

Abstract
Currently, there is a renewed interest in common dietaries and plant-based traditional medicines for the prevention and treatment of cancer. In the search for potential anticancer agents from natural sources, ursolic acid (UA), a pentacyclic triterpenoid widely found in various medicinal herbs and fruits, exhibits powerful biological effects including its attractive anticancer activity against various types of cancer cells. However, the limited solubility, rapid metabolism and poor bioavailability of UA restricted its further clinical applications. In the past decade, with substantial progress toward the development of new chemical entities for the treatment of cancer, numerous UA derivatives have been designed and prepared to overcome its disadvantages. Despite extensive effort, discovery of effective UA derivatives has so far met with only limited success. This review summarizes the current status of the structural diversity and evolution in medicinal chemistry of UA analogues and provides a detailed discussion of future direction for further research in the chemical modifications of UA.
Keywords: Natural products, ursolic acid derivatives, anticancer agents, drug discovery, chemical biology
INTRODUCTION
Throughout the ages, natural products have served and continue to serve as an unparalleled source to develop novel effective therapeutic agents for the treatment of a wide spectrum of diseases [1,2]. In the plant kingdom, triterpenoids represent a broad family of widespread natural compounds containing more than 20,000 members [3,4]. Among them, pentacyclic triterpenes have emerged as a unique group of triterpenoid natural products with distinct biological properties as demonstrated by promising results in preclinical and clinical studies [5,6]. Ursolic acid (UA, 3β-hydroxy-12-urs-12-ene-28-oic acid, Figure 1) as a hydroxy pentacyclic triterpene acid, is a constituent of certain medicinal herbs and the main component of wax-like protective coatings of various fruits including apples, pears, olives, prunes, cranberries and figs [7]. UA possesses considerable pharmacological effects including hepatoprotective [8,9], immunomodulatory [10], anti-inflammatory [11,12], antidiabetic [13], antibacterial [14,15], antiviral [16,17], antiulcer [18] and anticancer activities [19]. Recently, UA has been attracting a rising attention for its multifunctional anticancer activities [19,20]. Moreover, as an integral part of the human diet, UA is implicated in protection and prevention against human cancers [21,22].
Figure 1.
Structure of ursolic acid (UA, 3β-hydroxy-12-urs-12-ene-28-oic acid, 1).
Accumulating mechanistic studies indicate that the anticancer effect of UA is attributed to its ability to induce cancer cell apoptosis, prevent tumorigenesis, and inhibit cancer cell proliferation. UA triggers autophagy, cell cycle arrest, and apoptosis in different cancer cell lines through several signaling pathways [19], including NF-κB [23], STAT3 [24,25], and TRAIL [26]. For instance, UA has demonstrated to block the NF-κB pathway through suppression of p65 phosphorylation, leading to down-regulation of the expression of several downstream oncogenes such as Bcl2 and Bcl-XL [23]. Nevertheless, the precise molecular mechanisms of UA involved in apoptosis induction and proliferation inhibition in cancer remains to be elucidated [19]. The associated signaling pathways may be substantially different in various cancer cell lines, and partial signaling pathways may be regulated successively or simultaneously and synergized to contribute to UA treatment [27]. Further mechanistic elucidations and pathway signaling investigations are imperative to identify novel targets and provide useful insights into the molecular basis for the beneficial effects produced by UA and the relevant drug discovery of new chemical entities.
Despite its significant profiles of safety and efficacy in cancer treatment, its limited solubility, rapid metabolism and poor bioavailability of UA resulted in low therapeutic potential and restricted its further clinical applications [28]. Due to its naturally abundant supply from common sources and inexpensive availability, UA could be used as an attractive starting point for further structural modifications [29]. This article seeks to review the evolution in medicinal chemistry of UA derivatives to improve anticancer activity for preclinical development. Based on structural properties, we clustered most of reported UA analogues into two categories: 1) modifications on the positions of C-3/C-28, C-11, C-17 and C-28; and 2) modifications on C-2/C-3 positions and ring A.
MODIFICATONS ON THE POSITIONS OF C-3/C-28, C-11, C-17 AND C-28
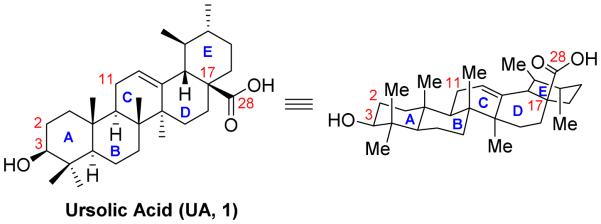
Over the past decade, numerous attempts have been made to develop more potent UA derivatives. Daneshtalab and co-workers [30] isolated UA and five triterpenes from apple peels. They found that UA was present in the largest quantity (76% in crude triterpenes, 17 g from 27 kg of fresh apples). After that, a series of UA analogues were prepared with the modifications on the C-3, C-11, C-17 and C-28 positions [30]. As shown in Figure 2, 3β-amino derivative 2exhibited 20-fold more potent than 3α-formin cytotoxicity on all tested cancer cell lines, indicating that the configuration at C-3 is a critical factor on the antiproliferative activity. Similarly, UA analogues with β-oriented hydrogen-bond forming groups at C-3 displayed more potent inhibition than their α-counterparts. All these results reinforced the notion of the important role of the configurations. Compound 3 bearing a hydroxymethyl group at C-17 displayed no activity, suggesting that the carbonyl group at C-17 is essential for the potency. In addition, introduction of an additional oxo moiety at C-11 position (compound 4) is intolerable. Notably, introduction of amino alkyl groups at C-28 position (compound 5) resulted in significant improvement of cytotoxicity. Despite the lack of detailed mechanistic and pharmacological studies, the general structure-activity relationship (SAR) described in this work served as the basis for further structural modifications on UA.
Figure 2.
Daneshtalab’s work on UA derivatives.
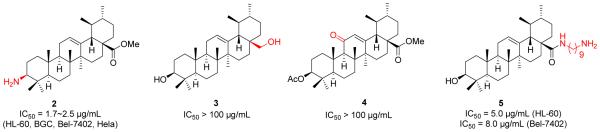
Following upon the initial findings discussed above, several research groups reported their drug discovery efforts and several interesting UA analogues have been discovered to show good in vitro anticancer activity. Meng et al. [31] prepared a series of UA derivatives (Figure 3) in order to investigate which positions are important for the activity. In consistent with the previous results, they also found that introduction of a substituted acetyl group at C-3 hydroxyl group together with amino alkyl moiety at C-28 could significantly enhance the anticancer activity [31]. The UA derivatives with acetyl group at C-3 exhibited more potent antiproliferative effects than unsubstituted analogues. To further explore the benefit of the modifications at the C-3 and C-28 positions, they designed, synthesized and biologically evaluated another series of UA derivatives featured with UA-amino acid conjugates or related amino alcohol moieties [32]. The preliminary SAR studies also suggested that both the 3-O-acetyl moiety and a 28-amido group appeared to be essential for improving anticancer activity. Meanwhile, UA derivatives with free hydroxyl moiety at the C-28 amide branched side chain displayed similar inhibitory activity to those compounds without the hydroxyl group on the amide side chain, indicating that a hydroxyl group at the C-28 amide branched side chain has little influence on the antiproliferative activity. Taken together, the general SAR from their work suggested that 1) acetylation of C-3 position and formation of an amide by coupling with an amino alcohol acetate or amino acid methyl ester at C-28 position resulted in UA derivatives showing enhanced anticancer activity; 2) the C-3 free hydroxyl may decrease anticancer effects, while the free hydroxyl at C-28 amide side chain may retain the activity; and 3) too many branched alkyl side chains at C-28 amide chain could decrease anticancer effects. The preliminary antiproliferative mechanisms of these UA–amino alcohol conjugates were also explored. It was found that compounds 7 and 8 inhibited cell growth via the induction of apoptosis and cell cycle arrest at the S phase. Further comparison studies of UA derivatives by acetylated and butylated at the C-3 position demonstrated that acetylated compounds exhibited higher antiproliferative effects[33].
Figure 3.
Meng’s work on UA derivatives.
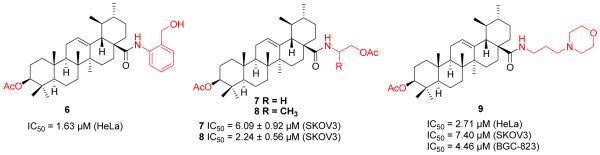
Shao et al. made a similar effort along these lines and synthesized another series of UA derivatives (Figure 4) by modifying at C-3 and C-28 positions [34]. Slight enhancement of anticancer activity was achieved by the introduction of an acetyl group at the C-3 position together with that of alkylamino and/or piperidine moieties at the C-28 position. Their SAR studies also revealed that 1) compounds with disubstituted amide displayed higher potency than those monosubstituted ones; and 2) the piperazine derivatives showed relatively lower potency than the piperidine analogues. Compound 10 induced cell apoptosis accompanied by cell cycle progression arrest at the S phase and the activation of caspase-3. However, this compound only demonstrated a slight reduction in tumor growth in hepatocellular carcinoma xenografts in nude mice even at a high dose of 150 mg/kg.
Figure 4.
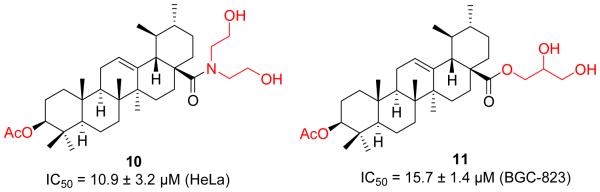
Shao’s and Guo’s work on UA derivatives.
The increasing studies support that the ester functionality at C-3 and acid moiety at C-17 are essential, while a hydrogen donor group at either C-3 position and/or C-28 position is favorable for the anticancer activity. Based upon available SAR information, Guo and his co-workers identified a novel derivative [3β-acetoxy-urs-12-en-28-oyl]-1-monoglyceride (11, Figure 4), which displayed significant anticancer effects without apparent cytotoxicity to human normal gastric cell line GES-1 [35]. Compound 11 was found to significantly induce apoptosis of BGC-823 cells by regulating the mitochondrial signaling pathway. Further extensive mechanism studies revealed that the activity of caspase-3 was up-regulated, while the expressions of Bcl-2 and Survivin were down-regulated. Intriguingly, compound 11 at 6.0 mg/kg exerted more efficacious antitumor effects than Taxol without apparent toxicity in nude mice bearing gastric tumor xenografts. The significant in vivo efficacy of this molecule may be attributed to its enhanced membrane permeability and favorable prodrug-like pharmaceutical properties.
Lin et al. also prepared and evaluated twenty-three new UA derivatives [36]. Several compounds exhibited significant anticancer effects against NTUB1 cells. Compound 12 (Figure 5) with the succinyl moiety at C-3 position or compound 13 with an isopropyl ester at C-28 position displayed significantly improved antiproliferative activity likely owing to their enhanced cell permeability of ester prodrug forms. Interestingly, these compounds mediated through generation of reactive oxygen species (ROS), inducing inhibition of tubulin polymerization, cell cycle arrest at G2/M and G1, and apoptosis. Chen et al. prepared a series of furoxan-based nitric oxide (NO)-donating UA analogues and the most potent NO-drug hybrid 14 exhibited significantly improved anticancer activity against HepG2 cells, providing a promising direction for further investigation [37].
Figure 5.
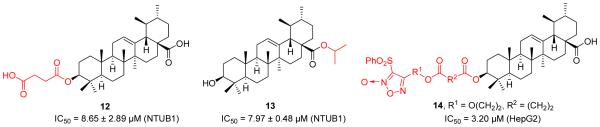
Lin’s and Chen’s work on UA derivatives.
Accumulating evidence in drug discovery and the previous observations from the structural modification on UA revealed that the incorporation of a piperazine moiety might provide unexpected biological enhancement [34,38]. In order to search for UA derivatives with higher anticancer activities, Yang et al. designed and synthesized a series of UA derivatives with an additional acyl piperazine moiety on C-28 position (Figure 6) [39]. They found that retaining the polar group at C-3 while incorporating a certain acyl piperazine motif at C-28 could significantly enhance anticancer activity against breast cancer and gastric cancer cell lines. Compound 15 was found to induce a higher cancer cell apoptosis ratio.
Figure 6.
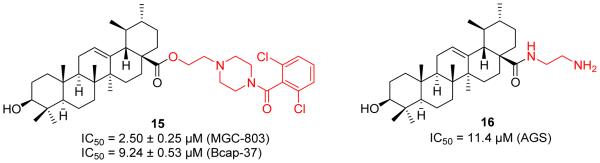
Yang’s and Guo’s work on UA derivatives.
Despite numerous efforts on the chemical modifications of UA at the C-3 and C-28 position, no systematical SAR studies were pursued to explore the role of electronic properties. To this end, Guo et al. synthesized a series of novel UA derivatives with distinct electronic property at these two interesting positions [40]. They divided these newly designed UA analogues into two groups, namely negatively charged group and positively charged group. The extensive SAR investigation revealed that the positively charged group exhibited more potent cytotoxicity. The improvement of the lipophilicity appeared to strengthen the anticancer activity. Among them, the representative compound 16 (Figure 6) not only displayed potent anticancer activity and the ability to induce the apoptosis, but also exhibited reasonable oil/water partition coefficient and enhanced aqueous solubility.
Inspired by such advantages mentioned above, Bhat et al. designed and synthesized a focused library of UA-triazolyl based congeners at C-28 position by utilizing click chemistry protocol in order to develop more effective anticancer agents (Figure 7) [41]. Intriguingly, most of these newly designed UA analogues demonstrated remarkable antiproliferative effects against all the tested cancer cell lines while exhibiting low toxicity to normal cells. The extensive SAR studies revealed that UA-triazolyl derivatives (17-19) with o-bromo, o-chloro or o-methoxy substitution at aromatic ring possessed highly potent anticancer activity with IC50 values less than 0.1 μM against MCF-7 (breast) and THP-1 (leukemia) cancer cell lines. It is worthy to note that the scaffold of this series has a 3-oxo functionality in the A ring, indicating that appropriate modifications on the A ring appear to be tolerable for anticancer activity enhancement. Further constructions with diverse functionality at the A-ring may open new doors to provide unique chemical entities with a promising pharmacological profile.
Figure 7.
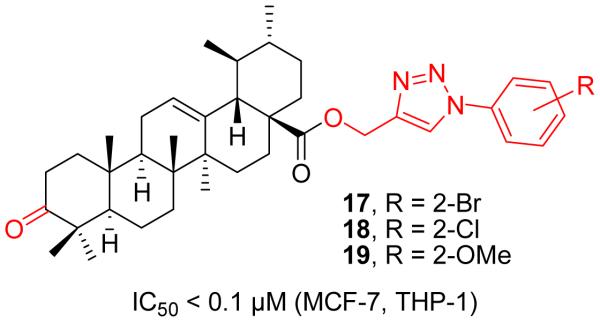
Bhat’s work on UA derivatives.
MODIFICATIONS ON THE C-2 POSITION
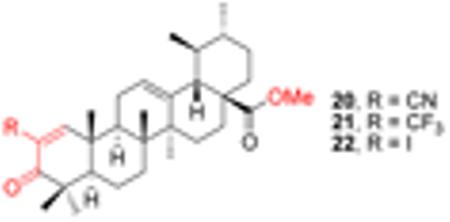
As discussed above, considerable structural modifications on UA performed so far are primarily on the hydroxyl group at C-3 position and on the carboxylic acid group at C-28 position. Esters and amides are the most common semisynthetic analogues and some derivatives exhibit significantly improved anticancer effects. In view of the above discussion, it is noteworthy to mention that the major advancement in triterpenoid research over the past decade was the synthesis of 2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid (CDDO, Figure 8) [42]. This oleanolic acid (OA) derivative, and its C-28 methyl ester (CDDO-Me) as well as its C-28 imidazole (CDDO-Im) showed significantly improved anti-inflammatory and antitumor activities. The enhanced effects are attributed to the 1-en-3-one and 9-en-12-one functionalities in the ring A and ring C, respectively. Inspired by the success of CDDO analogues in human clinical trials for cancer therapy, similar effort has been made on UA modifications since both UA and OA share a similar triterpenoid template. Interestingly, derivatives of UA substituted with cyano or trifluoromethyl group at the C-2 position in ring A containing the 1-en-3-one functionality also exhibited higher IC50 values than UA methyl ester (Table 1) [43]. On the other hand, C-2 cyano or trifluoromethyl analogues (compounds 20 and 21) also demonstrated higher potency than C-2 iodo-substituted compound (22).
Figure 8.
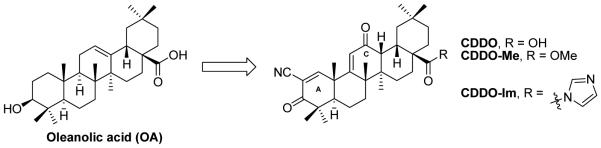
The chemical structures of oleanolic acid (OA) and its derivatives (CDDO, CDDO-Me and CDDO-Im).
Table 1.
The IC50 μM) of UA derivatives (Safe’s work)
| ||||
|---|---|---|---|---|
|
| ||||
| Compd | 235JB-V | KU7 | Panc-1 | Panc-28 |
| CDDO-Me | 0.03 | 0.12 | 0.27 | 0.29 |
| UA methyl ester | 6.13 | 8.95 | 11.75 | 10.58 |
| 20 | 0.17 | 0.30 | 0.53 | 0.97 |
| 21 | 0.17 | 0.47 | 0.65 | 1.13 |
| 22 | 4.90 | 6.02 | 6.91 | 13.49 |
To modify the A ring of UA, Lin et al. developed a promising compound 23 (Figure 9) with seco-structures [36]. This compound was prepared from UA by oxidation, lactonization and ring-opening reaction. Despite only two-fold enhancement in cytotoxicity against NTUB1 cells, this kind of scaffold provides a new skeleton with potential for further explorations.
Figure 9.
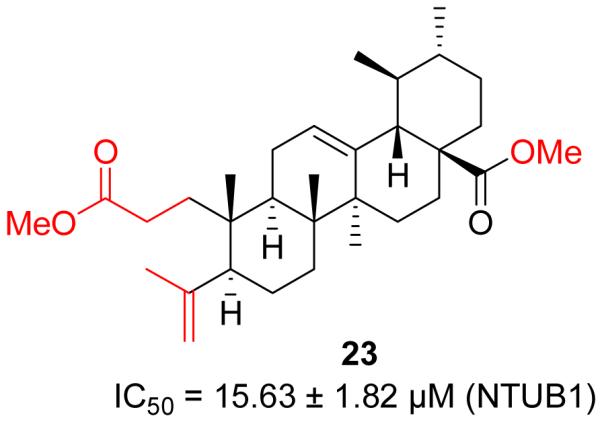
Lin’s work on UA derivatives.
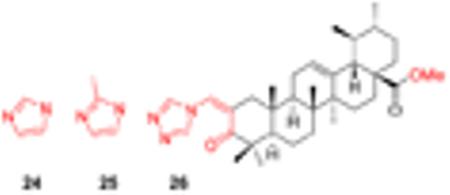
Jing et al. prepared a series of new N-acylimidazoles and N-alkylimidazoles of UA derivatives with an α,β-unsaturated ketone and found that most of these compounds exhibited significantly enhanced anticancer activity [44]. These novel UA derivatives (24-26, Table 2) displayed improved anticancer activity with IC50 values ranging from 1.9 to 7.3 μ M. The introduction of the N-alkyl heterocyclic motif in the ring A conjugated with an α,β-unsaturated ketone provides a better Michael acceptor and allows potential interaction with certain target proteins. Compound 26 arrests cell cycle in G1 phase and induces apoptosis in AsPC-1 cells by up-regulating p53, p21waf1 and NOXA. UA derivatives with an α,β-unsaturated ketone conjugated with an heterocyclic moiety present a new scaffold for further drug discovery in cancer treatment.
Table 2.
Cytotoxicity (μM) of UA heterocyclic derivatives (Jing’s work)
| |||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Compd | AsPC-1 | PANC-1 | MIA PaCa-2 | HepG2 | MCF7 | A549 | PC-3 |
| UA | 12.6 | 14.9 | 10.4 | 15.0 | 12.3 | 11.4 | 20.8 |
| 24 | 5.8 | 5.1 | 7.3 | 2.0 | 5.3 | 5.6 | 6.8 |
| 25 | 2.1 | 5.6 | 5.6 | 4.0 | 2.2 | 5.3 | 3.5 |
| 26 | 1.9 | 3.5 | 4.0 | 3.2 | 2.3 | 4.9 | 3.0 |
CONCLUSIONS AND PERSPECTIVES
Natural products continue to serve as an invaluable source of molecular diversity for drug discovery. In the past two decades, UA, a widespread occurrence throughout the plant kingdom, has attracted considerable interest due to its biological potential as an antiviral, anti-inflammatory, antioxidant, or anticancer agent. Its most prominent function is anticancer effect because accumulating studies have demonstrated that UA is capable of regulating cell cycle, autophagy, and apoptosis in various malignant cancer cells, including breast, leukemia, lung, endometrial, and melanoma cancers [45]. In a phase I clinical and pharmacokinetic study to evaluate its pharmacokinetics, tolerance and safety, UA was proven to be extremely safe [46]. Despite its great profile of safety and efficacy, UA has not been adopted widely into clinical practice due to limited aqueous solubility, short plasma half-life, and low bioavailability. Therefore, a variety of UA derivatives with diversified modifications have been designed and synthesized in the search of more effective and orally bioavailable UA-based chemotherapeutic agents for cancer therapy.
This review summarizes the current progress in medicinal chemistry of UA analogues (Table 3) as potential anticancer agents, which may serve as an important reference for further research on structural optimization, SAR and molecular biology studies of UA and other triterpenoid acids. The accumulated results on the structural variability and their anticancer effects against various malignant cancer cells have established some meaningful SAR which is depicted in Figure 10. By critical examination of all reported UA analogues with modifications either at the positions of C-3/C-28, C-11, C-17 and C-28 or at C-2/C-3 positions of ring A, most of UA derivatives appeared to be active in vitro against various cancer cell lines but unfortunately, did not exhibit satisfactory potency and efficacy in vivo. These findings warrant more comprehensive SAR study and further development of UA derivatives with enhanced druggability through alternative drug design strategies [47]. To advance UA and its analogues as a viable cancer therapy, there remain several issues and new directions on which we can take action. (1) From the above literature survey, modifications on the ring A may significantly increase efficacy of UA. Compared to the modifications at C3, C17 and C28 positions, which have already received tremendous attention in the past decade, ring A/C-based diverse construction is relatively less explored and may be a more effective strategy to yield highly potent UA derivatives [48]. More extensive synthetic efforts and efficient methodologies are needed due to the challenges in structural diversifications on the ring A/C. To this end, our previous work on the concise synthesis of novel oridonin A-ring structural diversification with the success to yield superior diterpenoids may provide some useful and efficient synthetic strategies [49-52]. (2) To increase the efficiency in terms of structural modifications, and especially the efficacy in animal models, the synthesis and extensive pharmacological evaluation of a larger library of UA derivatives are imperative to establish useful criteria for the further drug design and development. However, in most of the reported studies, only individual compounds of each series were tested in vitro against limited cancer cell lines [53] or in vivo in selected cancer models. Therefore, a more extensive investigation of UA derivatives using comprehensive cancer panel assays may provide a more detailed SAR. (3) Aqueous insolubility is one of the key restricting factors limiting clinical potential of many chemotherapeutic drugs. Although some of the UA analogues demonstrated potent effects in vitro, they are insoluble in aqueous media with poor efficacy in vivo [54]. More attention should be paid to enhance the aqueous solubility of UA analogues during structural modifications of UA by incorporation of hydrophilic fragments or moieties. One of the potential approaches to access better UA derivatives with improved aqueous solubility may take advantage of our published strategies by generating O-alkylamino-tethered derivatives, capable of having more favorable physicochemical properties [55-57]. Beneficial effects of making glucosides widely used in pharmaceutical industry may be another viable approach for the enhancement of solubility [58,59]. (4) Drug delivery systems are well known to improve aqueous solubility and bioavailability of many clinically used anticancer drugs [60,61]. Such delivery strategies may also prove useful for some potent UA derivatives to further improve their hydrophilicity and oral bioavailability. Formulations containing liposomes, cyclodextrin complexes, micelles, colloids and nanoparticles may be designed and generated to overcome such difficulties in clinical application of UA derivatives [62,63]. In addition, nanoparticulate drug delivery systems may provide unique approaches for cancer therapy [64,65]. The intelligently designed nanoparticles loaded with UA derivatives may enable targeting tumor cells passively or actively by enhanced permeation and retention (EPR) effect or by utilizing molecular targeting moieties. (5) Prodrug strategy has been developed to improve bioavailability of many conventional drugs [66]. By intelligent design, UA may be modified to be inactive in the gastrointestinal tract to avoid first-pass effect but active in tumor tissues after the targeted release of parent compound from the prodrug forms. (6) In most of the aforementioned literatures, the mechanisms of UA derivatives with respect to the activity enhancement in cancers are not in depth [67,68]. Many researchers have revealed that UA can inhibit cell proliferation, cause cell cycle arrest, induce apoptosis, as well as induce intracellular ROS formation in different cancer cell lines through several signaling pathways. However, the detailed action modes and potential novel targets of the UA derivatives remain to be unraveled. Once the exact targets of UA are identified, structure-based and fragment-based drug design can be applied in the refined structural modifications [69]. (7) Some natural products have been successfully used as potent adjunct drug to conventional chemotherapy although they are lack of efficacy as a single agent at low concentrations [70]. Combinations of UA derivatives with other conventional chemotherapeutic agents may offer the possibility of not only lowering their effective doses and thereby reducing unwanted adverse effects, but also acquiring unexpected attractive pharmacological properties as anticancer agents.
Table 3.
Summary of in vitro and in vivo studies with representative UA derivatives against various cancer cell lines.
| Compd | In vitro anti-proliferation (IC50) | In vivo study | Potential molecular mechanisms | Ref. |
|---|---|---|---|---|
| 2 | 2.0 μg/mL(HL-60) 2.5 μg/mL(BGC) 1.7 μg/mL (Bel-7402) 2.4 μg/mL (Hela) |
-a | - | [30] |
| 5 | 5.0 μg/mL(HL-60) 30.0 μg/mL(BGC) 8.0 μg/mL (MDA-MB-435) |
- | - | [30] |
| 6 | 1.63 μM (Hela) | - | - | [31] |
| 8 | 2.24 μM (SKOV3) | - | Induce apoptosis; block cell cycle in S phase (Hela cells) |
[32] |
| 9 | 2.71 μM (Hela) 7.40 μM (SKOV3) 4.46 μM (BGC-823) |
- | - | [33] |
| 10 | 20.25 μM (HepG2) 15.52 μM (BGC-823) 13.24 μM (SH-SY5Y) 10.87 μM (Hela) 38.06 μM (HELF) |
H22 xenografts in Kunming mice (50, 100, 150 mg/kg, i.p.) |
Induce apoptosis; arrest cell cycle progression at the S phase in HepG2 cells; up-regulation of caspase-3 |
[34] |
| 11 | 18.43 μM (HT-29) 27.46 μM (HepG2) 15.66 μM (BGC-823) |
BGC-823 cells xenograft nude mice (6.0 mg/kg, 30 mg/kg, i.v.) |
Induce apoptosis via the mitochondrial signaling pathway; up-regulation of caspase-3, down-regulation of Survivin and Bcl-2 (BGC-823 cells) |
[35] |
| 13 | 7.97 μM (NTUB1) | - | G2/M and G1 cell cycle arrest; enhancement of ROS; inhibition of tubulin polymerization (NTUB1 cells) |
[36] |
| 14 | 3.20 μM (HepG2) | - | - | [37] |
| 15 | 2.50 μM (MGC-803) 9.24 μM (Bcap-37) |
- | Induce cell apoptosis in MGC-803 cells |
[39] |
| 16 | 11.4 μM (AGC) 21.3 μM (HepG2) 14.3 μM (HT-29) < 10 μM (PC-3) |
- | Induce cell apoptosis in AGC cells |
[40] |
| 18 | <0.1μM (A-549, MCF-7, THP-1) 0.3 μM (HCT-116) |
- | - | [41] |
| 20 | 0.17 μM (235JB-V) 0.30 μM (KU7) 0.53 μM (Panc-1) 0.97 μM (Panc-28) |
- | - | [43] |
| 23 | 15.63 μM (NTUB1) | - | G2/M and G1 cell cycle arrest; enhancement of ROS; inhibition of tubulin polymerization (NTUB1 cells) |
[36] |
| 26 | 1.9 μM (AsPC-1) 3.5 μM (PANC-1) 4.0 μM (MIA PaCa-2) 3.2 μM (HepG2) 2.3 μM (MCF7) 4.9 μM (A549) 3.0 μM (PC-3) |
- | Arrest cell cycle in G1 phase and induce apoptosis in AsPc-1 cells, up-regulation of P53, P21waf1 and NOXA proteins |
[44] |
Not available.
Figure 10.
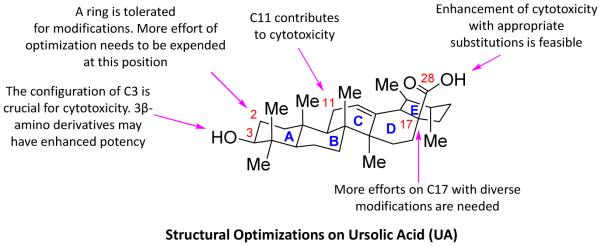
Graphical depiction of the general SAR for cell cytotoxicity based on available biological results of UA derivatives.
Highlights.
Recent developments on medicinal aspects of UA analogues were discussed.
Significant bioactive compounds as anticancer agents were documented.
Relevant SAR studies and potential mechanisms were summarized.
Future direction for further UA-based drug discovery was elaborated.
ACKNOWLEDGEMENTS
This work was supported by the National Natural Science Foundation of China (No. 81402781), the Technology Development Foundation of Fuzhou University, Fujian Natural Science Foundation of China (No. 2012J05155), grants from The State Oceanic Administration of China (No. 201205022), grants P50 CA097007, P30 DA028821, R21 MH093844 from the National Institutes of Health, Cancer Prevention and Research Institute of Texas (CPRIT) awards, R. A. Welch Foundation Chemistry and Biology Collaborative Grant from the Gulf Coast Consortia (GCC), John Sealy Memorial Endowment Fund, Institute for Translational Sciences (ITS), and the Center for Addiction Research (CAR) at UTMB.
ABBREVIATIONS USED
- UA
ursolic acid
- NF-κB
nuclear factor-κB
- STAT3
signal transducer and activator of transcription 3
- TRAIL
tumor necrosis factor (TNF)-related apoptosis inducing ligand
- Bcl2
B-cell lymphoma-2
- Bcl-XL
B-cell lymphoma-extra large
- SAR
structure-activity relationship
- ROS
reactive oxygen species
- NO
nitric oxide
- OA
oleanolic acid
- CDDO
2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid
- NOXA
phorbol-12-myristate-13-acetate-induced protein 1
- EPR
enhanced permeation and retention
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTEREST
The author(s) confirm that this article content has no conflicts of interest.
REFERENCES
- [1].Koehn FE, Carter GT. The evolving role of natural products in drug discovery. Nat Rev Drug Discov. 2005;4:206–220. doi: 10.1038/nrd1657. [DOI] [PubMed] [Google Scholar]
- [2].Cragg GM, Grothaus PG, Newman DJ. Impact of natural products on developing new anti-cancer agents. Chem Rev. 2009;109:3012–3043. doi: 10.1021/cr900019j. [DOI] [PubMed] [Google Scholar]
- [3].Dzubak P, Hajduch M, Vydra D, Hustova A, Kvasnica M, Biedermann D, Markova L, Urban M, Sarek J. Pharmacological activities of natural triterpenoids and their therapeutic implications. Nat Prod Rep. 2006;23:394–411. doi: 10.1039/b515312n. [DOI] [PubMed] [Google Scholar]
- [4].Hill RA, Connolly JD. Triterpenoids. Nat Prod Rep. 2013;30:1028–1065. doi: 10.1039/c3np70032a. [DOI] [PubMed] [Google Scholar]
- [5].Sheng H, Sun H. Synthesis, biology and clinical significance of pentacyclic triterpenes: a multi-target approach to prevention and treatment of metabolic and vascular diseases. Nat Prod Rep. 2011;28:543–593. doi: 10.1039/c0np00059k. [DOI] [PubMed] [Google Scholar]
- [6].Shanmugam MK, Nguyen AH, Kumar AP, Tan BK, Sethi G. Targeted inhibition of tumor proliferation, survival, and metastasis by pentacyclic triterpenoids: potential role in prevention and therapy of cancer. Cancer Lett. 2012;320:158–170. doi: 10.1016/j.canlet.2012.02.037. [DOI] [PubMed] [Google Scholar]
- [7].He X, Liu RH. Triterpenoids isolated from apple peels have potent antiproliferative activity and may be partially responsible for apple's anticancer activity. J Agric Food Chem. 2007;55:4366–4370. doi: 10.1021/jf063563o. [DOI] [PubMed] [Google Scholar]
- [8].Saravanan R, Viswanathan P, Pugalendi KV. Protective effect of ursolic acid on ethanol-mediated experimental liver damage in rats. Life Sci. 2006;78:713–718. doi: 10.1016/j.lfs.2005.05.060. [DOI] [PubMed] [Google Scholar]
- [9].Jin YR, Jin JL, Li CH, Piao XX, Jin NG. Ursolic acid enhances mouse liver regeneration after partial hepatectomy. Pharm Biol. 2012;50:523–528. doi: 10.3109/13880209.2011.611143. [DOI] [PubMed] [Google Scholar]
- [10].Saaby L, Jager AK, Moesby L, Hansen EW, Christensen SB. Isolation of immunomodulatory triterpene acids from a standardized rose hip powder (Rosa canina L.) Phytother Res. 2011;25:195–201. doi: 10.1002/ptr.3241. [DOI] [PubMed] [Google Scholar]
- [11].Ali MS, Ibrahim SA, Jalil S, Choudhary MI. Ursolic acid: a potent inhibitor of superoxides produced in the cellular system. Phytother Res. 2007;21:558–561. doi: 10.1002/ptr.2108. [DOI] [PubMed] [Google Scholar]
- [12].Zhang P, Cheng Y, Duan RD. Ursolic acid inhibits acid sphingomyelinase in intestinal cells. Phytother Res. 2013;27:173–178. doi: 10.1002/ptr.4709. [DOI] [PubMed] [Google Scholar]
- [13].Perez Gutierrez RM, Vargas Solis R, Garcia Baez E, Gallardo Navarro Y. Hypoglycemic activity of constituents from Astianthus viminalis in normal and streptozotocin-induced diabetic mice. J Nat Med. 2009;63:393–401. doi: 10.1007/s11418-009-0343-7. [DOI] [PubMed] [Google Scholar]
- [14].do Nascimento PG, Lemos TL, Bizerra AM, Arriaga AM, Ferreira DA, Santiago GM, Braz-Filho R, Costa JG. Antibacterial and antioxidant activities of ursolic acid and derivatives. Molecules. 2013;19:1317–1327. doi: 10.3390/molecules19011317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kurek A, Nadkowska P, Pliszka S, Wolska KI. Modulation of antibiotic resistance in bacterial pathogens by oleanolic acid and ursolic acid. Phytomedicine. 2012;19:515–519. doi: 10.1016/j.phymed.2011.12.009. [DOI] [PubMed] [Google Scholar]
- [16].Wu HY, Chang CI, Lin BW, Yu FL, Lin PY, Hsu JL, Yen CH, Liao MH, Shih WL. Suppression of hepatitis B virus x protein-mediated tumorigenic effects by ursolic Acid. J Agric Food Chem. 2011;59:1713–1722. doi: 10.1021/jf1045624. [DOI] [PubMed] [Google Scholar]
- [17].Kong L, Li S, Liao Q, Zhang Y, Sun R, Zhu X, Zhang Q, Wang J, Wu X, Fang X, Zhu Y. Oleanolic acid and ursolic acid: novel hepatitis C virus antivirals that inhibit NS5B activity. Antiviral Res. 2013;98:44–53. doi: 10.1016/j.antiviral.2013.02.003. [DOI] [PubMed] [Google Scholar]
- [18].Ishikawa T, Donatini Rdos S, Diaz IE, Yoshida M, Bacchi EM, Kato ET. Evaluation of gastroprotective activity of Plinia edulis (Vell.) Sobral (Myrtaceae) leaves in rats. J Ethnopharmacol. 2008;118:527–529. doi: 10.1016/j.jep.2008.05.007. [DOI] [PubMed] [Google Scholar]
- [19].Shanmugam MK, Dai X, Kumar AP, Tan BK, Sethi G, Bishayee A. Ursolic acid in cancer prevention and treatment: molecular targets, pharmacokinetics and clinical studies. Biochem Pharmacol. 2013;85:1579–1587. doi: 10.1016/j.bcp.2013.03.006. [DOI] [PubMed] [Google Scholar]
- [20].Kuttan G, Pratheeshkumar P, Manu KA, Kuttan R. Inhibition of tumor progression by naturally occurring terpenoids. Pharm Biol. 2011;49:995–1007. doi: 10.3109/13880209.2011.559476. [DOI] [PubMed] [Google Scholar]
- [21].Aggarwal BB, Shishodia S. Molecular targets of dietary agents for prevention and therapy of cancer. Biochem Pharmacol. 2006;71:1397–1421. doi: 10.1016/j.bcp.2006.02.009. [DOI] [PubMed] [Google Scholar]
- [22].Ovesna Z, Vachalkova A, Horvathova K, Tothova D. Pentacyclic triterpenoic acids: new chemoprotective compounds. Minireview. Neoplasma. 2004;51:327–333. [PubMed] [Google Scholar]
- [23].Shishodia S, Majumdar S, Banerjee S, Aggarwal BB. Ursolic acid inhibits nuclear factor-kappaB activation induced by carcinogenic agents through suppression of IkappaBalpha kinase and p65 phosphorylation: correlation with down-regulation of cyclooxygenase 2, matrix metalloproteinase 9, and cyclin D1. Cancer Res. 2003;63:4375–4383. [PubMed] [Google Scholar]
- [24].Wang W, Zhao C, Jou D, Lu J, Zhang C, Lin L, Lin J. Ursolic acid inhibits the growth of colon cancer-initiating cells by targeting STAT3. Anticancer Res. 2013;33:4279–4284. [PubMed] [Google Scholar]
- [25].Pathak AK, Bhutani M, Nair AS, Ahn KS, Chakraborty A, Kadara H, Guha S, Sethi G, Aggarwal BB. Ursolic acid inhibits STAT3 activation pathway leading to suppression of proliferation and chemosensitization of human multiple myeloma cells. Mol Cancer Res. 2007;5:943–955. doi: 10.1158/1541-7786.MCR-06-0348. [DOI] [PubMed] [Google Scholar]
- [26].Prasad S, Yadav VR, Kannappan R, Aggarwal BB. Ursolic acid, a pentacyclin triterpene, potentiates TRAIL-induced apoptosis through p53-independent up-regulation of death receptors: evidence for the role of reactive oxygen species and JNK. J Biol Chem. 2011;286:5546–5557. doi: 10.1074/jbc.M110.183699. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [27].Wang J, Liu L, Qiu H, Zhang X, Guo W, Chen W, Tian Y, Fu L, Shi D, Cheng J, Huang W, Deng W. Ursolic acid simultaneously targets multiple signaling pathways to suppress proliferation and induce apoptosis in colon cancer cells. PLoS One. 2013;8:e63872. doi: 10.1371/journal.pone.0063872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Yin MC, Lin MC, Mong MC, Lin CY. Bioavailability, distribution, and antioxidative effects of selected triterpenes in mice. J Agric Food Chem. 2012;60:7697–7701. doi: 10.1021/jf302529x. [DOI] [PubMed] [Google Scholar]
- [29].Sultana N. Clinically useful anticancer, antitumor, and antiwrinkle agent, ursolic acid and related derivatives as medicinally important natural product. J Enzyme Inhib Med Chem. 2011;26:616–642. doi: 10.3109/14756366.2010.546793. [DOI] [PubMed] [Google Scholar]
- [30].Ma CM, Cai SQ, Cui JR, Wang RQ, Tu PF, Hattori M, Daneshtalab M. The cytotoxic activity of ursolic acid derivatives. Eur J Med Chem. 2005;40:582–589. doi: 10.1016/j.ejmech.2005.01.001. [DOI] [PubMed] [Google Scholar]
- [31].Liu D, Meng Y-Q, Zhao J, Chen L-G. Synthesis and anti-tumor activity of novel amide derivatives of ursolic acid. Chem Res Chinese U. 2008;24:42–46. [Google Scholar]
- [32].Meng YQ, Liu D, Cai LL, Chen H, Cao B, Wang YZ. The synthesis of ursolic acid derivatives with cytotoxic activity and the investigation of their preliminary mechanism of action. Bioorg Med Chem. 2009;17:848–854. doi: 10.1016/j.bmc.2008.11.036. [DOI] [PubMed] [Google Scholar]
- [33].Meng Y, Song Y, Yan Z, Xia Y. Synthesis and in vitro cytotoxicity of novel ursolic acid derivatives. Molecules. 2010;15:4033–4040. doi: 10.3390/molecules15064033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Shao JW, Dai YC, Xue JP, Wang JC, Lin FP, Guo YH. In vitro and in vivo anticancer activity evaluation of ursolic acid derivatives. Eur J Med Chem. 2011;46:2652–2661. doi: 10.1016/j.ejmech.2011.03.050. [DOI] [PubMed] [Google Scholar]
- [35].Bai KK, Chen FL, Yu Z, Zheng YQ, Li YN, Guo YH. Synthesis of [3beta-acetoxy-urs-12-en-28-oyl]-1-monoglyceride and investigation on its anti tumor effects against BGC-823. Bioorg Med Chem. 2011;19:4043–4050. doi: 10.1016/j.bmc.2011.05.017. [DOI] [PubMed] [Google Scholar]
- [36].Tu HY, Huang AM, Wei BL, Gan KH, Hour TC, Yang SC, Pu YS, Lin CN. Ursolic acid derivatives induce cell cycle arrest and apoptosis in NTUB1 cells associated with reactive oxygen species. Bioorg Med Chem. 2009;17:7265–7274. doi: 10.1016/j.bmc.2009.08.046. [DOI] [PubMed] [Google Scholar]
- [37].Chen L, Qiu W, Tang J, Wang ZF, He SY. Synthesis and bioactivity of novel nitric oxide-releasing ursolic acid derivatives. Chin Chem Lett. 2011;22:413–416. [Google Scholar]
- [38].Patel RV, Park SW. An evolving role of piperazine moieties in drug design and discovery. Mini Rev Med Chem. 2013;13:1579–1601. doi: 10.2174/13895575113139990073. [DOI] [PubMed] [Google Scholar]
- [39].Liu MC, Yang SJ, Jin LH, Hu DY, Xue W, Song BA, Yang S. Synthesis and cytotoxicity of novel ursolic acid derivatives containing an acyl piperazine moiety. Eur J Med Chem. 2012;58:128–135. doi: 10.1016/j.ejmech.2012.08.048. [DOI] [PubMed] [Google Scholar]
- [40].Bai KK, Yu Z, Chen FL, Li F, Li WY, Guo YH. Synthesis and evaluation of ursolic acid derivatives as potent cytotoxic agents. Bioorg Med Chem Lett. 2012;22:2488–2493. doi: 10.1016/j.bmcl.2012.02.009. [DOI] [PubMed] [Google Scholar]
- [41].Rashid S, Dar BA, Majeed R, Hamid A, Bhat BA. Synthesis and biological evaluation of ursolic acid-triazolyl derivatives as potential anti-cancer agents. Eur J Med Chem. 2013;66:238–245. doi: 10.1016/j.ejmech.2013.05.029. [DOI] [PubMed] [Google Scholar]
- [42].Shanmugam MK, Dai X, Kumar AP, Tan BK, Sethi G, Bishayee A. Oleanolic acid and its synthetic derivatives for the prevention and therapy of cancer: Preclinical and clinical evidence. Cancer Lett. 2014;346:206–216. doi: 10.1016/j.canlet.2014.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Chadalapaka G, Jutooru I, McAlees A, Stefanac T, Safe S. Structure-dependent inhibition of bladder and pancreatic cancer cell growth by 2-substituted glycyrrhetinic and ursolic acid derivatives. Bioorg Med Chem Lett. 2008;18:2633–2639. doi: 10.1016/j.bmcl.2008.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Leal AS, Wang R, Salvador JA, Jing Y. Synthesis of novel ursolic acid heterocyclic derivatives with improved abilities of antiproliferation and induction of p53, p21waf1 and NOXA in pancreatic cancer cells. Bioorg Med Chem. 2012;20:5774–5786. doi: 10.1016/j.bmc.2012.08.010. [DOI] [PubMed] [Google Scholar]
- [45].Zang LL, Wu BN, Lin Y, Wang J, Fu L, Tang ZY. Research progress of ursolic acid's anti-tumor actions. Chin J Integr Med. 2014;20:72–79. doi: 10.1007/s11655-013-1541-4. [DOI] [PubMed] [Google Scholar]
- [46].Zhu Z, Qian Z, Yan Z, Zhao C, Wang H, Ying G. A phase I pharmacokinetic study of ursolic acid nanoliposomes in healthy volunteers and patients with advanced solid tumors. Int J Nanomedicine. 2013;8:129–136. doi: 10.2147/IJN.S38271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Kalani K, Yadav DK, Singh A, Khan F, Godbole MM, Srivastava SK. QSAR guided semi-synthesis and in-vitro validation of anticancer activity in ursolic acid derivatives. Curr Top Med Chem. 2014;14:1005–1013. doi: 10.2174/1568026614666140324121606. [DOI] [PubMed] [Google Scholar]
- [48].Siewert B, Wiemann J, Kowitsch A, Csuk R. The chemical and biological potential of C ring modified triterpenoids. Eur J Med Chem. 2014;72:84–101. doi: 10.1016/j.ejmech.2013.11.025. [DOI] [PubMed] [Google Scholar]
- [49].Ding C, Zhang Y, Chen H, Yang Z, Wild C, Ye N, Ester CD, Xiong A, White MA, Shen Q, Zhou J. Oridonin ring A-based diverse constructions of enone functionality: identification of novel dienone analogues effective for highly aggressive breast cancer by inducing apoptosis. J Med Chem. 2013;56:8814–8825. doi: 10.1021/jm401248x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Ding C, Zhang Y, Chen H, Yang Z, Wild C, Chu L, Liu H, Shen Q, Zhou J. Novel nitrogen-enriched oridonin analogues with thiazole-fused A-ring: protecting group-free synthesis, enhanced anticancer profile, and improved aqueous solubility. J Med Chem. 2013;56:5048–5058. doi: 10.1021/jm400367n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Ding C, Zhang Y, Chen H, Wild C, Wang T, White MA, Shen Q, Zhou J. Overcoming synthetic challenges of oridonin A-ring structural diversification: regio- and stereoselective installation of azides and 1,2,3-triazoles at the C-1, C-2, or C-3 position. Org Lett. 2013;15:3718–3721. doi: 10.1021/ol4015865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Ding C, Wang L, Chen H, Wild C, Ye N, Ding Y, Wang T, White MA, Shen Q, Zhou J. ent-Kaurane-based regio- and stereoselective inverse electron demand hetero-Diels-Alder reactions: synthesis of dihydropyran-fused diterpenoids. Org Biomol Chem. 2014;12:8442–8452. doi: 10.1039/c4ob01040j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Mazumder K, Tanaka K, Fukase K. Cytotoxic activity of ursolic acid derivatives obtained by isolation and oxidative derivatization. Molecules. 2013;18:8929–8944. doi: 10.3390/molecules18088929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Chen Q, Luo S, Zhang Y, Chen Z. Development of a liquid chromatography-mass spectrometry method for the determination of ursolic acid in rat plasma and tissue: application to the pharmacokinetic and tissue distribution study. Anal Bioanal Chem. 2011;399:2877–2884. doi: 10.1007/s00216-011-4651-x. [DOI] [PubMed] [Google Scholar]
- [55].Chen H, Yang Z, Ding C, Chu L, Zhang Y, Terry K, Liu H, Shen Q, Zhou J. Discovery of -Alkylamino Tethered Niclosamide Derivatives as Potent and Orally Bioavailable Anticancer Agents. ACS Med Chem Lett. 2013;4:180–185. doi: 10.1021/ml3003082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Chen H, Mrazek AA, Wang X, Ding C, Ding Y, Porro LJ, Liu H, Chao C, Hellmich MR, Zhou J. Design, synthesis, and characterization of novel apigenin analogues that suppress pancreatic stellate cell proliferation in vitro and associated pancreatic fibrosis in vivo. Bioorg Med Chem. 2014;22:3393–3404. doi: 10.1016/j.bmc.2014.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Chen H, Yang Z, Ding C, Xiong A, Wild C, Wang L, Ye N, Cai G, Flores RM, Ding Y, Shen Q, Zhou J. Discovery of potent anticancer agent HJC0416, an orally bioavailable small molecule inhibitor of signal transducer and activator of transcription 3 (STAT3) Eur J Med Chem. 2014;82:195–203. doi: 10.1016/j.ejmech.2014.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Chen H, Gao Y, Wu J, Chen Y, Chen B, Hu J, Zhou J. Exploring therapeutic potentials of baicalin and its aglycone baicalein for hematological malignancies. Cancer Lett. 2014;354:5–11. doi: 10.1016/j.canlet.2014.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Chen H, He G, Li C, Dong L, Xie X, Wu J, Gao Y, Zhou J. Development of a Concise Synthetic Approach to Access Oroxin A. RSC Adv. 2014;4:45151–45154. doi: 10.1039/C4RA08573F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Chen H, Khemtong C, Yang X, Chang X, Gao J. Nanonization strategies for poorly water-soluble drugs. Drug Discov Today. 2011;16:354–360. doi: 10.1016/j.drudis.2010.02.009. [DOI] [PubMed] [Google Scholar]
- [61].Merisko-Liversidge E, Liversidge GG. Nanosizing for oral and parenteral drug delivery: a perspective on formulating poorly-water soluble compounds using wet media milling technology. Adv Drug Deliv Rev. 2011;63:427–440. doi: 10.1016/j.addr.2010.12.007. [DOI] [PubMed] [Google Scholar]
- [62].Yang G, Yang T, Zhang W, Lu M, Ma X, Xiang G. In vitro and in vivo antitumor effects of folate-targeted ursolic Acid stealth liposome. J Agric Food Chem. 2014;62:2207–2215. doi: 10.1021/jf405675g. [DOI] [PubMed] [Google Scholar]
- [63].Xia Y, Wei G, Si D, Liu C. Quantitation of ursolic acid in human plasma by ultra performance liquid chromatography tandem mass spectrometry and its pharmacokinetic study. J Chromatogr B Analyt Technol Biomed Life Sci. 2011;879:219–224. doi: 10.1016/j.jchromb.2010.11.037. [DOI] [PubMed] [Google Scholar]
- [64].Jain RK, Stylianopoulos T. Delivering nanomedicine to solid tumors. Nat Rev Clin Oncol. 2010;7:653–664. doi: 10.1038/nrclinonc.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Parhi P, Mohanty C, Sahoo SK. Nanotechnology-based combinational drug delivery: an emerging approach for cancer therapy. Drug Discov Today. 2012;17:1044–1052. doi: 10.1016/j.drudis.2012.05.010. [DOI] [PubMed] [Google Scholar]
- [66].Zhong Y, Dai Z, Xu Y, Teng Y, Wu B. Synthesis, stability and pharmacological evaluation of a novel codrug consisting of lamivudine and ursolic acid. Eur J Pharm Sci. 2012;45:110–115. doi: 10.1016/j.ejps.2011.10.028. [DOI] [PubMed] [Google Scholar]
- [67].Liu J. Pharmacology of oleanolic acid and ursolic acid. J Ethnopharmacol. 1995;49:57–68. doi: 10.1016/0378-8741(95)90032-2. [DOI] [PubMed] [Google Scholar]
- [68].Liu J. Oleanolic acid and ursolic acid: research perspectives. J Ethnopharmacol. 2005;100:92–94. doi: 10.1016/j.jep.2005.05.024. [DOI] [PubMed] [Google Scholar]
- [69].Chen H, Zhou X, Wang A, Zheng Y, Gao Y, Zhou J. Evolutions in fragment-based drug design: the deconstruction-reconstruction approach. Drug Discov Today. 2014 doi: 10.1016/j.drudis.2014.09.015. DOI: 10.1016/j.drudis.2014.1009.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Naus PJ, Henson R, Bleeker G, Wehbe H, Meng F, Patel T. Tannic acid synergizes the cytotoxicity of chemotherapeutic drugs in human cholangiocarcinoma by modulating drug efflux pathways. J Hepatol. 2007;46:222–229. doi: 10.1016/j.jhep.2006.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]