized hybrid 22. When compound 22 was tested in vivo it showed an activity in the acute pain model (i.v. administration) which appeared to be similar to the previously determined activity of hybrid 4,29 and approximately 3 times higher than the one of morphine (Figure 4). Both bifunctional compounds were also tested in two in vivo models for neuropathic pain (tactile and thermal stimuli). Both ligands were significantly more potent than morphine in neuropathic rats at day 7 after chronic constriction injury (CCI) in the von Frey test (tactile allodynia). The effect was even more pronounced in the cold plate test (thermal hyperalgesia), where 22 was more potent than 4, and much more potent than morphine. Moreover, it is remarkable that the most active compound in the neuropathic pain models (22) is not the most potent NK1 antagonist (KiNK1R (4) 0.5 nM vs. KiNK1R (22) 13 nM and pA2 (4) 7.8 vs. pA2 (22) 6.4), yet it is more active than 4. This finding is however in agreement with prior work.43 When testing a combination of opioid peptides with a tachykinin antagonist we have shown that a low dose (not antinociceptive when applied individually) of tachykinin antagonist strongly potentiated opioid antinociception. Increasing the dose of the SP antagonist resulted in neurotoxic side effects rather than potentiation of antinociception.
Also, an important result in this work is that in naïve rats the antinociceptive potencies of the hybrid compounds are weaker than those of the parent compounds (e.g. 0.1 nmol of 22 = 8.9 %MPE vs 33 = 99.7 %MPE in i.t. dosed rats), but in the neuropathic pain model, the potencies of the two parent compounds, lacking the NK-1R pharmacophore, were much lower, as was also the case with morphine. This confirms the known effect of a decrease in efficacy of opioids in the treatment of neuropathic pain and points to the potential value of hybrids that act more strongly in neuropathic pain attenuation. Although the NK-1 pharmacophores in hybrid 4 and 22 could not attenuate analgesic tolerance development, the results suggest that antagonism at pronociceptive systems is a very important factor in improving the analgesic effects of opioids in neuropathic pain. The data of compound 6 (i.e. parent NK-1 pharmacophore of hybrid 4) confirmed this assumption, but also earlier studies demonstrating that NK-1R antagonists are highly effective in attenuating hyperalgesia in experimental rodent neuropathy models.44, 45 We demonstrated in the present study that the parent NK-1 antagonist pharmacophore was efficient in reversing neuropathic pain behavioral effects in CCI rats and only showed a minimal effect in the acute pain model (tail-flick) in naïve rats after intrathecal administration. Additionally, the NK-1R pharmacophore 6 was not active at all in the latter assay after i.v. administration in mice, indicating that the parent NK-1R pharmacophore alone is not able to cross the BBB, in contrast to the hybrids in which the opioid unit may serve as a BBB transport vector (cf. activity of 4 versus 6). Despite promising preclinical results on rodents, NK-1 antagonists have been shown to lack efficacy in human neuropathy.46, 47 Nonetheless, NK-1 antagonists may prove useful in combination therapies or by use of hybrid compounds that contain both opioid and NK-1 pharmacophores, as this may ensure simultaneous drug action and may improve therapy as compared to the administration of a single general analgesic.
4 Materials and methods
4.1 General
Thin-layer chromatography (TLC) was performed on glass plates precoated with silica gel 60F254 (Merck, Darmstadt, Germany) using the mentioned solvent systems. Mass Spectrometry (MS) was done on a Micromass Q-Tof Micro spectrometer with electrospray ionisation (ESI). Data collection and spectrum analysis was done with Masslynx software. Analytical RP-HPLC was performed using a Waters 717plus Autosampler, a Waters 1525 Binary HPLC Pump and a Waters 2487 Dual Absorbance Wavelength Detector (Milford, MA), with a Grace (Deerfield, IL) Vydac RP C18 column (25 cm × 4.6 mm × 5 m) using UV detection at 215 nm. The mobile phase is a mixture of water and acetonitrile and contains 0.1% TFA. The used gradient runs from 3 to 100% acetonitrile in 20 minutes at a flow rate of 1 ml/min. Preparative RP-HPLC purification was done on a Gilson (Middleton, WI) HPLC system with Gilson 322 pumps, controlled by the software package Unipoint, and a reversed phase C18 column (Discovery®BIO SUPELCO Wide Pore C18 column, 25 cm × 2.21 cm, 5 m) with the same conditions as the analytical RP-HPLC but with a flow rate of 20 ml/min. After purification, the purity of all compounds was evaluated as being more than 95% by analytical RP-HPLC. All fractions were lyophilised using a Flexy-Dry lyophilizer (FTS Systems, Warminster, PA). 1H and 13C NMR spectra were recorded at 250 and 63 MHz on a Bruker Avance 250 spectrometer or at 500 and 125 MHz on a Bruker Avance II 500 (Bruker Corp, Billerica, MA). Trimethylsilane (TMS) or residual solvent signals are used as internal standard. The solvent used is mentioned in all cases, and the abbreviations used are as follows: s (singlet), d (doublet), dd (double doublet), t (triplet) and m (multiplet).
4.2 General Peptide Synthesis
All peptides were synthesized manually by Fmoc-based solid phase peptide synthesis (SPPS) on 2-chlorotritylchloride resin (0.15 mmol scale). The first amino acid (Fmoc-Gly-OH or Fmoc-β-Ala-OH) was loaded onto the resin by use of 2 eq. Fmoc-protected amino acid with 4 eq. DIPEA in CH2Cl2 for 2 hours. The remaining chlorines were substituted by treatment of the resin with a mixture of MeOH/CH2Cl2/DIPEA (2:17:1) during 4 times 5 minutes. For normal couplings, a 3-fold excess of the Fmoc-protected amino acids (Fmoc-D-Arg(Pbf)-OH, Fmoc-D-Cit-OH) and 3-fold excess of coupling reagent (TBTU) in 0.4 NMM in DMF was used for 1.5 hours. Fmoc deprotection was carried out by treatment of the resin with 20% 4-methylpiperidine in DMF for 5 and 15 minutes. After every reaction step, the resin was washed with DMF (3× 1 min), iPrOH (3× 1 min) and CH2Cl2 (3× 1 min).
4.3 Peptide Synthesis including Aba structures
4.3.1 Reductive amination
The reductive amination was executed after Fmoc deprotection of the first amino acid. The resin was first swollen in 0.5% AcOH in TMOF/CH2Cl2 for 30 minutes and filtered. Two equivalents of Phth-ortho-formyl phenylalanine32 were dissolved in the same mixture and 4 eq. of NaBH3CN was dissolved in a minimum volume of DMF. Both solutions were added to the resin and the reaction vessel was shaken for 30 minutes. The course of the reaction was monitored after this time by the Kaiser test. When the test was positive, the reaction was left for another 30 minutes. The monitoring is repeated until the reaction remains complete, which is indicated by a light red colour of the Kaiser test due to the presence of the secondary amine.
4.3.2 Cyclisation towards the Aba building block (13)
An excess of 3 eq. TBTU is added to the resin and shaken for 3 hours.
4.3.3 Phthaloyl deprotection to 14
The resin was treated with 6 eq of hydrazine monohydrate in DMF for 18 hours
4.3.4 Boc-Dmt-OH coupling
An excess of 2 eq. Boc-Dmt-OH and 2 eq DIC/HOBt in DMF was added to the peptide-resin. The reaction vessel was shaken for 3 hours.
4.3.5 Cleavage from the solid support
The fully protected peptide was cleaved from the resin with 1% TFA in DMF for 30 minutes. The filtrate was concentrated and added to cold ether. The precipitated peptide was then dissolved in acetonitrile/H2O and lyophilised to get the compounds as a powder.
4.4 Coupling of the benzylamines to the peptide acids and final deprotection
The crude protected peptide was dissolved in a minimal amount of CH2Cl2. The solution was cooled in an ice-bath and DIPEA (2 eq.), DIC/HOBt (1.5 eq. each) are added. The mixture was stirred for 30 min at 0°C. Then the amine (1.5 eq.) was added to the solution and the reaction mixture stirred again for 30 min with cooling. The reaction was left to warm up to room temperature. When the mixture of all the reagents does not dissolve sufficiently, some drops of DMF are added and a clear solution is obtained. The reaction mixture was left to react for 15 hours. At completion, the solvent was evaporated and the residue was treated with TFA/TES/H2O (95:2.5:2.5) for 3 hours, after which the solvent was removed in vacuo.
4.5 Purification
The crude peptides were dissolved in H2O and acetonitrile was added until complete dissolving was observed. The solution was injected on a Gilson preparative RP-HPLC. Fractions were collected and combined and lyophilised. The peptides were obtained as white powders with a purity of >95% as determined by analytical HPLC. The structures were confirmed by high-resolution electrospray mass spectrometry.
4.6 Synthesis of benzylamine derivates
4.6.1 N-isobutyl benzylamine (18a)
Isobutylamine (1.5 ml, 5 eq.) is dissolved in 16 ml methanol in a two-headed round bottom flask equiped with magnetic stirrer, reflux column and septum. The reaction mixture is heated to 50°C in an oil bath. Next, benzyl chloride (347 μl, 1 eq.) is dissolved in 16 ml methanol and added drop wise (during 1 hour). The mixture is stirred overnight at 50°C and the solvent is evaporated. The residue is dissolved in 28 ml CH2Cl2 and washed with a 20% NaOH-solution (3 × 60 ml) and water (2 × 60 ml). The organic phase is dried with MgSO4, filtered en evaporated. After evaporation, a pale yellow oil is obtained. The purification is done with flash chromatography (Davisil LC60A, 40-63μm) with hexane/ethyl acetate 9:1. After evaporation of the solvent, 1 eq. of TFA is added to make a salt and the solution was lyophilised. A white to yellow powder is obtained with a yield of 43%. N-isobutylbenzylamine. Yield: 48% (504 mg, TFA salt); Formula: C11H17N; MW: 163.26 g/mol; TLC Rf = 0.26 (EtOAc/chex 1:1); HPLC: tR = 9.4 min; MS (ES+): 164 [M+H]+; 1HNMR (250 MHz, CDCl3): δ (ppm) 0.92 (6H, d, CH3, J=6.7 Hz), 1.97 (1H, m, CH), 2.64 (2H, m, CH-CH2), 3.90 (2H, s, NH-CH2-Bn), 7.30-7.42 (5H, m, arom. H). 13CNMR (63 MHz, CDCl3): δ (ppm) 20.0 (CH3), 26.0 (CH), 51.5 (CH2-Bn), 53.9 (CH-CH2-NH), 129.0 (CH arom.), 129.4 (CH arom.), 130.2 (CH arom.), 130.4 (Cq arom.).
4.6.2 N-isobutyl-3’,5’-bistrifluoromethyl benzylamine (18b)
Isobutylamine (1.5 ml, 5 eq.) is dissolved in 24 ml methanol in a two-headed round bottom flask equiped with magnetic stirrer, reflux column and septum. The reaction mixture is heated to 50°C in an oil bath. Next, 3’,5’-trifluoromethylbenzylchloride (1 eq., 528 mg) is dissolved in 20 ml methanol and added drop wise (during 1 hour). The mixture is stirred overnight at 50°C and the solvent is evaporated. The residue is dissolved in 150 ml dichloromethane and washed with a 20% NaOH-solution (3 × 90 ml) and water (2 × 90 ml). The organic phase is dried with MgSO4, filtered en evaporated. After evaporation, a light yellow oil is obtained. The purification is done with flash chromatography (Davisil LC60A, 40-63μm) with a gradient going from hexane/ethyl acetate 9:1 to 7.5:2.5. After evaporation of the solvent, TFA is added to make a salt and lyophilised. A white to yellow powder is obtained with a yield of 48%. N-isobutyl-3’,5’-bistrifluoromethyl benzylamine. Yield: 43% (354 mg, TFA salt); Formula: C13H15F6N; MW: 299.26 g/mol; Rf: = 0.65 (EtOAc/petroleum ether 1:1), TLC: Rf = 0.17 (EtOAc/hexane 1:9); HPLC: tR = 13.3 min; MS (ES+): 300 [M+H]+; 1HNMR (250 MHz, CD3OD): δ (ppm) 1.06 (3H, d, CH3, J= 6.7 Hz), 2.08 (1H, m, CH), 2.99 (2H, m, CHCH2), 4.43 (2H, s, NH-CH2-Bn), 8.11-8.20 (3H, m, arom. H). 13CNMR (63 MHz, CD3OD): δ (ppm) 20.3 (CH3), 27.3 (CH), 51.3 (CH2-Bn), 56.5 (CH-CH2-NH), 124.6 (q, CF3, 1J = 272.0 Hz), 132.1 (CH arom.), 133.4 (q, Cq arom., 2J = 33.7 Hz), 135.7 (CH arom.).
4.7 Peptide characterization
H-Dmt-D-Arg-Aba-Gly-NMe-3’,5’-(CF3)2-Bn (4)
Preparative HPLC yielded the desired compound (white powder, 34%). HPLC: tR = 14.0 min. TLC Rf 0.72 (EBAW). HRMS (ESP+) found m/z 821.3536 [M + H]+, C39H46F6N8O5 requires 821.3568.
H-Dmt-D-Arg-Aba-β-Ala-NMe-3’,5’-(CF3)2-Bn (21)
Preparative HPLC yielded the desired compound (white powder, 17%). HPLC: tR = 14.0 min. TLC Rf 0.67 (EBAW). HRMS (ESP+) found m/z 835.3708 [M + H]+, C40H48F6N8O5 requires 835.3724.
H-Dmt-D-Arg-Aba-β-Ala-NMe-Bn (22)
Preparative HPLC yielded the desired compound (white powder, 34%). HPLC: tR = 11.7 min. TLC Rf 0.64 (EBAW). HRMS (ESP+) found m/z 699.3955 [M + H]+, C38H50N8O5 requires 699.3977.
H-Dmt-D-Cit-Aba-β-Ala-NMe-3’,5’-(CF3)2-Bn (23)
Preparative HPLC yielded the desired compound (white powder, 21%). HPLC: tR = 14.6 min. TLC Rf 0.72 (EBAW). HRMS (ESP+) found m/z 836.3515 [M + H]+, C40H47F6N7O6 requires 836.3565.
H-Dmt-D-Cit-Aba-β-Ala-NMe-Bn (24)
Preparative HPLC yielded the desired compound (white powder, 34%). HPLC: tR = 12.1 min. TLC Rf 0.70 (EBAW). HRMS (ESP+) found m/z 700.3790 [M + H]+, C38H49N7O6 requires 700.3817.
H-Dmt-D-Arg-Aba-Gly-NMe-Bn (25)
Preparative HPLC yielded the desired compound (white powder, 63%). HPLC: tR = 11.7 min. TLC Rf 0.67 (EBAW). HRMS (ESP+) found m/z 685.3788 [M + H]+, C37H48N8O5 requires 685.3820.
H-Dmt-D-Arg-Aba-Gly-NH-Bn (26)
Preparative HPLC yielded the desired compound (white powder, 22%). HPLC: tR = 11.2 min. TLC Rf 0.66 (EBAW). HRMS (ESP+) found m/z 671.3694 [M + H]+, C36H46N8O5 requires 671.3664.
H-Dmt-D-Arg-Aba-Gly-N-i-Bu-3’,5’-(CF3)2-Bn (27)
Preparative HPLC yielded the desired compound (white powder, 32%). HPLC: tR = 14.3 min. TLC Rf 0.62 (EBAW). HRMS (ESP+) found m/z 863.4044 [M + H]+, C42H52F6N8O5 requires 863.4037.
H-Dmt-D-Arg-Aba-Gly-N-i-Bu-Bn (28)
Preparative HPLC yielded the desired compound (white powder, 15%). HPLC: tR = 12.5 min. TLC Rf 0.61 (EBAW). HRMS (ESP+) found m/z 727.4304 [M + H]+, C40H54N8O5 requires 727.4290.
H-Dmt-D-Arg-Aba-β-Ala-N-i-Bu-3’,5’-(CF3)2-Bn (29)
Preparative HPLC yielded the desired compound (white powder, 12%). HPLC: tR = 14.6 min. TLC Rf 0.64 (EBAW). HRMS (ESP+) found m/z 877.4240 [M + H]+, C43H54F6N8O5 requires 877.4194.
H-Dmt-D-Arg-Aba-β-Ala-N-i-Bu-Bn (30)
Preparative HPLC yielded the desired compound (white powder, 21%). HPLC: tR = 12.8 min. TLC Rf 0.57 (EBAW). HRMS (ESP+) found m/z 741.4465 [M + H]+, C41H56N8O5 requires 741.4446.
H-Dmt-D-Arg-Phe-Sar-NMe-3’,5’-(CF3)2-Bn (31)
Preparative HPLC yielded the desired compound (white powder, 24%). HPLC: tR = 13.7 min. TLC Rf 0.70 (EBAW). HRMS (ESP+) found m/z 823.3728 [M + H]+, C39H48F6N8O5 requires 823.3724.
H-Dmt-D-Arg-Phe-Sar-NMe-Bn (32)
Preparative HPLC yielded the desired compound (white powder, 31%). HPLC: tR = 11.5 min. TLC Rf 0.66 (EBAW). HRMS (ESP+) found m/z 687.3949 [M + H]+, C37H50N8O5 requires 687.3983.
H-Dmt-D-Arg-Aba-β-Ala-NH2 (33)
Preparative HPLC yielded the desired compound (white powder, 49.5%). HPLC: tR = 10.1 min. TLC Rf 0.44 (EBAW). HRMS (ESP+) found m/z 595.3394 [M+H]+, C30H43N8O5 requires 595.3351.
4.8 Functional NK1R Assay48. Cell Line and Cell Culture Conditions
The Chinese hamster ovary K1 (CHO-K1) cell line, stably expressing human NK1 receptor (hereafter referred to as CHO-NK1 cells), was transfected with an apoaequorin expression vector (pER2) using Fugene6 (Roche Applied Science). The cell line and expression vector were obtained from Euroscreen (Belgium). The CHO-NK1 cells were cultured in sterile DMEM/HAM's F12 medium (Sigma) supplemented with 10% fetal bovine serum, 100 IU/mL penicillin, 100 g/mL streptomycin, and 400 g/mL G 418 (Geneticin, Gibco) at 37 °C with 5% CO2 and were trypsinized every 3 days.
4.8.1 Aequorin Charging Protocol
Transfected cells in the midlog phase were detached by changing the growth medium for PBS buffer supplemented with 5 mM EDTA (pH 8). The cells were spun down and incubated for 4 h at a concentration of 5 × 106 cells/mL in DMEM-F12 medium without phenol red (Gibco) supplemented with 0.1% BSA (BSA medium) and 5 M coelenterazine h (Molecular Probes). After coelenterazine loading, the cells were diluted 10-fold in the same medium and incubated for an additional period of 30 min. The cells were mildly shaken during the incubation periods.
4.8.2 Aequorin Luminescence Assay
A dilution series of peptide agonist (SP was purchased from Sigma) ranging from 10−11 to 10−4 M was distributed in a white 96-well plate. For investigating antagonism, the synthetic compounds were added to these wells to obtain the desired concentrations (ranging from 10−8 to 10−4 M). One negative control sample (BSA medium only) was included in each row of the 96-well plate. The plate was loaded in a “Multimode Reader Mithras, LB940” (Berthold). The wells were screened one by one, and each measurement started at the moment of injection of 50 L of the coelenterazine-loaded cell suspension, containing 2.5 ×104 cells. Light emission was measured every second for 30 s after which 50 L of 10 nM ATP solution (positive control) was injected. Each measurement was carried out in duplicate. Light emission was recorded for an additional period of 10 s per well, and the data were presented in relative light units (RLU).
4.8.3 Data Analysis
Luminescence data (peak integration) were calculated using MikroWin 2000 software (Berthold), which was linked to the Microsoft Excel program. All statistical and curve-fitting analyses were performed using Prism 4.0 (GraphPad) software. Data are expressed in percentage (% RLU) of the maximal luminescence that was detected with 10−4 M SP (without antagonist). The competitive nature of antagonism was evaluated using the Schild plot method.40 All antagonists analyzed in this study provided linear regression plots and were considered competitive. The pA2 values were calculated using Schild's equation.49
4.9 hNK1/CHO Cell Membrane Preparation and Radioligand Binding Assay
Recombinant hNK1/CHO cells were grown to confluency in 37 °C, 95% air and 5% CO2, humidified atmosphere, in a Forma Scientific (Thermo Forma, OH) incubator in Ham's F12 medium supplemented with 10% fetal bovine serum, 100 IU/mL penicillin,100 g/mL streptomycin, and 500 g/mL geneticin. The confluent cell monolayers were then washed with Ca2+, Mg2+-deficient phosphate-buffered saline (PD buffer) and harvested in the same buffer containing 0.02% EDTA. After centrifugation at 2700 rpm for 12 min, the cells were homogenized in ice-cold 10 mM Tris-HCl and 1 mM EDTA, pH 7.4, buffer. A crude membrane fraction was collected by centrifugation at 18000 rpm for 12 min at 4 °C, the pellet was suspended in 50 mM Tris-Mg buffer, and the protein concentration of the membrane preparation was determined by using Bradford assay. Six different concentrations of the test compound were each incubated, in duplicates, with 20 g of membrane homogenate, and 0.4 nM [3H]SP (135 Ci/mmol, Perkin-Elmer, United States) in 1 mL final volume of assay buffer (50 mM Tris, pH 7.4, containing 5mMMgCl2, 50 g/mL bacitracin, 30 M bestatin, 10 M captopril, and 100 M phenylmethylsulfonylfluoride) SP at 10 M was used to define the nonspecific binding. The samples were incubated in a shaking water bath at 25 °C for 20 min. The [3H]SP concentration and the incubation time were selected based on the studies of Yamamoto et al.20 The reaction was terminated by rapid filtration through Whatman grade GF/B filter paper (Gaithersburg, MD) presoaked in 1% polyethyleneimine, washed four times each with 2 mL of cold saline, and the filter bound radioactivity was determined by liquid scintillation counting (Beckman LS5000 TD). The media and chemicals listed above were purchased from Sigma (Sigma-Aldrich, St. Louis, MO) unless otherwise stated.
4.9.1 Data Analysis
Analysis of data collected from three independent experiments performed in duplicates is done using GraphPad Prizm 4 software (GraphPad, San Diego, CA). Log IC50 values for each test compound were determined from nonlinear regression. The inhibition constant (Ki) was calculated from the antilogarithmic IC50 value by the Cheng and Prusoff equation.50, 51
4.10 Functional GPI and Mouse Vas Deferens (MVD) Assays
The GPI and MVD bioassays were carried out as described in detail elsewhere.52, 53 A dose-response curve was determined with [Leu5]enkephalin as standard for each ileum and vas preparation, and IC50 values of the compounds being tested were normalized according to a published procedure.54
4.11 Opioid Receptor Binding Assays
Opioid receptor binding studies were performed as described in detail elsewhere.52 Binding affinities for and δ opioid receptors were determined by displacing, respectively, [3H]DAMGO (Multiple Peptide Systems, San Diego, CA) and [3H]DSLET (Multiple Peptide Systems) from rat brain membrane binding sites. Incubations were performed for 2 h at 0 °C with [3H]DAMGO and [3H]DSLET at respective concentrations of 0.72 and 0.78 nM. IC50 values were determined form log-dose displacement curves, and Ki values were calculated from the IC50 values by means of the equation of Cheng and Prusoff,50 using values of 1.3 and 2.6 nM for the dissociation constants of [3H]DAMGO and [3H]DSLET, respectively.
4.12 In vivo analgesic test in naive rats and mice
The pain threshold to a thermal stimulus was assessed using tail-flick latency evoked by noxious thermal stimulation as determined with a tail-flick analgesic meter (Analgesia Meter; Ugo Basile, Comerio, Italy) as described previously. 55, 56, 57 The tail-flick was used to measure spinal nociceptive responses to thermally-induced pain using male C57BI6 mice and Wistar rats. Mice (weighing 25-28 g) or rats (250-300 g) were maintained on a normal light-dark cycle and testing occurred during the light phase. Different doses of the drug were dissolved in saline or water for injection and injected intravenously (i.v.) in mice or intrathecally (i.t.) in rats. The effect was assessed at the following time point post-injection: 5, 15, 30, 60, 120, 180 minutes. The thermal stimulus emitted by a light bulb was applied to the tail at two-thirds of its length until the animal vigorously withdrew the tail. The baseline response was approx. 2 s and the maximal exposure was set to 7 s (mice) and 9 s (rats) to avoid tissue damage. All experimental procedures used in this animal testing followed the guidelines on ethical standards for the investigation of experimental pain in animals and were approved by the Animal Research Committees of the Medical Research Centre, Polish Academy of Sciences and were approved by the local Bioethics Committee (Krakow, Poland). All in vivo activity was determined as a percentage of the maximal possible effect (%MPE) and was calculated as: %MPE = [posttreatment latency / cut-off latency] × 100.
4.13 In vivo analgesic tests in neuropathic pain model in rats
4.13.1 Animals
Male Wistar rats (300-350 g) from Charles River (Sulzfeld, Germany) were housed in cages lined with sawdust under a standard 12/12 h light/dark cycle (lights on at 08.00 h) with food and water available ad libitum. All experiments were performed according to the recommendations of the International Association for the Study of Pain (IASP)58 and the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the local Bioethics Committee (Krakow, Poland).
4.13.2 Implantation of intrathecal cannulas
The rats were chronically implanted with i.t. catheters according to Yaksh and Rudy59 under pentobarbital anaesthesia (60 mg/kg; ip) as described previously.56, 57 The rats were placed on a stereotaxic table (David Kopf), and a sterile catheter (PE 10, INTRAMEDIC, Clay Adams, Becton Dickinson and Company, Rutherford, NJ, USA.), flushed with sterile water prior to insertion, was carefully introduced through the atlanto-occipital membrane to the subarachnoid space at the rostral level of the spinal cord lumbar enlargement (L4-L6).
4.13.3 Chronic constriction injury
A chronic constriction injury (CCI) was produced according to Bennett and Xie60 as described previously.46, 47 The right sciatic nerve was exposed under sodium pentobarbital anaesthesia (60 mg/kg; ip). Four ligatures (4/0 silk) were made around the nerve distal to the sciatic notch with 1 mm spacing until a brief twitch in the respective hind limb was observed. After CCI, all animals developed allodynia and hyperalgesia.
4.13.4 Behavioural tests in neuropathic pain model
Two different behavioral tests were used to assess analgesic potency in neuropathic pain. The von Frey test evaluates tactile (mechanical) allodynia. After placing the animals in a plastic cage with the floor made from metal mesh, a mechanical stimulus is applied to the surface of the hind paw sole by use of the von Frey filament. In the cold plate test which measures thermal hyperalgesia, the rats are placed on a cold plate of 5°C and latency of reaction is assessed. In both methods, the time of reaction was measured automatically. The obtained signal is expressed as a percentage of the maximal potential effect (%MPE).
4.13.4.1 von Frey test
Mechanical allodynia in rats with CCI was measured using an automatic von Frey apparatus (Dynamic Plantar Anesthesiometer Cat. No. 37400, Ugo Basile Italy). The animals were placed in plastic cages with wire net floors. They were acclimatised to this environment for approximately 3 min prior to testing. The von Frey filament was applied to the midplantar surface of the hind paw, and the measurements were taken automatically, as described previously by Mika et al.57 The strength of the von Frey stimuli ranged from 0.5 to 26 g in the rats.
4.13.4.2 Cold plate test
To assess the threshold to a cold stimulus in rats with CCI, the Cold/Hot Plate Analgesia Meter (No. 05044, Columbus Instruments, USA) was used, as described previously by Mika et al.57 The rats were placed on the cold plate (5 °C), and the time until the shake of the hind paw was recorded. The cut-off latency for this test was 30 s. In all cases, the injured paw was the first to react.
4.13.4.3 Tolerance development
Tolerance development was carried out in neuropathic pain model. Day seven after CCI was the first day of tested drugs i.t. injections which were continued up to 13th day. In this study morphine (3.5 nmol), 4 (1 nmol) and 22 (1 nmol) were administered i.t. once a day and the behavioural tests were carried out 30 min after drug administration.
4.13.4.4 Cross tolerance
Cross tolerance measurement was conducted one day after tolerance development completion. When the animals developed tolerance to analgesic effect of 4 and 22 a single dose of morphine was injected to check if there is a cross tolerance between these compounds and morphine. We also checked whether the hybrid 22 will be effective in rats in whom tolerance to morphine was induced.
4.14 Chemicals administration
The chemicals used were obtained from the following sources: morphine hydrochloride (Polfa Kutno, Poland). All drugs were dissolved in sterile water (water for injection). All drugs were injected i.t. at a volume of 5 μl, followed by an injection of 10 μl of distilled water to flush the catheter. For acute pain, studies were carried out on day 7 after catheter implantation and 30 min after drug administration. In rats drugs were administered in a single i.t. injection at the following doses: morphine (0.35, 3.5, 35 nmol), 22 (0.1, 1, 11 nmol), 33 (0.001, 0.01, 0.1 nmol), 4 (0.1, 0.5, 1 nmol), 7 (0.001, 0.01, 0.1 nmol), and 6 (1, 10, 50 nmol). In mice drugs were administered in a single i.v. injection at the following doses: morphine 12.5 μmol/kg, 22 1.08 and 10.8 μmol/kg, 33 1.21 μmol/kg. In neuropathic pain model morphine (0.35, 3.5, 35 nmol), 22 (0.1, 0.4, 1 nmol), 33 (0.001, 0.01, 0.1, 1 nmol), 4 (0.1, 0.5, 1 nmol), 7 (0.001, 0.01, 0.1, 1 nmol) and 6 (0.1, 0.5, 1 nmol) were administered in a single i.t. injection on day 7-14 after CCI and the measurements were performed 30 min after drug administration. The control groups received vehicle (water for injection) injections according to the same schedule. The CCI to the sciatic nerve was performed 5-7 days after catheter implantation. After completion of the experiment, the animals were killed with an overdose of pentobarbital (i.p.).
4.15 Data analysis
The behavioural data (6-16 rats per group) are presented as the percentage of the maximal possible antinociceptive effect (% MPE ± SEM), which was calculated according to the following equation: % MPE=[(TL-BL)/(CUT-OFF-BL)]×100%, where BL was the baseline latency and TL was the latency obtained after drug injection. The results of the experiments were statistically evaluated using one-way analysis of variance (ANOVA). The differences between the treatment groups throughout the study were further analysed with Bonferroni post-hoc tests.
Highlights.
Hybrid opioid agonist – neurokinin 1 receptor antagonist ligands
In vitro and in vivo evaluation for treatment of acute and neuropathic pain
Hybrid 22 performs well in both acute and neuropathic pain models
Acknowledgements
Dr. Cecilia Betti is acknowledged for in-house reproduction of the total solid phase assembly methodology depicted in Scheme 2. We thank Dr. Emeric Miclet and Dr. Isabelle Correia for the NMR analysis of hybrid 4.
Funding Sources
The work of SB, DT and PWS was supported by a collaboration convention between the Ministère du Développement Economique, de l’Innovation et de l’Exportation du Québec and the Research Foundation – Flanders (FWO Vlaanderen) (PSR-SIIRI-417). The research of PWS was also supported by grants CIHR (MOP-89716), and the NIH (DA-004443). Supported by Institute of Pharmacology statutory founds (WM) and grant from National Science Center Poland NCN2012/06/A/NZ4/00028 for (BP and JM).
Abbreviations
- %MPE
percent of maximal possible effect
- Aba
4-amino-2-benzazepinone scaffold
- CCI
chronic constriction injury
- CNS
central nervous system
- D-Cit
D-citrulline
- DIC
N,N’-dicyclohexylcarbodiimide
- DIPEA
diisopropylethylamine
- DMF
N,N-dimethylformamide
- DML
designed multiple ligand
- Dmt
2’,6’-dimethyl-L-tyrosine
- DOR
δ opioid receptor
- GPI
guinea pig ileum
- HOBt
1-hydroxybenzotriazole
- i-Bu
isobutyl; intraperitoneal
- i.p.
intraperitoneal
- i.t.
intrathecal
- i.v.
intravenous
- MOR
μ opioid receptor
- MPE
maximal possible effect
- MVD
mouse vas deferens
- NK1
neurokinin-1
- NMM
N-methylmorpholine
- Pbf
2,2,4,6,7-pentamethyldihydrobenzofuran-5-sulfonyl
- RP-HPLC
reverse phase high pressure liquid chromatography
- SP
substance P
- SPPS
solid phase peptide synthesis
- TBTU
O-(Benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium tetrafluoroborate
- TES
triethylsilane
- TMOF
trimethyl orthoformate
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
5.1 Author Contributions
KG, OVDP and IVDE were in charge of ligand synthesis. AK, EV, JL, FP, JVD, JV contributed by providing the NK1 binding and antagonism data. NNC, CL and PWS provided in vitro opioid data. PK, AL and AWL performed the tail-flick test in mice and JM, ER, WM and BP performed acute and neuropathic pain study in rats. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
References
- 1.Pasternak GW. Opioids and their receptors: Are we there yet? Neuropharmacol. 2014;76:198–203. doi: 10.1016/j.neuropharm.2013.03.039. doi: 10.1016/j.neuropharm.2013.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Labuz D, Machelska H. Stronger antinociceptive efficacy of opioids at the injured nerve trunk than at its peripheral terminals in neuropathic pain. J. Pharmacol. Exp. Ther. 2013;346:535–544. doi: 10.1124/jpet.113.205344. doi: 10.1124/jpet.113.205344. [DOI] [PubMed] [Google Scholar]
- 3.Ossipov MH, Lopez Y, Nichols ML, Bian D, Porreca F. The loss of antinociceptive efficacy of spinal morphine in rats with nerve ligation injury is prevented by reducing spinal afferent drive. Neurosci. Lett. 1995;199:87–90. doi: 10.1016/0304-3940(95)12022-v. doi: 10.1016/0304-3940(95)12022-V. [DOI] [PubMed] [Google Scholar]
- 4.Evans CJ, Keith DE, Jr., Morrison H, Magendzo K, Edwards RH. Cloning of a delta opioid receptor by functional expression. Science. 1992;258:1952–1955. doi: 10.1126/science.1335167. doi: 10.1126/science.1335167. [DOI] [PubMed] [Google Scholar]
- 5.Kieffer BL, Befort K, Gaveriaux-Ruff C, Hirth CG. The delta-opioid receptor: isolation of a cDNA by expression cloning and pharmacological characterization. Proc. Natl. Acad. Sci. U.S.A. 1992;89:12048–12052. doi: 10.1073/pnas.89.24.12048. doi: 10.1073/pnas.89.24.12048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen Y, Mestek A, Liu J, Yu L. Molecular cloning of a rat kappa opioid receptor reveals sequence similarities to the mu and delta opioid receptors. Biochem. J. 1993;295:625–628. doi: 10.1042/bj2950625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Costigan M, Scholz J, Woolf CJ. Neuropathic pain: a maladaptive response of the nervous system to damage. Annu. Rev. Neurosci. 2009;32:1–32. doi: 10.1146/annurev.neuro.051508.135531. doi: doi:10.1146/annurev.neuro.051508.135531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Payne B, Norfleet MA. Chronic pain and the family: a review. Pain. 1986;26:1–22. doi: 10.1016/0304-3959(86)90169-7. [DOI] [PubMed] [Google Scholar]
- 9.Chaplan SR, Malmberg AB, Yaksh TL. Efficacy of spinal NMDA receptor antagonism in formalin hyperalgesia and nerve injury evoked allodynia in the rat. J. Pharmacol. Exp. Ther. 1997;280:829–838. [PubMed] [Google Scholar]
- 10.King T, Gardell L, Wang R, Vardanyan A, Ossipov M, Malan T, Vanderah T, Hunt S, Hruby V, Lai J, Porreca F. Role of NK-1 neurotransmission in opioid-induced hyperalgesia. Pain. 2005;116:276–288. doi: 10.1016/j.pain.2005.04.014. doi: 10.1016/j.pain.2005.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yamamoto T, Nair P, Jacobsen N, Kulkarni V, Davis P, Ma S-W, Navratilova E, Yamamura H, Vanderah T, Porreca F, Lai J, Hruby V. Biological and conformational evaluation of bifunctional compounds for opioid receptor agonists and neurokinin 1 receptor antagonists possessing two penicillamines. J. Med. Chem. 2010;53:5491–5501. doi: 10.1021/jm100157m. doi: 10.1021/jm100157m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Costantino L, Barlocco D. Designed multiple ligands: basic research vs clinical outcomes. Curr. Med. Chem. 2012;19:3353–3387. doi: 10.2174/092986712801215883. doi: 10.2174/092986712801215883. [DOI] [PubMed] [Google Scholar]
- 13.Morphy R, Rankovic Z. Lead Generation Approaches in Drug Discovery. John Wiley & Sons, Inc; Hoboken: 2010. Design of multitarget ligands. pp. 141–164. [Google Scholar]
- 14.Gentilucci L. New trends in the development of opioid peptide analogues as advanced remedies for pain relief. Curr. Top. Med. Chem. 2004;4:19–38. doi: 10.2174/1568026043451663. doi: 10.2174/1568026043451663. [DOI] [PubMed] [Google Scholar]
- 15.Lipkowski AW. Cooperative reinforcement of opioid pharmacophores. Pol. J. Pharmacol. Pharm. 1987;39:585–596. [PubMed] [Google Scholar]
- 16.Lipkowski AW, Carr DB, Misicka A, Misterek K. Biological activities of a peptide containing both casomorphin-like and substance P antagonist structural characteristics. In: Brantl V, Teschemacher H, editors. BCasomorphins and Related Peptides: Recent Developments. VCH; Weinheim: 1994. pp. 113–118. [Google Scholar]
- 17.Bonney IM, Foran SE, Marchand JE, Lipkowski AW, Carr DB. Spinal antinociceptive effects of AA501, a novel chimeric peptide with opioid receptor agonist and tachykinin receptor antagonist moieties. Eur. J. Pharmacol. 2004;488:91–99. doi: 10.1016/j.ejphar.2004.02.023. doi: 10.1016/j.ejphar.2004.02.023. [DOI] [PubMed] [Google Scholar]
- 18.Foran SE, Carr DB, Lipkowski AW, Maszczynska I, Marchand JE, Misicka A, Beinborn M, Kopin AS, Kream RM. A substance P-opioid chimeric peptide as a unique nontolerance-forming analgesic. Proc. Natl. Acad. Sci. U.S.A. 2000;97:7621–7626. doi: 10.1073/pnas.130181897. doi: 10.1073/pnas.130181897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamamoto T, Nair P, Vagner J, Largent-Milnes T, Davis P, Ma S.-w., Navratilova E, Moye S, Tumati S, Lai J, Yamamura HI, Vanderah TW, Porreca F, Hruby VJ. A structure– activity relationship study and combinatorial synthetic approach of C-terminal modified bifunctional peptides that are δ/μ opioid receptor agonists and neurokinin 1 receptor antagonists. J. Med. Chem. 2008;51:1369–1376. doi: 10.1021/jm070332f. doi: 10.1021/jm070332f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamamoto T, Nair P, Davis P, Ma S.-w., Navratilova E, Moye S, Tumati S, Lai J, Vanderah TW, Yamamura HI, Porreca F, Hruby VJ. Design, synthesis, and biological evaluation of novel bifunctional C-terminal-modified peptides for δ/μ opioid receptor agonists and neurokinin-1 receptor antagonists. J. Med. Chem. 2007;50:2779–2786. doi: 10.1021/jm061369n. doi: 10.1021/jm061369n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takaya T. Discovery of neurokinin antagonists. Pure Appl. Chem. 1996;68:875–880. [Google Scholar]
- 22.Longmore J, Hill RG, Hargreaves RJ. Neurokinin-receptor antagonists: pharmacological tools and therapeutic drugs. Can. J. Physiol. Pharm. 1997;75:612–621. doi: 10.1139/cjpp-75-6-612. doi: 10.1139/y97-069. [DOI] [PubMed] [Google Scholar]
- 23.Gonzalez MI, Field MJ, Hughes J, Singh L. Evaluation of selective NK1 receptor antagonist CI-1021 in animal models of inflammatory and neuropathic pain. J. Pharmacol. Exp. Ther. 2000;294:444–450. [PubMed] [Google Scholar]
- 24.Ballet S, Feytens D, Buysse K, Chung NN, Lemieux C, Tumati S, Keresztes A, Van Duppen J, Lai J, Varga E, Porreca F, Schiller PW, Vanden Broeck J, Tourwé D. Design of novel neurokinin 1 receptor antagonists based on conformationally constrained aromatic amino acids and discovery of a potent chimeric opioid agonist-neurokinin 1 receptor antagonist. J. Med. Chem. 2011;54:2467–2476. doi: 10.1021/jm1016285. doi: 10.1021/jm1016285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schiller P, Nguyen T, Berezowska I, Dupuis S, Weltrowska G, Chung N, Lemieux C. Synthesis and in vitro opioid activity profiles of DALDA analogues. Eur. J. Med. Chem. 2000;35:895–901. doi: 10.1016/s0223-5234(00)01171-5. doi: 10.1016/S0223-5234(00)01171-5. [DOI] [PubMed] [Google Scholar]
- 26.Van den Eynde I, Laus G, Schiller PW, Kosson P, Chung NN, Lipkowski AW, Tourwé D. A new structural motif for μ-opioid antagonists. J. Med. Chem. 2005;48:3644–3648. doi: 10.1021/jm0491795. doi: 10.1021/jm0491795. [DOI] [PubMed] [Google Scholar]
- 27.Ballet S, Feytens D, Wachter RD, Vlaeminck MD, Marczak ED, Salvadori S, Graaf C. d., Rognan D, Negri L, Lattanzi R, Lazarus LH, Tourwé D, Balboni G. Conformationally constrained opioid ligands: The Dmt-Aba and Dmt-Aia versus Dmt-Tic scaffold. Bioorg. Med. Chem. Lett. 2009;19:433–437. doi: 10.1016/j.bmcl.2008.11.051. doi: 10.1016/j.bmcl.2008.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ballet S, De Wachter R, Van Rompaey K, Tömböly C, Feytens D, Töth G, Quartara L, Cucchi P, Meini S, Tourwé D. Bradykinin analogs containing the 4-amino-2-benzazepin-3-one scaffold at the C-terminus. J. Pept. Sci. 2007;13:164–170. doi: 10.1002/psc.827. doi: 10.1002/psc.827. [DOI] [PubMed] [Google Scholar]
- 29.Guillemyn K, Kleczkowska P, Novoa A, Vandormael B, Van den Eynde I, Kosson P, Asim M, Schiller P, Spetea M, Lipkowski A, Tourwe D, Ballet S. In vivo antinociception of potent mu opioid agonist tetrapeptide analogues and comparison with a compact opioid agonist -neurokinin 1 receptor antagonist chimera. Mol. Brain. 2012;5:4. doi: 10.1186/1756-6606-5-4. doi: 10.1186/1756-6606-5-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Largent-Milnes T, Yamamoto T, Nair P, Moulton J, Hruby V, Lai J, Porreca F, Vanderah T. Spinal or systemic TY005, a peptidic opioid agonist/neurokinin 1 antagonist, attenuates pain with reduced tolerance. Br. J. Pharmacol. 2010;161:986–1001. doi: 10.1111/j.1476-5381.2010.00824.x. doi: 10.1111/j.1476-5381.2010.00824.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ballet S, Frycia A, Piron J, Chung N, Schiller P, Kosson P, Lipkowski A, Tourwe D. Synthesis and biological evaluation of constrained analogues of the opioid peptide H-Tyr-D-Ala-Phe-Gly-NH2 using 4-amino-2-benzazepin-3-one scaffold. J. Pept.. Res. 2005;66:222–230. doi: 10.1111/j.1399-3011.2005.00291.x. doi: 10.1111/j.1399-3011.2005.00291.x. [DOI] [PubMed] [Google Scholar]
- 32.Van Rompaey K, Van den Eynde I, De Kimpe N, Tourwé D. A versatile synthesis of 2-substituted 4-amino-1,2,4,5-tetrahydro-2-benzazepine-3-ones. Tetrahedron. 2003;59:4421–4432. doi: 10.1016/S0040-4020(03)00583-0. [Google Scholar]
- 33.Mondini S, Dall Avo M, Guerrato A. Process for the preparation of 3,5-bistrifluoromethyl-N-Methylbenzylamine. 2007 WO2007107818 A2.
- 34.Sasaki Y, Ambo A, Suzuki K. Studies on analgesic oligopeptides. VII. Solid phase synthesis and biological properties of Tyr-D-Arg-Phe-ß-Ala-NH2 and its fluorinated aromatic amino acid derivatives. Chem. Pharm. Bull. 1991;39:2316–2318. doi: 10.1248/cpb.39.2316. doi: 10.1248/cpb.39.2316. [DOI] [PubMed] [Google Scholar]
- 35.Ananthan S. Opioid ligands with mixed mu/delta opioid receptor interactions: an emerging approach to novel analgesics. AAPS J. 2006;8:E118–E125. doi: 10.1208/aapsj080114. doi: 10.1208/aapsj080114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abdelhamid E, Sultana M, Portoghese P, Takemori A. Selective blockage of delta opioid receptors prevents the development of morphine tolerance and dependence in mice. J. Pharmacol. Exp. Ther. 1991;258:299–303. [PubMed] [Google Scholar]
- 37.Dietis N, Guerrini R, Calo G, Salvadori S, Rowbotham D, Lambert D. Simultaneous targeting of multiple opioid receptors: a strategy to improve side-effect profile. Br. J. Anaesth. 2009;103:38–49. doi: 10.1093/bja/aep129. doi: 10.1093/bja/aep129. [DOI] [PubMed] [Google Scholar]
- 38.Takeuchi Y, Shands EFB, Beusen DD, Marshall GR. Derivation of a three-dimensional pharmacophore model of substance P antagonists bound to the neurokinin-1 receptor. J. Med. Chem. 1998;41:3609–3623. doi: 10.1021/jm9700171. doi: 10.1021/jm9700171. [DOI] [PubMed] [Google Scholar]
- 39.Novoa A, Van Dorpe S, Wynendaele E, Spetea M, Bracke N, Stalmans S, Betti C, Chung NN, Lemieux C, Zuegg J, Cooper MA, Tourwé D, De Spiegeleer B, Schiller PW, Ballet S. Variation of the net charge, lipophilicity, and side chain flexibility in Dmt1-DALDA: effect on opioid activity and biodistribution. J. Med. Chem. 2012;55:9549–9561. doi: 10.1021/jm3008079. doi: 10.1021/jm3008079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schild H. pA2, a new scale for the measurement of drug antagonism. Br. J. Pharmacol. 1947;2:189–206. doi: 10.1111/j.1476-5381.1947.tb00336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fundytus ME, Schiller PW, Shapiro M, Weltrowska G, Coderre TJ. Attenuation of morphine tolerance and dependence with the highly selective δ-opioid receptor antagonist TIPP[ψ]. Eur. J. Pharmacol. 1995;286:105–108. doi: 10.1016/0014-2999(95)00554-x. doi: 10.1016/0014-2999(95)00554-X. [DOI] [PubMed] [Google Scholar]
- 42.Schiller PW, Fundytus ME, Merovitz L, Weltrowska G, Nguyen TMD, Lemieux C, Chung NN, Coderre TJ. The opioid μ agonist/δ antagonist DIPP-NH2[Ψ] produces a potent analgesic effect, no physical dependence, and less tolerance than morphine in rats. J. Med. Chem. 1999;42:3520–3526. doi: 10.1021/jm980724+. doi: 10.1021/jm980724+. [DOI] [PubMed] [Google Scholar]
- 43.Misterek K, Maszczynska I, Dorociak A, Gumulka SW, Carr DB, Dzyfelbein SK, Lipkowski AW. Spinal co-administration of peptide substance P antagonist potentiates antinociceptive effect of opioid peptide. Life Sci. 1994;54:939–944. doi: 10.1016/0024-3205(94)00494-3. [DOI] [PubMed] [Google Scholar]
- 44.Cahill CM, Coderre TJ. Attenuation of hyperalgesia in a rat model of neuropathic pain after intrathecal pre- or post-treatment with a neurokinin-1 antagonist. Pain. 2002;95:277–285. doi: 10.1016/S0304-3959(01)00410-9. doi: 10.1016/S0304-3959(01)00410-9. [DOI] [PubMed] [Google Scholar]
- 45.Cumberbatch MJ, Carlson E, Wyatt A, Boyce S, Hill RG, Rupniak NM. Reversal of behavioural and electrophysiological correlates of experimental peripheral neuropathy by the NK1 receptor antagonist GR205171 in rats. Neuropharmacol. 1998;37:1535–1543. doi: 10.1016/s0028-3908(98)00125-7. doi: 10.1016/S0028-3908(98)00125-7. [DOI] [PubMed] [Google Scholar]
- 46.Goldstein DJ, Wang O, Gitter BD, Iyengar S. Dose-response study of the analgesic effect of lanepitant in patients with painful diabetic neuropathy. Clin. Neuropharmacol. 2001;24:16–22. doi: 10.1097/00002826-200101000-00004. doi: 10.1097/00002826-200101000-00004. [DOI] [PubMed] [Google Scholar]
- 47.Sindrup SH, Graf A, Sfikas N. The NK1-receptor antagonist TKA731 in painful diabetic neuropathy: a randomised, controlled trial. Eur. J. Pain. 2006;10:567–571. doi: 10.1016/j.ejpain.2005.08.001. doi: 10.1016/j.ejpain.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 48.Janecka A, Poels J, Fichna J, Studzian K, Vanden Broeck J. Comparison of antagonist activity of spantide family at human neurokinin receptors measured by aequorin luminescence-based functional calcium assay. Regul. Peptides. 2005;131:23–28. doi: 10.1016/j.regpep.2005.05.006. doi: 10.1016/j.regpep.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 49.Arunlakshana O, Schild HO, Jenkinson DH. Some quantitative uses of drug antagonists. Br. J. Pharmacol. 1997;120:148–150. doi: 10.1111/j.1476-5381.1997.tb06793.x. doi: 10.1111/j.1476-5381.1997.tb06792.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yung-Chi C, Prusoff WH. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharm. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 51.Paton WDM. The action of morphine and related substances on contraction and on acetylcholine output of coaxially stimulated guinea-pig ileum. Br. J. Pharmacol. Chemother. 1957;12:119–127. doi: 10.1111/j.1476-5381.1957.tb01373.x. doi: 10.1111/j.1476-5381.1957.tb01373.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.DiMaio J, Nguyen T, Lemieux C, Schiller P. Synthesis amd pharmacological characterization in vitro of cyclic enkephalin analogues: effect of the conformational constraints on opioid receptor selectivity. J. Med. Chem. 1982;25:1432–1438. doi: 10.1021/jm00354a008. doi: 10.1021/jm00354a008. [DOI] [PubMed] [Google Scholar]
- 53.Waterfield AA, Leslie FM, Lord JAH, Ling N, Kosterlitz HW. Opioid activities of fragments of β-endorphin and of ites leucine65-analogue. Comparison of the binding properties of methionine- and leucine-enkephalin. Eur. J. Pharmacol. 1979;58:11–18. doi: 10.1016/0014-2999(79)90334-0. doi: 10.1016/0014-2999(79)90334-0. [DOI] [PubMed] [Google Scholar]
- 54.Mohamadi F, Richards NGJ, Guida WC, Liskamp R, Lipton M, Caufield C, Chang G, Hendrickson T, Still WC. Macromodel - an integrated software system for modeling organic and bioorganic molecules using molecular mechanics. J. Comput. Chem. 1990;11:440–467. doi: 10.1002/jcc.540110405. [Google Scholar]
- 55.D'Amour F, Smith D. A method for determining loss of pain sensation. J. Pharmacol. Exp. Ther. 1941;72:74–79. [Google Scholar]
- 56.Makuch W, Mika J, Rojewska E, Zychowska M, Przewlocka B. Effects of selective and non-selective inhibitors of nitric oxide synthase on morphine- and endomorphin-1-induced analgesia in acute and neuropathic pain in rats. Neuropharmacol. 2013;75:445–457. doi: 10.1016/j.neuropharm.2013.08.031. doi: 10.1016/j.neuropharm.2013.08.031. [DOI] [PubMed] [Google Scholar]
- 57.Mika J, Osikowicz M, Makuch W, Przewlocka B. Minocycline and pentoxifylline attenuate allodynia and hyperalgesia and potentiate the effects of morphine in rat and mouse models of neuropathic pain. Eur. J. Pharmacol. 2007;560:142–149. doi: 10.1016/j.ejphar.2007.01.013. doi: 10.1016/j.ejphar.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 58.Zimmermann M. Ethical guidelines for pain research in humans. International Association for the Study of Pain. 1983 [Google Scholar]
- 59.Yaksh TL, Rudy TA. Chronic catheterization of the spinal subarachnoid space. Physiol. Behav. 1976;17:1031–1036. doi: 10.1016/0031-9384(76)90029-9. doi: 10.1016/0031-9384(76)90029-9. [DOI] [PubMed] [Google Scholar]
- 60.Bennett GJ, Xie YK. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain. 1988;33:87–107. doi: 10.1016/0304-3959(88)90209-6. doi: 10.1016/0304-3959(88)90209-6. [DOI] [PubMed] [Google Scholar]